
Abstract
The PDCD1-encoded immune checkpoint receptor PD-1 is a key tumor suppressor in T cells that is recurrently inactivated in T cell non-Hodgkin lymphomas (T-NHLs). The highest frequencies of PDCD1 deletions are detected in advanced disease, predicting inferior prognosis. However, the tumor-suppressive mechanisms of PD-1 signaling remain unknown. Here, using tractable mouse models for T-NHL and primary patient samples, we demonstrate that PD-1 signaling suppresses T cell malignancy by restricting glycolytic energy and acetyl coenzyme A (CoA) production. In addition, PD-1 inactivation enforces ATP citrate lyase (ACLY) activity, which generates extramitochondrial acetyl-CoA for histone acetylation to enable hyperactivity of activating protein 1 (AP-1) transcription factors. Conversely, pharmacological ACLY inhibition impedes aberrant AP-1 signaling in PD-1-deficient T-NHLs and is toxic to these cancers. Our data uncover genotype-specific vulnerabilities in PDCD1-mutated T-NHL and identify PD-1 as regulator of AP-1 activity.
Similar content being viewed by others


Main
T cell non-Hodgkin lymphomas (T-NHLs) represent a heterogeneous group of highly aggressive cancers that typically originate from mature CD4+ T cells1. The therapeutic options for these malignancies are limited, which is largely due to their ill-defined molecular pathogenesis1. However, recent genomic analyses of large cohorts of patients with T-NHL revealed numerous oncogenic, gain-of-function alterations in T cell antigen receptor (TCR) signaling pathways. In addition, PDCD1, which encodes the inhibitory immune receptor PD-1, emerged as a key tumor suppressor in T-NHL2. Inactivating mutations in PDCD1 predict an aggressive clinical phenotype, and they portend poor overall survival in patients2. In addition, anti-PD-1 checkpoint inhibitors have been correlated with the emergence of secondary T-NHLs in patients with other primary malignancies3,4. Moreover, clinical trials in patients with T-NHL have reported hyperprogression of individual T cell lymphomas, which were apparently still under PD-1 control, after anti-PD-1 treatment (NCT02631746 (refs. 5,6), NCT03075553 (ref. 7)). While all these clinical observations highlight the critical importance of inhibitory PD-1 signaling in T cell malignancies, the tumor-suppressive mechanisms of PD-1 remain unknown.
In non-transformed T cells, acute PD-1 engagement leads to inhibition of the (phosphoinositide 3-kinase (PI3K))–AKT axis8, and persistent PD-1 signaling triggers T cell exhaustion, a dysfunctional state with specific metabolic and epigenetic characteristics9,10. In addition, RNA-sequencing (RNA-seq) analysis from primary human and murine PD-1-deficient T-NHLs2 demonstrates that these tumors are characterized by distinct gene expression that is presumably also shaped by epigenetic mechanisms. Nevertheless, the molecular link between PD-1 signaling and metabolic and epigenetic reprogramming during lymphomagenesis remains unclear.
Here, we identify PD-1 as a major gatekeeper for the oncogene-triggered metabolic switch to glycolysis. We show that PDCD1 mutations enforce the induction of key metabolic molecules for glucose uptake, metabolization and generation of energy carriers. In addition, PD-1 regulates ACLY, which uses extramitochondrial citrate to fuel acetyl-CoA pools for histone acetylation and enables aberrant AP-1 activity in the tumor cells. These AP-1-inducing mechanisms are hijacked in aggressive PD-1-deficient lymphomas but not in their PD-1-competent counterparts and link PD-1 signaling to epigenetic reprogramming in T-NHL.
Results
To dissect T cell lymphoma pathogenesis, we previously engineered a genetically controllable murine model of human T-NHL based on tamoxifen-inducible irreversible expression of the T-NHL oncogene ITK-SYK, which enforces strong oncogenic TCR signaling11, together with an enhanced green fluorescent protein (eGFP) reporter in individual mature CD4+ T cells in vivo (ITK-SYKCD4-CreERT2 mice; for the experimental system, see Fig. 1a,b)11,12. While acute expression of interleukin (IL)-2-inducible T cell kinase (ITK)–spleen-associated tyrosine kinase (SYK) (together with eGFP) in otherwise unperturbed primary T cells can trigger lymphocyte proliferation for a few days, it is unable to induce overt malignancy, which requires the acquisition of additional genetic hits (Extended Data Fig. 1a)11,12. However, loss of the tumor-suppressor gene Pdcd1 is sufficient to enable immediate unrestricted clonal expansion of lymphomatous T cells upon single oncogene expression that is lethal to the host (Extended Data Fig. 1a)12. These cells can transmit the malignant disease to secondary recipients12. We leveraged this genetically tractable mouse model for human T-NHL to investigate the very early events of T cell transformation.

a, Transgenic ITK-SYK allele with eGFP reporter sequence and Cre-induced excision of the stop cassette. IRES, internal ribosome entry site; TAM, tamoxifen. b, Experimental strategy to explore early molecular events upon ITK–SYK induction in the presence or absence of Pdcd1. c, Top 20 differentially expressed genes between ITK-SYKCD4-CreERT2 (‘Pdcd1+/+’)- and ITK-SYKCD4-CreERT2;Pdcd1−/− (‘Pdcd1−/−’)-derived ITK–SYK-expressing T cells. RNA-seq was performed using spleen-derived eGFP+ T cells, sorted by flow cytometry on day 5 after tamoxifen injection (n = 4 mice per group). Expression values are normalized z scores. d, GSEA of the indicated signatures. FDR, color intensity of circles; NES, circle diameter. Blue and red indicate the group in which the signature was positively enriched; NES, normalized enrichment score; FDR, false discovery rate. e, Flow cytometry for HIF1α in ITK–SYK-expressing T cells. Spleen-derived single-cell suspensions were generated from ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice on day 5 after tamoxifen injection. Max, maximum. f, Western blot analysis from lysates of ITK–SYK-expressing eGFP+ T cells, sorted by flow cytometry from ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice on day 5 after tamoxifen injection. g, ECAR metabolic flux analysis of ITK–SYK-expressing CD4+ T cells isolated from ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice on day 5 after tamoxifen injection (n = 3 biological replicates per group). Data were normalized using total cellular protein. P, two-sided Student’s t-test. Middle line denotes the median, the top and bottom box edges denote 0.25 and 0.75 quantiles, respectively, and the whiskers denote the minimum and maximum values. h, OCR metabolic flux analysis from the same experiment as in g. P, two-sided Student’s t-test. i, Lactate concentration in cell culture supernatants. ITK–SYK-expressing eGFP+ cells were sorted by flow cytometry from ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice on day 5 after tamoxifen injection and incubated overnight in vitro (n = 3 and n = 4 biological replicates per group). Data were normalized using viable cell numbers. P, two-sided Student’s t-test. Shown are the mean ± s.d. and individual data points. c,d, Data from one experiment. e,f, Representative data from two independent experiments with two biological replicates per group. g,h, Representative data from two independent experiments with three biological replicates per group. i, Representative data from two independent experiments.
Loss of Pdcd1 enables oncogene-enforced glycolysis
To identify the potent PD-1-controlled tumor-suppressor programs that prevent T cell transformation upon oncogenic T cell signaling, we induced ITK–SYK expression in vivo in primary CD4+ T cells with and without Pdcd1 by injecting tamoxifen into ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice. Three days after ITK–SYK expression, we purified oncogene-expressing cells by flow cytometry for RNA-seq. Among the top 20 differentially expressed genes, Hif1a, encoding a master regulator of cellular metabolism, was specifically upregulated in the absence of Pdcd1 (Fig. 1c; adjusted P < 0.0001). In addition, gene set enrichment analysis (GSEA) revealed an enrichment of multiple gene expression signatures that indicate global activation of the PI3K–AKT–mammalian target of rapamycin (mTOR)–hypoxia-inducible factor (HIF)1α axis13 upon PD-1 loss. Furthermore, enrichment of multiple gene expression signatures suggests enhanced glucose metabolism in the PD-1-deficient lymphoma cells compared to their premalignant ITK–SYK+PD-1+ counterparts (Fig. 1d). These gene expression data were corroborated by protein expression analysis of HIF1α and several enzymes that mediate glycolysis, including hexokinase 2, phosphofructokinase 1, aldolase A and enolase 1 in ex vivo isolated, acute oncogene-expressing T cells from tamoxifen-injected ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice (Fig. 1e,f)14,15.
Based on these findings, we next performed direct ex vivo functional analyses of glucose metabolism of acute ITK–SYK-expressing T cells with and without PD-1 function. Upon oncogenic T cell signaling, we detected enhanced glucose uptake in PD-1-deficient T cells compared to that of wild-type cells (Extended Data Fig. 1b; P = 0.0017). Moreover, extracellular flux analysis revealed a greater extracellular acidification rate (ECAR) in transformed ITK–SYK+PD-1− T cells than in their premalignant ITK–SYK+PD-1+ T cell counterparts, whereas the oxygen-consumption rate (OCR) remained unchanged (Fig. 1g,h; P < 0.0001 and P, not significant (NS)). Because the increase in ECAR implies aerobic glycolytic conversion of pyruvate to lactate, we next measured the production of this metabolite. Lactate generation was enhanced in the transformed ITK–SYK+PD-1− T cells compared to in their premalignant counterparts (Fig. 1i; P = 0.034). To study glucose metabolism in vivo, we used the glucose analog tracer [18F]fluorodeoxyglucose ([18F]FDG) and positron emission tomography (PET). The maximum [18F]FDG uptake in individual mice was measured in the spleens of both ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice (Fig. 2a and Extended Data Fig. 2a). The overall [18F]FDG uptake and maximum uptake in single voxels was significantly higher in ITK-SYKCD4-CreERT2;Pdcd1−/− animals than in PD-1-proficient ITK-SYKCD4-CreERT2 mice, demonstrating augmented glucose uptake in vivo (Fig. 2b; P = 0.005). To measure glycolytic conversion, we infused hyperpolarized [1-13C]pyruvate intravenously into tamoxifen-injected animals and compared [1-13C]lactate/[1-13C]pyruvate signal ratios in the spleens using hyperpolarized 13C magnetic resonance spectroscopic imaging (MRI)16. The [1-13C]lactate/[1-13C]pyruvate signal ratio was substantially higher in oncogene-expressing ITK-SYKCD4-CreERT2;Pdcd1−/− animals than in ITK-SYKCD4-CreERT2 mice, confirming enhanced lactate production in vivo (Fig. 2c,d and Extended Data Fig. 2a,b; P = 0.0284).

a, Whole-body standardized uptake value (SUV) maps from [18F]FDG PET measurements of C57BL/6 mice, ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice together with anatomical MRI acquisition. The [18F]FDG PET measurements were conducted on day 5 after tamoxifen application. Magnified [18F]FDG PET signals from lymph nodes are shown (insets). White arrows indicate spleens. Color intensity represents the calculated uptake of [18F]FDG in units of MBq ml−1. b, SUVmax for the indicated genotypes (n = 3 for C57BL/6, n = 9 for Pdcd1+/+ and n = 6 for Pdcd1−/− mice) from the experiment shown in a. SUVmax denotes the maximum signal found in a single voxel across the entire tumor lesion. P, one-way ANOVA and two-sided Student’s t-test. Shown are the mean ± s.e.m. and individual data points. c, Exemplary 7-T MRI chemical shift imaging (CSI)-derived 13C magnetic resonance spectra showing [1-13C]pyruvate and [1-13C]lactate amplitudes in the splenic region of interest of ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice following injection of hyperpolarized [1-13C]pyruvate on day 5 after tamoxifen injection. AU, arbitrary units. d, Ratio of [1-13C]pyruvate and [1-13C]lactate amplitudes in spleens measured by 7-T MRI in ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice on day 5 after tamoxifen injection (n = 3 mice per genotype). P, two-sided Student’s t-test. Shown are the mean ± s.d. and individual data points. e, Flow cytometry for p-AKTS473, p-mTORS2448, HIF1α, GLUT1 and hexokinase 2 in ITK–SYK-expressing T cells. Spleen-derived single-cell suspensions were generated from ITK-SYKCD4-CreERT2 mice on day 5 after tamoxifen injection. Before fixation, anti-PD-L1 or control antibodies were added for 4 h in vitro. f, Survival of ITK–SYK+PD-1− lymphoma-bearing NSG mice treated with torin-1 or vehicle. On day 0, NSG mice received 1,000 ITK–SYK-expressing eGFP+ cells from an ITK-SYKCD4-CreERT2;Pdcd1−/− mouse, sorted by flow cytometry on day 5 after tamoxifen injection. Mice received 6 mg per kg torin-1 or vehicle daily via intraperitoneal injection (n = 6 mice per group) from day 3 post tamoxifen on. P, two-sided log-rank test. a, Representative mice from two independent experiments. b, Pooled data from two independent experiments. c, Representative amplitudes from one experiment. d, Pooled data from two independent experiments. e, Representative data from two independent experiments with two biological replicates per group. f, Pooled data from two independent experiments.
Altogether, these sets of genetically controlled in vivo and ex vivo experiments demonstrate that deficiency in PD-1 forces T cells with oncogenic signaling to adapt their metabolism to aerobic glycolysis. This is characteristic of the Warburg effect that promotes the initiation and progression of most cancer types17,18,19.
PD-1 represses mTOR and HIF1α in premalignant cells
To determine how PD-1 inactivation promotes glycolysis, we acutely blocked PD-1 function in ITK–SYK-expressing T cells with anti-programmed cell death ligand 1 (PD-L1) checkpoint inhibitors as experimental tools in vivo or ex vivo. This strategy allowed us to control the inactivation of PD-1 signaling in oncogene-expressing T cells in a time-dependent manner. Similar to genetic deletion of Pdcd1, injection of anti-PD-L1 antibody into tamoxifen-treated ITK-SYKCD4-CreERT2 mice induced unrestricted expansion of oncogene-expressing T cells, which rapidly killed the host (Extended Data Fig. 2c)12. These acutely PD-1-inhibited cells also switched to glycolysis with an increase in ECAR and no change in their basal OCR (Extended Data Fig. 2d–f; P < 0.0001 and P, NS). Intracellular flow cytometric analysis directly after acute PD-1 blockade demonstrated that inactivation of the PD-1 signal resulted in prompt activation of the AKT and mTOR pathways within oncogene-expressing T cells (Fig. 2e) and in direct upregulation of HIF1α expression20. Furthermore, we observed an induction of HIF1α transcriptional targets, including the glucose transporter GLUT1 and hexokinase 2, which are rate-limiting factors for glucose uptake and glucose utilization, respectively, in normal and malignant lymphocytes (Fig. 2e)21. Thus, these key metabolic switches in premalignant cells are under the direct negative control of PD-1 tumor-suppressor signaling.
To test the dependency of fully transformed lymphomas with Pdcd1 deletion on the released mTOR–HIF1α–glycolysis cascade, we incubated ITK–SYK+PD-1− cells with small-molecule inhibitors of mTOR, HIF1α or glycolysis22,23. Direct inhibition of glycolysis with 2-deoxy-d-glucose (2-DG) reduced the production of lactate, similar results were observed with the inhibition of mTOR or HIF1α (Extended Data Fig. 3a–c), and all three treatments were toxic to PD-1-deficient lymphoma cells (Extended Data Fig. 3d–f). Moreover, treatment of ITK–SYK+PD-1− lymphoma-bearing mice with the mTOR inhibitor torin-1, which blocks mTOR activity within tumor cells in vivo (Extended Data Fig. 3g), significantly prolonged survival (Fig. 2f and Extended Data Fig. 3h; P = 0.0057). Together, these pharmacological studies demonstrate that glycolytic reprogramming is key for the transformation and survival of oncogene-expressing PD-1-deficient T cells.
PD-1 limits oncogene-triggered energy metabolism
In general, cancer cells use the metabolism of glucose to generate ATP for energy supply, rapidly assimilate biomass and generate signaling molecules that can regulate gene expression18. To identify glycolysis-dependent lymphoma-enabling mechanisms in an unbiased manner, we incubated acute oncogene-expressing ITK–SYK+PD-1− T cells and their premalignant ITK–SYK+PD-1+ counterparts with uniformly labeled [U-13C]glucose ex vivo and performed LC–MS/MS analysis to trace [U-13C]glucose-derived metabolites24. The amount of 13C-labeled glycolysis intermediates, fructose 1,6-bisphosphate, dihydroxyacetone phosphate, glyceraldehyde 3-phosphate, 3-phosphoglycerate, phosphoenolpyruvate and lactate, was elevated in PD-1-deficient lymphoma cells (Fig. 3a), indicating enhanced glucose usage within the canonical upper glycolytic pathway. This pathway is particularly important for energy production25. Indeed, inactivation of PD-1 signaling boosts ATP levels (Fig. 3b; P < 0.0003), indicating that loss of PD-1 function can overcome the energy deficit in premalignant T cells that is required to fuel overt malignancy.

a, Intracellular 13C abundance within the glycolysis pathways detected by targeted LC–MS/MS relative to intracellular [U-13C]glucose in ITK–SYK+ T cells with or without PD-1, cultured with uniformly labeled [U-13C]glucose for 3 h in vitro. Data are normalized to viable cell number and z scores per metabolite. Min, minimum. b, Time-dependent ATP measurement in ITK–SYK+ T cells with or without PD-1. P, two-sided Student’s t-test. Data are presented as the mean ± s.e.m. and individual data points for each animal. The cross indicates death of the animal. c, Ion count for intracellular 13C-labeled metabolites in the pentose phosphate pathway (PPP). d, Ion count for intracellular 13C-citrate (m + 2) isotopomers. P, two-sided Student’s t-test. Shown are the mean ± s.d. and individual data points. e, Western blot analysis of acetylated histones in ITK–SYK+ T cells with or without PD-1. H4ac, H4 acetylation. f, Flow cytometry for H3K27ac in wild-type CD4+ or ITK–SYK+ T cells after a 4-h in vitro incubation with different glucose concentrations. r, Pearson’s correlation coefficient. Data are presented as mean ± s.d. and individual data points. g, Flow cytometry for H3K27ac in ITK–SYK+PD-1− cells after a 4-h in vitro culture with 2-DG (1 mM) or DMSO. P, paired two-sided Student’s t-test. Data are presented as mean ± s.d. h, Levels of [U-13C]glucose-derived intracellular [1,2-13C]acetyl-CoA determined by targeted LC–MS/MS in ITK–SYK+ T cells with or without PD-1 cultured with uniformly labeled [U-13C]glucose (11 mM) for 4 h in vitro (n = 4 mice per genotype). Data were normalized as in a. P, two-sided Student’s t-test. Data are presented as mean ± s.e.m. i, Levels of [U-13C]glucose-derived 13C-H3K27ac (m + 2) determined by orbitrap LC–MS/MS. Experiment as in h (with n = 3 mice per genotype). Data were normalized as in a. P, two-sided Student’s t-test. Data are presented as mean ± s.d. a–d, Representative data from two independent experiments with n = 3 biological replicates per group. e, Representative data from two independent experiments with two biological replicates per group. f, Representative data from two independent experiments. g, Representative data from two independent experiments with three biological replicates per group. h, Representative data from one experiment with four biological replicates per group. i, Representative data from one experiment with three biological replicates per group.
In addition, we detected increased glucose utilization in the pentose phosphate pathway in ITK–SYK+PD-1− lymphoma cells, with augmented generation of [U-13C]glucose-derived glucono-lactone 6-phosphate, 6-phospho-d-gluconate, ribose 5-phosphate, sedoheptulose 7-phosphate and erythrose 4-phosphate (Fig. 3c). The overall abundance of [U-13C]glucose-derived tricarboxylic acid cycle (TCA) metabolites only slightly differed between transformed ITK–SYK+PD-1− T cells and their premalignant ITK–SYK+PD-1+ counterparts, although the specific levels of citrate (m + 2) showed a more than twofold increase in the absence of PD-1 (Fig. 3d; P = 0.0045).
PD-1 facilitates glycolysis-dependent histone acetylation
There is increasing evidence that metabolite abundance is also highly relevant for regulation of the tumor epigenome26,27. In particular, histone acetylation is extremely sensitive to the availability of extramitochondrial acetyl-CoA28. In this context, glucose-derived citrate, after export from the mitochondrion, is converted by ACLY to oxalacetate and cytosolic acetyl-CoA, which is subsequently used by acetyltransferases to mediate the acetylation of target proteins. After entering the nucleus, this pool of acetyl-CoA is critical for histone acetyltransferases to mediate histone acetylation for chromatin opening29,30. Because these pathways can link glucose metabolism to epigenetic regulation, which is frequently altered in T cell malignancy1, we next studied the effects of PD-1 inactivation on histone acetylation in ITK–SYK oncogene-expressing primary CD4+ T cells. Intriguingly, upon oncogenic T cell signaling, both the genetic deletion of Pdcd1 and acute pharmacological blockade of PD-1 triggered a significant increase in histone H4 and H3 lysine 27 (H3K27) acetylation (H3K27ac; Fig. 3e and Extended Data Fig. 4a). Ex vivo incubation of ITK–SYK+PD-1− lymphoma cells with glucose at increasing concentrations demonstrated that the level of H3K27ac depends directly on glucose availability (Fig. 3f; r = 0.9339)31. Conversely, inhibition of glycolysis with 2-DG resulted in a reduction in H3K27ac in PD-1-deficient lymphoma cells (Fig. 3g; P = 0.0003)32, demonstrating a link between PD-1 inactivation and glucose-dependent histone acetylation in oncogene-expressing T cells.
To explore whether this link involves glycolysis-dependent de novo generation of acetyl-CoA, we directly incubated ITK–SYK-expressing T cells from tamoxifen-injected ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice ex vivo with [U-13C]glucose and studied acetyl-CoA production by targeted LC–MS/MS analysis. Indeed, in the transformed ITK–SYK+PD-1− T cells, there was a significant increase in [1,2-13C]acetyl-CoA and total acetyl-CoA ([1,2-12C]acetyl-CoA + [1,2-13C]acetyl-CoA) generation (Fig. 3h and Extended Data Fig. 4c; P = 0.0002 and P = 0.0206), whereas the levels of [1,2-12C]acetyl-CoA did not differ (Extended Data Fig. 4b; P, NS). Next, we isolated histones from these oncogene-expressing primary T cells incubated with [U-13C]glucose for high-resolution LC–MS/MS analysis. We observed higher de novo acetylation of histones H3 and H4 with [U-13C]glucose-derived [13C]acetyl marks at H3K27, H3 lysine 9 (H3K9), H3 lysine 14 (H3K14), H3 lysine 18 (H3K18) and H3 lysine 23 (H3K23) in transformed ITK–SYK+PD-1− T cells than in PD-1-proficient ITK–SYK+PD-1+ cells (Fig. 3i and Extended Data Fig. 4d; P = 0.047)33. These data indicate that histone acetylation requires glycolysis-dependent acetyl-CoA synthesis after PD-1 inactivation in T cells with oncogenic T cell signaling.
ACLY is a critical effector molecule downstream of PD-1
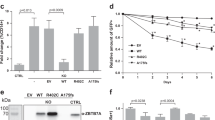
As indicated above, ACLY is the key enzyme that mediates the generation of glucose-derived extramitochondrial acetyl-CoA (Fig. 4a)34. To explore the function of ACLY in PD-1-regulated histone acetylation, we treated tamoxifen-injected ITK-SYKCD4-CreERT2 mice with anti-PD-L1 or isotype control antibodies and isolated oncogene-expressing CD4+ T cells. Next, we incubated these cells in vitro with the small-molecule ACLY inhibitor BMS-303141 for 3 h32. This treatment reduced histone acetylation in ITK–SYK+ cells in comparison to the dimethyl sulfoxide (DMSO) vehicle control (Fig. 4b). Notably, the dependence of H3K27ac on ACLY function was significantly higher in ITK–SYK-expressing cells with pharmacologically inactivated PD-1 pathway (Fig. 4b; P = 0.0131). Consistent with published results, exogenous supplementation of acetate at physiological levels (100 µM) could not restore decreased H3K27ac signals in ACLY inhibitor-treated lymphoma cells (Extended Data Fig. 4e).

a, Schematic of glucose flux to histone acetyl groups. b, Flow cytometry for H3K27ac in ITK–SYK+ cells after 4 h of in vitro culture with the ACLY inhibitor BMS-303141 (‘iACLY’, 15 μM) or DMSO. ITK-SYK+ cells were sorted by flow cytometry from ITK-SYKCD4-CreERT2 mice on day 5 after tamoxifen injection and 12 h of anti-PD-L1 or isotype control antibody treatment (intraperitoneal, 200 μg). P, paired two-sided Student’s t-test. Data are presented as the mean ± s.d. c, Western blot analysis in lysates from ITK–SYK+PD-1+ cells. Spleen-derived single-cell suspensions were sorted by flow cytometry from ITK-SYKCD4-CreERT2 mice on day 5 after tamoxifen injection and cultured in vitro overnight with anti-PD-L1 or isotype control antibodies and the PI3K inhibitors (‘iPI3K’) wortmannin (20 nM), LY294002 (20 μM) or DMSO. d, Flow cytometry for CD4 and eGFP in viable lymphocytes derived from spleens of acutely induced ITK-SYKCD4-CreERT2;Pdcd1−/− mice after 3 d of in vitro culture with BMS-303141 (5 μM) or DMSO. e, Fold change in viable ITK–SYK-expressing eGFP+ cells in medium supplemented with BMS-303141 (5 μM) or DMSO. Spleen-derived lymphocytes, sorted by flow cytometry from ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice on day 5 after tamoxifen injection, were incubated for 3 d in vitro. P, two-sided Student’s t-test. Data are presented as mean ± s.d. f, Western blot for ACLY in ITK–SYK+PD-1− cells electroporated with Acly-targeting (sgACLY) or non-targeting control (sgMOCK)RNP complexes. Protein lysates were generated after 48 h of in vitro culture. g, Survival of recipient mice transplanted with Acly (‘sgACLY’) or mock-edited (‘sgMOCK’) ITK–SYK+PD-1− cells. P, two-sided log-rank test. h, Gene-wise global H3K27ac scores from anti-H3K27ac ChIP–seq in a 1.5-kb window around the TSS. ITK–SYK+ cells were sorted by flow cytometry from ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice on day 5 after tamoxifen injection, fixed and processed for anti-H3K27ac ChIP–seq analysis. b, Representative data from three independent experiments with three biological replicates per group. c, Representative data from two independent experiments with two biological replicates. d,e, Representative data from three independent experiments. f,g, Data from one experiment with n = 5 mice per group. h, Data from a single experiment with two biological replicates per group.
The activity of ACLY itself can in principle be regulated by AKT, which can phosphorylate ACLY at serine 455, a key point of control for this enzyme35,36,37,38,39. As PD-1 inactivation results in (PI3K)–AKT signaling in oncogene-expressing T cells (Fig. 2e), we hypothesized that these events could additionally trigger ACLY activation39. To test this hypothesis, we induced ITK–SYK expression in CD4+ cells by injecting tamoxifen into ITK-SYKCD4-CreERT2 mice and acutely blocked PD-1 function with anti-PD-L1 treatment. Inactivation of PD-1 signaling resulted in direct activation of ACLY, as measured by phosphospecific phosphorylated (p)-ACLYS455 antibodies (Fig. 4c), and this phosphorylation could be reversed by PI3K inhibition with wortmannin or LY294002 (Fig. 4c)40.
To determine whether ACLY activity is required for PD-1-deficient lymphoma cell survival, we next incubated transformed ITK–SYK+PD-1− T cells from tamoxifen-injected ITK-SYKCD4-CreERT2;Pdcd1−/− mice with an ACLY inhibitor. This treatment killed ITK–SYK+PD-1− lymphoma cells (Extended Data Fig. 4f; P = 0.0037). Importantly, co-cultures with splenocytes demonstrated that ITK–SYK+PD-1− T cells were highly sensitive to ACLY inhibition, whereas ITK–SYK+PD-1+ T cells and normal T cells were less affected (Fig. 4e,f; P = 0.019 and P = 0.010).
To genetically define whether ACLY is necessary for PD-1-deficient lymphoma growth in vivo, we disrupted Acly in transformed ITK–SYK+PD-1− T cells using CRISPR–Cas9-mediated gene editing and transplanted these cells into syngeneic wild-type hosts (Fig. 4f). While the control-edited ITK–SYK+PD-1− cells proliferated rapidly in vivo, as measured by eGFP monitoring, and killed all recipient animals as expected in less than 80 d, ACLY-deficient ITK–SYK+PD-1− T cells were unable to expand (Extended Data Fig. 4g and Fig. 4i; P = 0.0072). The acetyl-CoA pool can be used for both histone acetylation and other acetyl-CoA dependent pathways such as fatty acid synthesis. To distinguish the importance of these pathways, we used pharmacological inhibitors. Our experiments demonstrated that ITK–SYK+PD-1− cells were more sensitive to direct inhibition of histone acetylation with inhibitors of histone acetyltransferases p300 and CBP than their PD-1-competent counterparts. By contrast, inhibition of fatty acid synthase did not reveal a significant difference (Extended Data Fig. 4h,i; P = 0.0156).
Together, these functional and genetic data demonstrate that loss of PD-1 tumor-suppressor function results in enhanced ACLY activity via enforced (PI3K)–AKT signaling and that this mechanism is critical for both glucose-dependent histone acetylation and the malignant expansion of PD-1-deficient lymphomatous T cells.
PD-1 controls epigenetic reprogramming and AP-1 activity
After establishing enhanced histone acetylation as a consequence of Pdcd1 inactivation in oncogene-expressing T cells, we next tested the epigenetic effects of this mechanism on genome regulation using genome-wide assays. Because H3K27 showed the greatest dependence on glucose-dependent de novo acetylation, we immunoprecipitated H3K27-acetylated histones from ITK–SYK-expressing T cells with or without PD-1 activity and performed high-throughput chromatin immunoprecipitation followed by sequencing (ChIP–seq41). Overall, deletion of Pdcd1 increased H3K27ac in oncogene-expressing T cells, particularly within promoter regions and around gene transcription start sites (TSS), indicating direct effects on transcriptional regulation (Fig. 4h and Extended Data Fig. 4j,k)42,43. To assess how these events would affect chromatin accessibility for transcription factor occupancy, we conducted a genome-wide assay for transposase-accessible chromatin using sequencing (ATAC-seq44) and performed transcription factor footprint analysis45,46,47. Intriguingly, in PD-1-deficient ITK–SYK-expressing T cells, we observed increased activity scores for the AP-1 family transcription factors c-FOS, FOSL1, FOSL2, c-JUN, JUNB and BATF (Fig. 5a,b). Next, we tested whether the histones at the AP-1 family target gene sequences are hyperacetylated in PD-1-deficient ITK–SYK-expressing T cells by performing GSEA using the H3K27ac ChIP–seq dataset (Fig. 4h; ChIP-Enrich analysis48) and the transcription factor target gene sets from the ENCODE, JASPAR and ChEA databases46,49,50,51. We detected a significant enrichment of c-FOS, FOSL1, FOSL2, c-JUN, and BATF target genes in H3K27ac ChIP–seq peaks upregulated in ITK–SYK+PD-1− lymphoma cells compared to their premalignant ITK–SYK+PD-1+ counterparts (Fig. 5c). Furthermore, global expression of AP-1 target mRNA species also increased with positive enrichment of the c-FOS, FOSL1, FOSL2, c-JUN and BATF gene sets in transformed ITK–SYK+PD-1− cells (Fig. 5d).

a, Differential transcription factor footprint analysis of ATAC-seq data. ITK–SYK+ cells were sorted by flow cytometry from ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice on day 5 after tamoxifen injection and immediately processed for ATAC-seq. JASPAR database motif names for the selected AP-1 family transcription factors are indicated. Enrichment of a transcription factor motif is indicated by motif score, motif rank and color. b, Binding profiles of selected transcription factors for the indicated genotypes from the experiment shown in a. c, ChIP-Enrich analysis of ChIP–seq data shown in Fig. 4h for the indicated gene sets. FDR, color intensity of the circles; ChIP–seq peaks in gene set, circle diameter. Blue and red indicate the genotype in which a signature was positively enriched. d, GSEA for the indicated gene sets. FDR, color intensity of circles; NES, circle diameter. Blue and red indicate the group in which a signature was positively enriched. e, Phosflow analysis of p-c-FOSS32 in ITK–SYK+ cells. Single-cell suspensions were generated from lymph nodes of ITK-SYKCD4-CreERT2 mice on day 5 after tamoxifen injection and 12 h of anti-PD-L1 or isotype control antibody treatment (intraperitoneal, 200 μg). P, paired two-sided Student’s t-test. Data are presented as mean ± s.d. f, Phosflow analysis of p-c-JUNS73. Same experiment as in e. g,h, Differential transcription factor footprint analysis of ATAC-seq data from ITK–SYK+PD-1+ and ITK–SYK+PD-1- cells after 3 h of in vitro incubation with BMS-303141 (5 μM, h) or DMSO (g). Spleen-derived eGFP+ cells were sorted by flow cytometry from ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice on day 5 after tamoxifen injection, incubated in iACLY- or DMSO-containing medium and processed for ATAC-seq. Legend as in a. a,b, Data from a single experiment with three biological replicates per genotype. c, Data from one experiment with two biological replicates per group. d, Data from a single experiment with four biological replicates per genotype. e,f, Representative data from two independent experiments with three biological replicates per group. g,h, Data from one experiment with three biological replicates per genotype. var., variant.
To investigate the underlying mechanism for AP-1 hyperactivation, we focused on the top-ranking motif in PD-1-deficient lymphoma cells, which was the c-FOS–c-JUN heterodimer (Fig. 5a). We performed additional ex vivo experiments to determine the PD-1-dependent phosphorylation status of c-FOS at serine 32 and of c-JUN at serine 73, respectively. These post-translational modifications are known to increase protein stability, nuclear localization and transcriptional activity52,53. To capture immediate changes upon PD-1 pathway inactivation, we used pharmacological blockade of PD-1 as introduced above (Fig. 2e). Our Phosflow data revealed a significant increase in p-c-FOSS32 and p-c-JUNS73 signals in ITK–SYK-expressing cells isolated from animals that had been treated with anti-PD-L1 checkpoint inhibition, suggesting that PD-1 indeed controls the transduction of oncogenic signals to AP-1 family members (Fig. 5e,f; P = 0.0317 and P = 0.0412).
We asked whether AP-1 hyperactivity in ITK–SYK+PD-1− lymphoma cells is induced by ACLY activity and subsequently inflated acetyl-CoA pools. To address this question, we again isolated ITK–SYK+PD-1+ and ITK–SYK+PD-1− cells from tamoxifen-treated ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− animals. This time, we incubated the cells in vitro in the presence of the ACLY inhibitor BMS-303141 or DMSO for 3 h. Next, we sorted eGFP+ cells by flow cytometry and performed ATAC-seq and transcription factor footprint analysis. In agreement with the ex vivo results (Fig. 5a), we detected AP-1 family members as top motifs enriched in PD-1-deficient cells (Fig. 5g). Strikingly, upon pharmacological inhibition of ACLY, AP-1 footprints were de-enriched in Pdcd1-deleted cells in comparison to their counterparts with intact PD-1 pathway (Fig. 5h).
Altogether, these results from our genetically defined mouse models demonstrate that the loss of PD-1 signaling unleashes ACLY activity and thereby triggers glucose-dependent production of extramitochondrial acetyl-CoA to mediate de novo acetylation of histones to open chromatin and enable the enhanced activity of oncogenic AP-1 family transcription factors. Importantly, AP-1 hyperactivation in Pdcd1-deleted lymphoma cells was critically dependent on ACLY activity. Thus, our results indicate a link between glucose-derived acetyl-CoA availability and selective opening of compact chromatin at AP-1-binding sites, which is mediated by the PD-1 pathway (for a schematic model, see Extended Data Fig. 5a).
Glycolysis and AP-1 activation in human PDCD1-mutated T-NHL
To elucidate whether these PD-1-dependent metabolic and epigenetic changes are also present in human T cell lymphoma, we analyzed leukemic stage primary patient samples from cohorts of clinically annotated T-NHLs of cutaneous origin by isolating malignant lymphoma cells by flow cytometry. We first examined a cohort of patients who experienced rapid disease progression, where the blood tumor burden increased by >400% in less than a 1-month period, which we termed ‘hyperprogression’ (Fig. 6a). TCR clonotyping confirmed that post-hyperprogression samples originated from identical T cell clones as the pre-hyperprogression samples (Extended Data Fig. 5b). Intriguingly, in all three instances, hyperprogressive disease was associated with profound downregulation of PDCD1 transcripts (Fig. 6b,c; P = 4.6 × 10−4, log2 (fold change) = −3.3). Notably, one of these cancers acquired a genetic deletion of PDCD1 upon disease progression (Fig. 6d), whereas the others lost PDCD1 expression by still uncharacterized mechanisms. These pre- and post-hyperprogression lymphomas were used for comparative RNA-seq and GSEA analysis. Consistent with our mouse model, we detected a significant enrichment of all signatures that represent enhanced activity of the (PI3K)–AKT–mTOR pathway, enforced HIF1α activity and increased glucose metabolism in post-hyperprogression samples with impaired PDCD1 expression compared to pre-hyperprogression samples (Fig. 6e).

a, Serial longitudinal analysis of the tumor burden index and white blood cell (WBC) count for three individual patients with CTCL and hyperprogressive disease. Hyperprogression, rapid increase in tumor burden index (‘after’, shaded in red) after an extended period of stable disease course (‘before’, shaded in blue). b, Differential gene expression analysis of RNA-seq data derived from the three patients with CTCL in a before and after hyperprogression. Data points for selected glycolysis-related genes and PDCD1 are indicated. P, two-sided Wald test. c, RNA-seq read count of PDCD1 transcripts before and after hyperprogression shown for each patient individually. d, Whole-genome sequencing-based copy number aberration analysis of PDCD1-containing chromosomal region 2q36.3–2q37.3. The time point of genomic DNA isolation relative to hyperprogression is indicated (‘before’ versus ‘after’). The patient shown corresponds to the first patient in a. Top: the dashed box indicates the genomic region on human chromosome 2 that is shown in detail at the bottom. Bottom: copy number aberration analysis. The location of the PDCD1 locus is shown. e, GSEA for the indicated gene sets based on RNA-seq data derived from three patients with hyperprogression (a). The time point of RNA extraction relative to hyperprogression is indicated (‘before’ versus ‘after’). FDR, color intensity of circles; NES, circle diameter. Blue and red indicate the group in which a signature was positively enriched. a, Longitudinally collected data from three individual patients. b–e, Data from a single experiment with three patients.
In addition, we studied a second cohort of patients with cutaneous T cell lymphoma (CTCL) that were either PDCD1 wild type or PDCD1 mutant at initial diagnosis. In line with previously reported frequencies2,12, we detected PDCD1 deletions in seven of the 21 specimens (Fig. 7a). RNA-seq and GSEA revealed a significant enrichment of the key gene sets for (PI3K)–AKT–mTOR signaling and HIF1α activation (Fig. 7b) in PDCD1-mutant patients. Again, we also observed enhanced activity of glycolysis gene sets in PDCD1-mutant lymphomas compared to PDCD1-wild-type lymphomas (Fig. 7b). None of the analyzed primary patient samples harbored the rare ITK–SYK fusion oncogene that we used in the mouse to model oncogenic TCR signaling. Instead, PDCD1-mutant human tumors harbored diverse and numerous oncogenic TCR signaling mutations including in CD28, PLCG1 and RHOA, suggesting that transcriptional effects are not tied to the identity of the oncogene (Extended Data Fig. 6a). Interestingly, multiple PDCD1-mutant lymphomas carried mutations in another tumor suppressor encoded by CDKN2A (Extended Data Fig. 6a).

a, Genomic region within q37.3 on chromosome 2 in 21 patients with CTCL. CNA, copy number aberrations. PDCD1mut, PDCD1 mutant; PDCD1WT, PDCD1 wild type. b, GSEA for the indicated gene sets based on RNA-seq data from the 21 patients with CTCL shown in a. c, Phosflow analysis of serine 240 and 244 of S6 ribosomal protein in PDCD1-mutant (n = 3) and PDCD1-wild-type (n = 3) primary CTCL cells and in CD4+ T cells from healthy donors (n = 3). P, one-way ANOVA and two-sided Student’s t-test. Shown are the mean ± s.d. and individual data points. d, Uptake of the glucose analog 2-NBDG in PDCD1-mutant (n = 3) and PDCD1-wild-type (n = 3) primary CTCL cells and in CD4+ T cells from healthy donors (n = 3). Malignant cells sorted by flow cytometry were stimulated with anti-CD3 and anti-CD28 beads, and 2-NBDG uptake was determined at the indicated time points. P, two-sided Student’s t-test. Shown are the mean ± s.e.m. and individual data points. e, Relative division index of malignant T cells from three patients with mutated PDCD1 or reduced PD-1 expression after hyperprogression compared to three PDCD1-wild-type samples and healthy CD4+ T cells (n = 3) in the presence of 2-DG (1 mM), everolimus (0.1 μM) or BMS-303141 (iACLY, 10 μM). P, two-sided Student’s t-test. Data are presented as the mean ± s.e.m. and individual data points. f, Differential transcription factor footprint analysis of ATAC-seq data from three patients with CTCL with (‘PDCD1mut’) and three patients without (‘PDCD1WT’) mutated PDCD1. Legend as in Fig. 5a. g, Binding profile for the JASPAR FOS–JUN heterodimer motif for the indicated genotypes from the experiment shown in f. h, GSEA for the indicated gene sets with RNA-seq data from 21 patients with CTCL who were PDCD1 mutant or PDCD1 wild type and three patients with hyperprogression. a,b, Data from a single experiment with 21 patients. c,d, Data from one experiment with three patients per group and three healthy donors. e, Data from one experiment with three patients and three healthy donors. f,g, Data from one experiment with three patients per group. h, Data from a single experiment.
Next, we used viable primary patient samples from these two cohorts to functionally assess our findings on the (PI3K)–AKT–mTOR axis, which is under PD-1 control in normal and malignant T cells, and our discovery of ACLY as a PD-1 target molecule in T-NHL. First, we probed the impact of PDCD1 deletions on (PI3K)–AKT–mTOR signaling and glucose uptake based on six individual patients, three with mutant PDCD1 and three with wild-type PDCD1. In line with our findings in the murine model, PDCD1-mutant lymphoma cells exhibited a significant increase in S6 ribosomal protein phosphorylation, demonstrating enhanced mTOR activity compared to PDCD1-wild-type cells (Fig. 7c; P = 0.0309)54. Moreover, uptake of the fluorescent glucose analog 2-NBDG was also significantly increased in PDCD1-mutant lymphoma cells (Fig. 7d; P = 0.03)55. To perturb (PI3K)–AKT–mTOR signaling, glycolysis or ACLY activity in viable PDCD1-wild-type and PDCD1-mutant lymphoma cells, we incubated the six primary patient samples with respective small-molecule inhibitors of these pathways. Treatment with the mTORC1 inhibitor everolimus, which is currently in clinical trials for T-NHL therapy (NCT00918333 and NCT01075321), reduced viability and division of PDCD1-mutant cancer cells (Fig. 7e). Similarly, inhibition of glycolysis with 2-DG or ACLY blockage with BMS-303141 also significantly impaired cell proliferation in PD-1-deficient lymphomas (Fig. 7e). Importantly, stage-matched PD-1-wild-type tumors were resistant to everolimus, 2-DG and inhibition of ACLY. These data suggest that PD-1 loss confers therapeutic vulnerability to these inhibitors (Fig. 7e).
Finally, we assessed chromatin accessibility in primary cells from three PDCD1-wild-type and three PDCD1-mutant CTCLs by ATAC-seq and performed transcription factor footprint analysis (Fig. 7f,g). Similar to the murine model, the top transcription factor motifs most enriched in the PDCD1 mutant compared to the wild type were AP-1 family members, including c-FOS, FOSL1, FOSL2, c-JUN, JUNB and BATF, which we corroborated with functional in vitro assays (Fig. 7f,g and Extended Data Fig. 6b,c). Furthermore, at the global transcript level, both PDCD1-mutant and post-hyperprogression T-NHL samples showed a significant enrichment in c-FOS, FOSL1, FOSL2, c-JUN and BATF target genes (Fig. 7h).
Discussion
Here, we identified the key components of the PD-1 tumor-suppressor program in T cell lymphoma. Using a combination of genetic mouse models and primary human T-NHL samples, our results highlight the role of PD-1 as a critical gatekeeper molecule curtailing the glycolytic switch during neoplastic transformation of a T cell. We provide evidence that metabolic reprogramming via the (PI3K)–AKT–mTOR–HIF1α axis leads to induction of rate-limiting factors for glucose uptake and metabolization to enforce tumor cell energy production. This tumor-suppressive activity of PD-1 specifically suppresses glycolytic reprogramming but not oxidative phosphorylation.
Interestingly, our data presented here and previous work2 indicate that PD-1-mutation status could serve as a potential biomarker for genotype-specific vulnerabilities in T cell lymphomas. For example, mTOR is currently being evaluated as a target in T-NHL with heterogeneous clinical results56. We provide evidence that molecular stratification based on genetic PDCD1 status could help to identify patients who will likely respond to mTOR pathway inhibition. Moreover, our data suggest enhanced ACLY activity as a selective vulnerability in PDCD1-mutant T-NHLs. Furthermore, we describe a previously unknown link between PD-1 signaling and metabolite-controlled epigenetic reprogramming. Upon PD-1 loss, increased pools of glycolysis-derived citrate permit enhanced acetyl-CoA production. This in turn enforces opening of compact chromatin at AP-1-binding sites and enhanced transcription of AP-1 target genes. Conversely, pharmacological inhibition of ACLY curtails AP-1 hyperactivation and is toxic to PDCD1-mutant lymphomas. Notably, antigenic stimulation of non-transformed T cells also induces AP-1 activity and enforces AP-1-directed chromatin remodeling57. However, in contrast to oncogenic AP-1 activation in T-NHL, this process strictly depends on external induction of co-stimulatory pathways such as CD28 (ref. 57) through still ill-defined mechanisms.
While our results identify PD-1 as a metabolic gatekeeper for T-NHL energy metabolism and induction of the epigenetic ACLY–AP-1 axis, several questions remain open. On the one hand, it is unclear whether unleashed ACLY activity in PDCD1-mutant lymphomas regulates acetyl-CoA-dependent metabolic pathways apart from histone acetylation, similar to de novo generation of lipids. On the other hand, it is undefined how PD-1 signaling regulates individual AP-1 family members. While our data show enhanced phosphorylation of key serine residues within the transactivation domains of c-JUN and c-FOS in the absence of PD-1 signaling, the precise mechanisms that underlie these effects remain to be uncovered. In this context, it will also be critical to elucidate how acetyl-CoA abundance can selectively enhance recruitment of AP-1 factors to AP-1–DNA binding sites and whether related pathways are also active in non-transformed T cells and during T cell exhaustion, which is frequently associated with chronic PD-1 signaling and dysregulated AP-1 activity58.
In sum, our findings establish PD-1 as a central regulator for tumor cell energy metabolism and AP-1 activity in T-NHL. We demonstrate that PD-1 restricts glycolysis, glucose-derived acetyl-CoA production and chromatin remodeling. PD-1-deficient T-NHLs acquire epigenetic changes that increase access for AP-1 factors in otherwise compact DNA. This mechanism is highly conserved between human and murine lymphomas. Our results link PD-1 mutations with AP-1 hyperactivation, which has been demonstrated to be a hallmark of aggressive, drug-resistant T-NHLs59. Moreover, our data highlight the (PI3K)–AKT–mTOR–HIF1α–ACLY-axis as a genotype-specific therapeutic vulnerability in aggressive T cell lymphomas with defective PD-1 function, which should be tested in future clinical interventions.
Methods
We confirm that all experiments with mice and human patient-derived material were performed in accordance with local animal-protection guidelines (Regierung von Oberbayern, Munich, Germany) or after approval by the Northwestern University Institutional Review Board, respectively.
Mice
Mice of both sexes aged 6–12 weeks were used for all experiments. Littermate controls were used whenever possible. Randomization and blinding were not performed. ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− animals were described earlier and maintained on a C57BL/6 genetic background12. Pdcd1−/− (028276), B6J.129(Cg)-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (Cas9, 026179) and NOD.CG-Prkdcscid IL2rgtm1Wjl/SzJ (NOD–SCID gamma (NSG), 005557) mice were purchased from Jackson Laboratory. All animal experiments were performed in accordance with local guidelines (Regierung von Oberbayern, Munich, Germany). Mice were euthanized if they exhibited signs of lymphoma (lymph node enlargement, palpable tumor, labored breathing, ascites) or if they lost 20% or more of their body weight. None of the approved thresholds were exceeded at any time. Mice were kept in greenline cages (Tecniplast Typ I superlong) at temperatures between 20 and 24 °C and at 45–60% humidity, receiving pelleted food (1324SP, Altromin) and autoclaved water ad libitum. A light–dark rhythm changed every 12 h with included dimming phases. For peripheral T cell lymphomas, both sexes showed nearly identical trends in survival, although incidence rates were higher in males than in females (https://seer.cancer.gov). In accordance with this, we used both female and male mice. We indicate also that our mouse models did not reveal any differences in PD-1 expression and survival after PD-1 inactivation based upon sex. We used the same numbers of male and female mice in each experiment whenever possible.
Induction of spontaneous ITK–SYK expression in peripheral T cells
Tamoxifen (T5648, Sigma) was dissolved in pure ethanol (with heating) and subsequently diluted in migliol (3274, Caesar & Loretz) to a final concentration of 10 mg ml−1. To induce expression of ITK–SYK in peripheral CD4+ T cells, ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice were intraperitoneally injected with tamoxifen at the indicated concentrations.
RNA-seq of mouse samples
ITK-SYKCD4-CreERT2 or ITK-SYKCD4-CreERT2;Pdcd1−/− mice received a single dose of 2 mg tamoxifen. Five days later, spleens were collected, and 1,000 eGFP+ ITK–SYK-expressing CD4+ T cells from both genotypes were directly sorted (FACSAria Fusion, BD Biosciences) into a 96-well PCR plate prefilled with 10 µl of 1× TCL buffer (1070498, Qiagen) containing 1% (vol/vol) β-mercaptoethanol (M6250, Sigma). Library preparation for bulk 3′ sequencing of poly(A) RNA was performed as outlined earlier60. Briefly, for each sample, barcoded full-length complementary DNA (cDNA) was generated with Maxima RT polymerase (EP0742, Thermo) using oligonucleotide-dT primer-containing barcodes, unique molecular identifiers (UMIs) and an adaptor. The addition of a template switch oligonucleotide resulted in extension of the 5′ ends of the cDNA, and full-length cDNA was amplified with a primer binding to the template switch oligonucleotide site and the adaptor. cDNA was fragmented using the Nextera XT kit (FC-131-1096, Illumina), and only the 3′ end fragments were amplified using primers with Illumina P5 and P7 overhangs. Compared to ref. 60, the P5 and P7 sites were exchanged to allow sequencing of the cDNA in read 1 and barcodes and UMIs in read 2 to achieve better cluster recognition. The library was sequenced on the NextSeq 500 platform (Illumina) with 75 cycles for cDNA and 16 cycles for barcodes and UMIs. Raw sequencing data were processed with DropSeq-tools (version 1.12) using gene annotations from the Ensembl GRCm38.87 database to generate sample- and gene-wise UMI tables61. Downstream analysis was conducted using R version 3.4.452 and DESeq2 version 1.18.153 (ref. 62). Technical replicates with <100,000 total UMIs were excluded before differential expression analysis, and the remaining replicates were collapsed. Genes with <10 reads across all conditions were excluded. Prior differential expression analysis and dispersion of the data were estimated using a parametric fit. GSEA 4.1.0 was used to calculate enrichment for the indicated signatures. All signatures were derived from MSigDB and Harmonizome51,63,64,65.
Western blot analysis of cytosolic proteins and histones
ITK-SYKCD4-CreERT2, ITK-SYKCD4-CreERT2;Pdcd1−/− or C57BL/6 mice were intraperitoneally injected with a single dose of 2 mg tamoxifen and received 200 μg anti-PD-L1 antibody (BE0101, BioXcell) or isotype control antibody (BE0090, BioXcell), depending on the experimental condition. At the indicated time points, single-cell suspensions were generated, and eGFP+ cells were sorted by flow cytometry and lysed on ice in protein lysis buffer (50 mM Tris, 150 mM NaCl, pH 8, 1% NP-40) supplemented with protease and phosphatase inhibitors (4906845001 and 539131, Sigma). The following antibodies were used at 1:1,000 or 1:10,000 (actin) dilutions for protein detection: anti-hexokinase 2 (ab209847, Abcam), anti-phosphofructokinase 1 (PFKM; MAB7687-SP, R&D Systems), anti-aldolase A (8060, CST), anti-enolase 1 (3810T, CST), anti-actin (3700, CST), anti-histone H3 (9715, CST), anti-acetyl-histone H3 Lys27 (8173, CST), anti-histone H4 (2935, CST), anti-acetyl-histone H4 (06-598, Millipore), anti-ACLY (4332, CST), anti-p-ACLY (4331, CST), anti-AKT (4691, CST) and anti-p-AKT (4060, CST).
Analysis of p-ACLYS455
For the analysis of PD-1-dependent ACLY phosphorylation at serine 455, single-cell suspensions from the spleens of acutely induced ITK-SYKCD4-CreERT2 mice were incubated overnight with anti-PD-L1 antibody (4 μg ml−1, BE0101, BioXcell), isotype control (4 μg ml−1, BE0090, BioXcell), DMSO, wortmannin (2 μM, 12-338, Sigma) or LY294002 (10 μM, S1105, SelleckChem). The next day, cells were sorted by flow cytometry for the eGFP+ phenotype and lysed as described above.
Analysis of histones
For western blotting of histones, single-cell suspensions were generated from the spleens of acutely induced ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice. Subsequently, eGFP+ cells sorted by flow cytometry were cultured for 4 h in vitro in glucose-free DMEM (11966025, Thermo) supplemented with 1% dialyzed fetal calf serum (FCS; A3382001, Thermo) and 1 mM glucose (G8270, Sigma). Next, cells were washed with ice-cold PBS, and acid-based histone extraction was performed according to published protocols66.
Glucose assay of cell culture supernatants
ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice were intraperitoneally injected with a single dose of 2 mg tamoxifen. On day five, single-cell suspensions were generated, and eGFP+ cells were sorted by flow cytometry. Two hundred thousand T cells were cultured in vitro in glucose-free DMEM (11966025, Thermo) supplemented with 10% dialyzed FCS (A3382001, Thermo), 2 mM glutamine (25030149, Thermo) and 10 mM glucose for 3 h. The remaining glucose in the supernatants was measured using the Amplex Red Glucose/Glucose Oxidase Assay Kit (A22189, Thermo).
Seahorse assays
ECAR and OCR were measured using a Seahorse XFe96 Analyzer (Agilent) in XF medium (non-buffered RPMI 1640 medium containing 2 mM glutamine and 1 mM sodium pyruvate (11360070, Thermo) and 25 mM glucose). Two hundred thousand eGFP+ ITK–SYK-expressing T cells per well were sorted by flow cytometry and centrifuged onto poly-d-lysine (P6403, Sigma)-coated 96-well plates (101085-004, Agilent) and pre-incubated at 37 °C for 45 min in the absence of CO2. Five days before the experiment, ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice received a single dose of 2 mg tamoxifen and, depending on the experiment, intraperitoneal injections of anti-PD-L1 antibody (200 μg, BE0101, BioXcell) or isotype control (200 μg, BE0090, BioXcell) as indicated in the figure legends.
In vivo metabolic imaging using PET–CT and hyperpolarized 13C magnetic resonance imaging
C57BL/6, ITK-SYKCD4-CreERT2 or ITK-SYKCD4-CreERT2;Pdcd1−/− mice received a single dose of 2 mg tamoxifen. For PET–CT imaging, mice were injected with [18F]FDG (~12 MBq), and tracer uptake was measured after 90 min using a preclinical Siemens Inveon PET–CT system. Splenic standardized uptake values were calculated using Inveon Research Workplace software. Hyperpolarized 13C MRI of the animals was performed using a preclinical 7-T magnetic resonance scanner (Agilent–GE Discovery MR901 magnet and gradient system, Bruker AVANCE III HD electronics). The animals were injected with hyperpolarized [1-13C]pyruvate (250 µl, 80 mM, HyperSense DNP, Oxford Instruments), and both [1-13C]pyruvate and [1-13C]lactate were recorded in vivo using a static 2D FID–CSI (matrix size, 18 × 12; FOV, 30 mm × 20 mm; slice thickness, 3 mm; flip angle, 12°; number of points, 128; bandwidth, 2,000 Hz). T2-weighted images (1H-RARE; matrix size, 150 × 100; FOV, 30 mm × 20 mm; slice thickness, 1 mm) were recorded for co-registration and determination of spleen size. Peak heights for [1-13C]pyruvate versus [1-13C]lactate were determined and summed over the spleen region of interest using MATLAB code developed in house.
Intracellular flow cytometry and Phosflow of murine cells
Ex vivo
TK-SYKCD4-CreERT2 or ITK-SYKCD4-CreERT2;Pdcd1−/− mice were intraperitoneally injected with a single dose of 2 mg tamoxifen. At the indicated time points, cells were isolated and immediately fixed in 4% PFA (28906, Thermo), permeabilized using methanol, stained for CD4 (100437, BioLegend), p-AKTS473 (48-9715-42, Thermo), p-mTORS2448 (2971, CST), hexokinase 2 (ab209847, Abcam), GLUT1 (PA1-46152, Thermo), p-S6S240/S244 (5364S, CST), acetyl-histone H3 Lys27 (13027, CST), HIF1α (36169, CST), Alexa Fluor 647 p-c-FOSS63 (8677, CST) and PE p-c-JUNS73 (8752, CST) and assessed by flow cytometry. Surface and intracellular antibodies were used at dilutions of 1:300 and 1:100, respectively. The following antibodies were used at a dilution of 1:300 as secondary antibodies: PE donkey anti-rabbit IgG (406421, BioLegend) or PE goat anti-mouse IgG (405307, BioLegend). Data were acquired using a FACSCanto II or an LSRFortessa flow cytometer (BD Biosciences). FlowJo software was used for data analyses (FlowJo).
In vitro
eGFP+ T cells sorted by flow cytometry from tamoxifen-injected C57BL/6 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice were cultured in vitro in glucose-free DMEM (11966025, Thermo) supplemented with 1% dialyzed FCS (A3382001, Thermo). After 4 h, the cells were fixed in 4% PFA and processed as described above. An exemplified gating strategy is provided in Supplementary Fig. 1.
Metabolomics
ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice were intraperitoneally injected with a single dose of 2 mg tamoxifen per mouse. Five days later, spleen- and lymph node-derived single-cell suspensions were sorted by flow cytometry for the eGFP+ phenotype. Subsequently, cells were incubated in glucose-free DMEM (11966025, Thermo) supplemented with 10% dialyzed FCS (A3382001, Thermo) and 11 mM uniformly labeled [U13C]glucose (CLM-1396, Cambridge Isotope Laboratories). After 3 h of in vitro culture, cells were washed with ice-cold 0.9% NaCl and resuspended in 1 ml of ice-cold 80% methanol–water solution (vol/vol; 51140, Thermo; AE71.1, Carl Roth), including internal standards, followed by incubation for 8 h at −80 °C. The cell suspension was centrifuged at 13,000g and 4 °C for 10 min, and then the supernatants were dried in a SpeedVac concentrator (Savant, Thermo). Lyophilized samples were reconstituted in 40 μl of LC–MS-grade water, vortexed and centrifuged again for 10 min at 13,000g and 4 °C. Samples were analyzed using an Agilent 1200 series HPLC system interfaced with an AB Sciex 5500 hybrid triple quadrupole/linear ion trap mass spectrometer equipped with an electrospray ionization source operating in positive or negative mode. Q1 (precursor ion) and Q3 (fragment ion) transitions, the metabolite identifier, dwell times and collision energies for both positive and negative ion modes were used according to published methods with additional transitions for our internal standards24,67. Five microliters of sample was injected onto an XBridge Amide HPLC column (3.5 μm; 4.6 mm × 100 mm, 186004868, Waters). Mobile phases were run at 400 μl min–1 and consisted of HPLC buffer A (pH 9.0, 95% (vol/vol) water, 5% (vol/vol) acetonitrile (ACN), 20 mM ammonium hydroxide, 20 mM ammonium acetate) and HPLC buffer B (100% ACN). The HPLC settings were as follows: from 0 to 0.1 min, the mobile phase was maintained at 85% buffer B; from 0.1 to 3 min, the percentage of buffer B was decreased from 85% to 30%; from 3 to 12 min, the percentage of buffer B was decreased from 30% to 2% and was maintained at 2% for an additional 3 min. At 15 min, the percentage of buffer B was increased again to 85%, and the column was flushed for an additional 8 min with 85% buffer B. MultiQuant (version 2.1.1, AB Sciex) software was used for data analysis. Metabolite peaks were normalized by cell number and internal standards before statistical analyses.
The retention times for all metabolites were verified using individual purified standards from Sigma: Glycolysis/Gluconeogenesis Metabolite Library (ML0013-1KT), Pentose Phosphate Metabolite Library (ML0012), TCA Cycle Metabolite Library (ML0010)), l-glutathione reduced (G4251), l-glutathione oxidized (G6654), l-serine (S4500) and α-d-glucose 1-phosphate (G6750) using the same chromatographic method. Metabolites were quantified by integrating the chromatographic peak area of the precursor ion.
For measurements without 13C labeling, eGFP+ cells sorted by flow cytometry from acutely induced ITK-SYKCD4-CreERT2;Pdcd1−/− mice were incubated overnight in vitro in glucose- and glutamine-free DMEM (A1443001, Thermo) supplemented with 10% dialyzed FCS (A3382001, Thermo) and 11 mM glucose (G8270, Sigma), 11 mM galactose (G0750, Sigma) or 11 mM galactose combined with 2 mM N-acetylcysteine (A7250, Sigma). The next day, metabolites were isolated and quantified as described above.
For measurement of acetyl-1,2-[13C2]CoA, cells from the indicated genotypes were prepared as previously described. LC–MS/MS analysis was performed with the following modifications: 10 µl of sample was injected onto an XSelect HSS T3 XP Column (100 Å, 2.5 µm, 2.1 mm × 100 mm, 186006151, Waters). Mobile phases were run at 350 μl min–1 and consisted of HPLC buffer A (20 mM ammonium hydroxide, 20 mM ammonium acetate in water (pH 9)) and HPLC buffer B (20 mM ammonium acetate in methanol). The HPLC settings were as follows: from 0 to 1 min, the mobile phase was maintained at 2% buffer B; from 1 to 6 min, the percentage of buffer B was increased from 2% to 80%; from 6 to 9 min, the percentage of buffer B was increased from 80% to 100% and was maintained at 100% for an additional 4 min. At 13 min, the percentage of buffer B was decreased again to 2%, and the column was flushed for an additional 5 min with 2% buffer B. The transitions used are listed in Supplementary Table 1.
To establish the method, the following purified standards were purchased: acetyl-1,2-[13C2]CoA lithium salt (658650, Sigma), acetyl-CoA sodium salt (A2056, Sigma) and CoA hydrate (C4282, Sigma).
LC–MS/MS analysis of histone post-translational modifications
ITK-SYKCD4-CreERT2 and ITK-SYKCD4-CreERT2;Pdcd1−/− mice received a single dose of 2 mg tamoxifen. Five days later, cells were isolated from the spleen, and eGFP+ cells were sorted by flow cytometry and cultured in glucose-free DMEM (Thermo, 11966025) supplemented with 10% dialyzed FCS (A3382001, Thermo) and 1 mM [U13C]glucose (CLM-1396, Cambridge Isotope Laboratories). After 4 h of in vitro culture, the cells were washed once with PBS and snap frozen in liquid nitrogen. For histone post-translational modification analysis, approximately 1 million cells were extracted with acid. The pelleted cells were resuspended in 100 μl of 0.2 M H2SO4, and histones were extracted by rotating overnight at 4 °C. Cell debris was removed by centrifugation at 20,817g for 10 min at 4 °C. Histones were precipitated by adding trichloroacetic acid (85183, Thermo) to a final concentration of 26%. The tubes were mixed and incubated at 4 °C for 2 h and centrifuged at 20,817g for 15 min. Pellets were washed three times with ice-cold 100% acetone (AA22928-K2, VWR) (5 min of rotation at 4 °C, 15 min of centrifugation at 20,817g and 4 °C between washes), dried for 15 min at room temperature, resuspended in 20 μl of 1× Laemmli sample buffer per million cells and boiled at 95 °C for 5 min. Samples were stored at −20 °C until further use. Precast polyacrylamide (4–20%) gels (43277.01, Serva) were used to separate the histones corresponding to 0.5 × 106 cells. Gels were briefly stained with InstantBlue Coomassie Protein Stain (ab119211, Abcam). For targeted mass spectrometry analysis, histone bands were excised, washed once with LC–MS-grade water (1153331000, Sigma) and destained twice (or until transparent) by incubating for 30 min at 37 °C with 200 μl of 50% ACN (8825.2, Carl Roth) in 50 mM NH4HCO3 (T871.1, Carl Roth). Gel pieces were then washed twice with 200 μl LC–MS-grade water and twice with 200 μl of 100% ACN to dehydrate them. Histones were acylated in gel by adding 20 μl of propionic anhydride (175641, Sigma) and 40 μl of 100 mM NH4HCO3. After 5 min, 140 μl of 1 M NH4HCO3 was slowly added to the reaction. The pH was confirmed to be approximately 7 for each sample (when the reaction was acidic, a few microliters of 1 M NH4HCO3 was added). The samples were incubated at 37 °C for 45 min at 550 r.p.m. Afterward, samples were washed five times with 200 μl of 100 mM NH4HCO3, four times with 200 μl MS-grade water and four times with 200 μl of 100% ACN. They were centrifuged briefly, and all the remaining ACN was removed. Gel pieces were rehydrated in 50 μl trypsin solution (25 ng ml−1 trypsin in 100 mM NH4HCO3) (V5111, Promega) with 1-μl spike tides (SPT-ME-TQL, JPT Peptide Technologies) and incubated at 4 °C for 20 min. After the addition of 150 μl of 50 mM NH4HCO3, histones were digested in gel overnight at 37 °C and 550 r.p.m. Peptides were sequentially extracted by incubating for 10 min at room temperature with 150 μl of 50 mM NH4HCO3, twice with 150 μl of 50% ACN (in LC–MS-grade water), 0.1% trifluoroacetic acid (TFA) and twice with 100 μl of 100% ACN. During each of the washing steps, the samples were sonicated for 3 min in a water bath followed by brief centrifugation. The obtained peptides were dried using a centrifugal evaporator and stored at −20 °C until resuspension in 30 μl of 0.1% TFA. For desalting, peptides were loaded in a C18 StageTip (prewashed with 20 μl of methanol followed by 20 μl of 80% ACN and 0.1% TFA and equilibrated with 20 μl of 0.1% TFA), washed twice with 20 μl 0.1% TFA and eluted three times with 10 μl of 80% ACN and 0.25% TFA. The flowthrough obtained from loading peptides in C18 was further desalted with TopTip Carbon (TT1CAR.96, GlyGen) by loading the flowthrough three times (prewashed three times with 30 μl of 100% ACN followed by equilibration three times with 30 μl of 0.1% TFA), washed five times with 30 μl of 0.1% TFA and eluted three times with 15 μl of 70% ACN and 0.1% TFA. Eluted peptides from both desalting steps were combined and evaporated in a centrifugal evaporator, resuspended in 17 μl of 0.1% TFA and stored at −20 °C. The resuspended samples were injected into an UltiMate 3000 RSLCnano system (Thermo) and separated in a 25-cm Aurora column (IonOpticks) with a 50-min gradient from 6% to 43% of 80% ACN in 0.1% formic acid and a 50-min gradient from 5% to 60% ACN in 0.1% formic acid. The effluent from the HPLC was directly electrosprayed into a Q Exactive HF system (Thermo) operated in data-dependent mode to automatically switch between full-scan MS and MS/MS acquisition. Survey full-scan MS spectra (from m/z 250 to 1,600) were acquired with a resolution of R = 60,000 at an m/z of 400 (AGC target of 3 × 106). The ten most intense peptide ions with charge states between 2 and 5 were sequentially isolated to a target value of 1 × 105 and fragmented at 27% normalized collision energy. Typical mass spectrometric conditions were as follows: spray voltage, 1.5 kV; no sheath and auxiliary gas flow; heated capillary temperature, 250 °C; ion-selection threshold, 33.000 counts. Peak integration was performed using Skyline68. Quantified data were further analyzed in R using a previously published formula69.
Chromatin immunoprecipitation followed by sequencing
ChIP–seq was performed as previously described70 but with the inclusion of a spike-in strategy to allow for relative quantification of the detected ChIP signal in control versus experimental conditions71. Briefly, ITK-SYKCD4-CreERT2 or ITK-SYKCD4-CreERT2;Pdcd1−/− mice were injected with 2 mg tamoxifen. Five days later, 300,000 cells from the spleens were sorted by flow cytometry for the eGFP+ phenotype, immediately cross-linked (1% formaldehyde, 10 min at room temperature), lysed in 100 μl Buffer-B-0.3 (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 0.3% SDS, 1× protease inhibitors, Roche) and sonicated in a microtube (C300010011, Diagenode) using a Bioruptor Pico device until most of the DNA fragments were 200–500 bp long (settings: temperature, 4 °C; 20 cycles with 30 s on and 30 s off). After shearing and centrifugation at 4 °C and 12,000g for 10 min, the supernatant was diluted 1:1 with dilution buffer (1 mM EGTA, 300 mM NaCl, 2% Triton X-100, 0.2% sodium deoxycholate, 1× protease inhibitors, Roche). Sonicated chromatin, supplemented with 50 ng of Drosophila spike-in chromatin (53083, Active Motif), was then incubated for 4 h at 4 °C on a rotating wheel with 2 μg anti-H3K27ac (C15410174, Diagenode) and 1 μg anti-H2Av (61686, Active Motif) antibodies conjugated to 15 µl Protein G Dynabeads (10003D, Thermo). Beads were washed five times with buffer A (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate, 140 mM NaCl, 1× protease inhibitors) and once with buffer C (10 mM Tris-HCl, pH 8.0, 10 mM EDTA). Beads were then incubated with 70 μl elution buffer (0.5% SDS, 300 mM NaCl, 5 mM EDTA, 10 mM Tris-HCl, pH 8.0) containing 2 μl proteinase K (20 mg ml−1) for 1 h at 55 °C and 8 h at 65 °C to reverse formaldehyde cross-linking, and the supernatant was transferred to a new tube. After adding 30 μl elution buffer to the beads again, the eluates were combined and incubated with another 1 μl of proteinase K for 1 h at 55 °C. Finally, DNA was purified using SPRI AMPure XP beads (A63880, Beckman Coulter) (1:2 sample-to-bead ratio). Purified DNA was used as input for library preparation using the MicroPlex Library Preparation Kit v2 (C05010012, Diagenode) and processed according to the manufacturer’s instructions. Libraries were controlled for quality with the Qubit and Agilent DNA Bioanalyzer. Deep sequencing was performed on HiSeq 1500 systems according to the standard Illumina protocol for 50-bp single-end reads. Reads were aligned to the mouse genome (mm9) and the Drosophila genome (dm6) using the Bowtie 2 alignment package72. Aligned reads were sorted and indexed using SAMtools (version 1.11)73. Sambamba (version 0.8.0) was used to filter out unmapped, multi-mapped and duplicate reads74. bamCoverage from deepTools (version 3.3.2) was used with spike-in (Drosophila) reads as a normalization factor to extract bigwig files for visualization of data75. For differential analysis, peaks were called using the MACS2 package (version 2.2.7.1)76. DiffBind (version 2.6.6.2) was used for differential analysis using the built-in spike-in (Drosophila) normalization option. GSEA for ChIP–seq peak data was performed using the ChIP-Enrich package (version 2.0.1)48.
Cell culture
Unless otherwise indicated, murine cells were cultured in DMEM containing 20% FCS. The following compounds were used for in vitro experiments: glycolysis inhibitor 2-DG (D8375, Sigma), mTOR inhibitor torin-1 (S2827, SelleckChem), HIF1α inhibitor PX-478 2HCl (S7612, SelleckChem), ACLY inhibitor BMS-303141 (4609, Tocris), fatty acid synthase inhibitor FT113 (S6666, SelleckChem) and BET-p300–CBP dual inhibitor NEO2734 (S9648, SelleckChem). Inhibitors were dissolved in DMSO or water, and the cell culture medium was supplemented with the indicated concentrations of the compound or DMSO or water (as a control).
Torin-1 inhibitor treatment in vivo
For in vivo treatment with the mTORC1 and mTORC2 inhibitor torin-1, NSG recipient mice received 1 × 103 eGFP+ T cells sorted by flow cytometry from ITK-SYKCD4-CreERT2;Pdcd1−/− mice, which had been injected with a single dose of 2 mg tamoxifen (T5648, Sigma) 5 d earlier. Three days after transplantation, NSG mice received torin-1 (S2226, SelleckChem) or vehicle (20% NMP and 50% PEG 400 in ultra-pure water, 494496 and 202398, Sigma) by intraperitoneal injection (10 mg per kg per day), 5 d per week.
CRISPR–Cas9-mediated deletion of Acly
CRISPR–Cas9 ribonucleoproteins (RNPs) were assembled from crRNA–tracrRNA duplexes and Alt-R S.p. Cas9 nuclease (1081059 and 1072534, IDT) according to the manufacturer’s recommendations. Briefly, equimolar amounts of crRNA and tracrRNA were mixed and resuspended in IDTE buffer at a concentration of 44 μM. This mixture was heated to 95 °C for 5 min and allowed to cool to room temperature before adding 36 μM Cas9 enzyme. A resting phase of 20 min at room temperature followed, before the RNPs were used for electroporation. ITK-SYKCD4-CreERT2;Pdcd1−/− mice received a single injection of 2 mg tamoxifen. Five days later, eGFP+ cells from the spleen were sorted by flow cytometry. A total of 2 × 106 cells were washed twice with PBS and resuspended in 80 μl P3 solution (V4XP-3032, Lonza), 20 μl RNPs and 1 μl Alt-R Cas9 Electroporation Enhancer (1075916, IDT). Cells were electroporated with a 4D-Nucleofector device (AAF-1002B, Lonza) with pulse code EH115. Immediately after electroporation, 1 ml prewarmed culture medium was added to the cuvettes, and cells were kept at 37 °C for 10 min. Afterward, cells were washed twice, and 1 × 106 eGFP+ cells were intravenously injected into wild-type recipient mice with constitutive Cas9 expression (026179, Jackson Laboratory) to avoid the immunogenicity of the Cas9 protein. The following crRNA species were used: GTTCAATGAGAAAGTTCTTG for Acly (Mm.Cas9.ACLY.1.AJ) and negative control crRNA 1 (1072544, IDT).
Isolation of primary human CTCL cells
This study was approved by the Northwestern University Institutional Review Board. Patients provided informed consent for inclusion in the study. Clinical and demographic information on human participants is listed in Supplementary Table 2. Peripheral blood mononuclear cells were isolated from the blood of patients with leukemic CTCL by Ficoll-Hypaque gradient centrifugation (17144003, Cytiva). Leukemic cells were sorted by flow cytometry (FACSAria 5, BD Biosciences) using cell surface markers that uniquely identified the neoplastic clones. If the antibody to TCRvβ was available, we isolated the CD3+TCRvβ+CD8− population. If not, we isolated CD3+CD26−CD8− cells. We found that the mutational spectra of cells were similar, regardless of the method of isolation. The resulting CTCL cells had a median purity of >90%. The following antibodies were used: Pacific Blue anti-CD3 (317313, BioLegend), APC anti-CD3 (317318, BioLegend), PerCP-Cy5.5 anti-CD8 (45-0088-42, eBioscience), PE anti-CD26 (302705, BioLegend), PE anti-TCRvβ2 (130-110-095, Miltenyi), FITC anti-TCRvβ13 (11-5792-41, eBioscience), PE anti-TCRvβ14 (130-108-804, Miltenyi) and PE anti-TCRvβ17 (1M2048, Beckman Coulter). For in vitro experiments, CTCL cells and T cells from healthy human donors were cultured in RPMI medium with 10% FCS (Gemini) and 1% penicillin–streptomycin (Gibco). The tumor burden for each patient was determined using well-validated clinical markers, taking whichever value was higher between Sezary cell counts or the CD26-negative cell number by clinical flow cytometry. These were analyzed in conjunction with white blood cell and absolute lymphocyte counts.
DNA sequencing
Genomic DNA was extracted using a QIAamp Micro Kit (56304, Qiagen). DNA libraries were prepared using a KAPA HyperPrep Kit (KK8504, KAPA Biosystems), and 150-bp paired-end sequencing was performed on an Illumina HiSeq 2000. Sequencing reads were aligned to the human genome (hg19) using the Burrows–Wheeler Aligner71.
Somatic copy number calling
To identify somatic copy number aberrations in whole-genome sequencing data from primary CTCL cells, we used Patchwork (version 2.4) with a window size of 10,000 bp. For quality control, we excluded calls with discordant log2 read ratios and delta B allele frequency or high-frequency calls in gnomAD, as previously described77,78,79.
RNA-seq human and TCR-α and TCR-β sequence analyses
RNA was extracted using the RNeasy Plus Micro Kit (74034, Qiagen). RNA quality was assessed with a Bioanalyzer 2100 (Agilent Technologies), and RNA was quantified using a Qubit RNA HS assay (Q3285, Thermo). cDNA libraries were generated using SMART-Seq version 4 (634891, Clontech) and Nextera XT (FC-131-1024, Illumina) and sequenced on an Illumina HiSeq with a read length of 150-bp paired-end reads. The sequencing reads were aligned using STAR (version 2.4.2), gene-specific transcripts were quantified using HTSeq (version 0.6.0), and differentially expressed transcripts were identified using DESeq2 (version 1.30.1)62. MiXCR software was used (version 2.1.10)80 to identify TCR sequences from RNA-seq data.
Phosflow analysis of human cells
Malignant CTCL cells were sorted by flow cytometry as described above. CD4+ T cells were immunomagnetically isolated from healthy donor-derived peripheral blood mononuclear cells (11331D, Thermo Scientific). Next, cells were stimulated with anti-CD3 and anti-CD28 beads (1:1 ratio, 11132D, Thermo) for 30 min, washed once with PBS and subsequently fixed and permeabilized (GAS004, Invitrogen). Phosflow staining was performed with anti-p-S6S240/S244 (5364S, CST) and secondary Alexa Fluor 647 anti-rabbit IgG Fab2 (4414S, CST) antibodies at dilutions of 1:100. Cells were analyzed using an LSR II flow cytometer (BD Biosciences).
Experiments with Jurkat T cells
Jurkat cells (clone E6-1, ATCC) and Raji cells (CCL-86, ATCC) were cultured in RPMI with 10% FCS (Gemini) and 1% penicillin–streptomycin (Gibco). For p-c-JUNS73 experiments, Jurkat cells transduced with a lentiviral vector encoding PDCD1 or an empty vector control were co-cultured at a 1:1 ratio with PD-L1-overexpressing Raji cells with or without anti-CD3 and anti-CD28 beads (11161D, Thermo) for 1 h. Fixable live–dead staining was used (L34960, Thermo), and the cells were fixed (554722, BD). Cells were then permeabilized with cold methanol, washed and stained with Alexa Fluor 488-conjugated anti-p-c-JUNS73 antibody (12714, CST) at a 1:100 dilution before flow cytometry analysis. For AP-1 reporter assays, cells were transduced with a retrovirus encoding an AP-1 fluorescent reporter construct (Addgene, 118095) with near-infrared fluorescent protein (iRFP) as the fluorescent reporter. AP-1 reporter Jurkat cells were then transduced with PDCD1 or an empty vector control and co-cultured at a 1:1 ratio with PD-L1 Raji cells with or without anti-CD3 and anti-CD28 beads. After 24 h, AP-1 reporter activity was determined by flow cytometry.
Glucose-uptake assay in human cells
Malignant CTCL cells were isolated, and CD4+ T cells from healthy donors were isolated as described above. Cells were cultured in vitro with anti-CD3 and anti-CD28 beads (1:1 ratio, 11132D, Thermo). The medium was supplemented with the fluorescent glucose analog 2-NBDG (100 μg ml−1, 600470, Cayman). At the indicated time points, cells were processed according to the manufacturer’s protocol (600470, Cayman) and analyzed on an LSR II flow cytometer (BD Biosciences).
Inhibitor treatment of human cells in vitro
Malignant CTCL cells and CD4+ T cells from healthy donors were isolated as previously described. Next, cells were stained with the Cell Division Tracker Kit (423801, BioLegend) and cultured in vitro with anti-CD3 and anti-CD28 beads (1:1 ratio, 11132D, Thermo) and 2-DG (S4701, SelleckChem) at the indicated concentrations, everolimus (HY-10218, MedChemExpress), BMS-303141 (SML0784, Sigma) or DMSO vehicle control. After 6 d, the cells were analyzed by flow cytometry, and the division index was determined using the FlowJo proliferation tool (version 10.6, FlowJo).
Preparation of mouse and human ATAC-seq libraries
Ex vivo
For mouse ATAC-seq, ITK-SYKCD4-CreERT2 or ITK-SYKCD4-CreERT2;Pdcd1−/− mice received a single dose of 2 mg tamoxifen. Five days after the injection, single-cell suspensions were generated from the spleens, and 50,000 eGFP+ cells sorted by flow cytometry were used for the transposase reaction. For human ATAC-seq, 50,000 malignant cells from patients with CTCL with and without the PDCD1 mutation were sorted by flow cytometry as described above. The transposase reaction was performed as previously described81. In brief, 50,000 cells were pelleted and resuspended in lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40 (11332473001, Roche), 0.1% Tween-20 (P1379, Sigma) and 0.01% digitonin (G944A, Sigma)). Lysis was performed on ice for 3 min before washing with 10 mM Tris-HCl, pH 7.4, 10 mM NaCl and 3 mM MgCl2 buffer containing 0.1% Tween-20. Nuclei were pelleted, and transposition (Tagment DNA Enzyme I, 15027865, Illumina) was performed at 37 °C for 30 min with shaking at 1,000 r.p.m. in 10 mM Tris-HCl, pH 7.6, 5 mM MgCl2, 10% dimethyl formamide (D4551, Sigma), PBS, 0.1% Tween-20, 0.01% digitonin. DNA was column purified (T1030L, NEB), and the transposed DNA was amplified as previously described81 in 50-μl reaction volumes with a modified version of custom-designed primers44. The reaction was monitored after four cycles with qPCR using a 5-μl PCR reaction mixture and the same primers in a total volume of 15 μl (KK4617, Roche). Cycles were added as calculated, and then the amplified samples were purified and size selected using SpeedBeads (GE65152105050250, Sigma) in 22% PEG. Libraries were quality controlled using the Qubit and Bioanalyzer (5067-4626, Agilent), and 76-bp paired-end sequencing was performed on an Illumina NextSeq 500.
In vitro
For ATAC-seq experiments involving inhibitor incubation, single-cell suspensions were generated from spleens of acutely induced ITK-SYKCD4-CreERT2 or ITK-SYKCD4-CreERT2;Pdcd1−/− mice. Next, cells were incubated in vitro for 3 h in the presence of 5 μM ACLY inhibitor BMS-303141 or DMSO. Finally, ITK–SYK-expressing eGFP+ cells were sorted by flow cytometry and immediately processed for ATAC-seq as described above.
ATAC-seq analysis in mice and humans
For mouse ATAC-seq analysis, Cutadapt (version 3.1) was used to remove adaptor sequences. Next, reads were aligned to the mm9 genome using Bowtie 2 (version 2.4.0)72. Afterward, duplicates were marked with MarkDuplicates (Picard, version 2.24.0) and removed with SAMtools (version 1.11). To filter properly mapped reads, we used SAMtools with the following options: exclude multi-mapped reads (MAPQ < 30) and reads with flag 1796 or 1804. Next, SAMtools with the option ‘-f 2’ was used to filter properly paired reads. Adjustment using the Tn5 shift was performed with alignmentSieve (deepTools, version 3.3.2). Next, we filtered for nucleosome-free reads (0–100 bp) as previously described44. Visual inspection after filtering was performed using the bamPEFragmentSize function from deepTools (version 3.3.2). Next, we used MACS2 (version 2.2.7.1) to call ATAC-seq peaks with the following parameters: ‘–nomodel –nolambda –keep-dup auto -call-summits’ (ref. 76). HINT-ATAC was used to calculate transcription factor profiles based on JASPAR version 2020 (refs. 45,46). chromVAR (version 1.14.0) was used to calculate motif scores47. Human ATAC-seq analysis was performed similarly with the following modifications: adaptor sequences were trimmed using Trimmomatic (version 0.36)82, and mapping was performed using the hg19 reference genome.
Statistical analysis of biological experiments and reproducibility
All statistical tests were performed using R software (version 3.5.0 or higher, R Foundation for Statistical Computing). In each experiment, appropriate statistical tests, including non-parametric tests, were used, as indicated in the figure legends. Multiple comparisons were performed using ANOVA, Tukey’s post hoc test and corrected P values. Parametric or non-parametric tests were used based on the data distribution. No specific test for normality or equal variances was conducted. No statistical methods were used to predetermine sample size estimates. No data were excluded from the analyses, except one animal that died before the respective time of analysis. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment. The statistical significance level was set at P < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available from the corresponding authors upon reasonable request. For mouse data, RNA-seq, ATAC-seq and ChIP–seq data have been deposited in the Gene Expression Omnibus database under accessions GSE212832, GSE213180 and GSE183530. For human data, whole-genome sequencing, RNA-seq and ATAC-seq data for consenting patients are deposited in the database of Genotypes and Phenotypes under accession codes phs002456.v1.p1 (for previously published data from ref. 2) and phs003312. Raw data from RNA-seq and metabolic experiments can be found in Supplementary Tables 3–14. All other data supporting the findings of this study are available from the corresponding authors on reasonable request. Source data are provided with this paper.
Code availability
The MATLAB code used to determine the [1-13C]pyruvate versus [1-13C]lactate peak heights is available from the corresponding authors upon reasonable request.
References
Fiore, D. et al. Peripheral T cell lymphomas: from the bench to the clinic. Nat. Rev. Cancer 20, 323–342 (2020).
Park, J. et al. Integrated genomic analyses of cutaneous T cell lymphomas reveal the molecular bases for disease heterogeneity. Blood 138, 1225–1236 (2021).
Anand, K. et al. T-cell lymphoma secondary to checkpoint inhibitor therapy. J. Immunother. Cancer 8, e000104 (2020).
Marks, J. A., Parker, D. C., Garrot, L. C. & Lechowicz, M. J. Nivolumab-associated cutaneous T-cell lymphoma. JAAD Case Rep. 9, 39–41 (2021).
Rauch, D. A. et al. Rapid progression of adult T-cell leukemia/lymphoma as tumor-infiltrating Tregs after PD-1 blockade. Blood 134, 1406–1414 (2019).
Ratner, L., Waldmann, T. A., Janakiram, M. & Brammer, J. E. Rapid progression of adult T-cell leukemia-lymphoma after PD-1 inhibitor therapy. N. Engl. J. Med. 378, 1947–1948 (2018).
Bennani, N. N. et al. A phase II study of nivolumab in patients with relapsed or refractory peripheral T-cell lymphoma. Blood 134, 467 (2019).
Patsoukis, N., Li, L., Sari, D., Petkova, V. & Boussiotis, V. A. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol. Cell. Biol. 33, 3091–3098 (2013).
Sen, D. R. et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169 (2016).
Franco, F., Jaccard, A., Romero, P., Yu, Y. R. & Ho, P. C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2, 1001–1012 (2020).
Pechloff, K. et al. The fusion kinase ITK–SYK mimics a T cell receptor signal and drives oncogenesis in conditional mouse models of peripheral T cell lymphoma. J. Exp. Med. 207, 1031–1044 (2010).
Wartewig, T. et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 552, 121–125 (2017).
Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 12, 325–338 (2012).
Luo, F. et al. Hypoxia-inducible transcription factor-1α promotes hypoxia-induced A549 apoptosis via a mechanism that involves the glycolysis pathway. BMC Cancer 6, 26 (2006).
Schodel, J. et al. High-resolution genome-wide mapping of HIF-binding sites by ChIP–seq. Blood 117, e207–e217 (2011).
Topping, G. J. et al. Acquisition strategies for spatially resolved magnetic resonance detection of hyperpolarized nuclei. MAGMA 33, 221–256 (2020).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Liberti, M. V. & Locasale, J. W. The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci 41, 211–218 (2016).
Warburg, O. Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften 12, 1131–1137 (1924).
Dodd, K. M., Yang, J., Shen, M. H., Sampson, J. R. & Tee, A. R. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 34, 2239–2250 (2015).
Muschen, M. Metabolic gatekeepers to safeguard against autoimmunity and oncogenic B cell transformation. Nat. Rev. Immunol. 19, 337–348 (2019).
Liu, Q. et al. Development of ATP-competitive mTOR inhibitors. Methods Mol. Biol. 821, 447–460 (2012).
Welsh, S., Williams, R., Kirkpatrick, L., Paine-Murrieta, G. & Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1α. Mol. Cancer Ther. 3, 233–244 (2004).
Yuan, M. et al. Ex vivo and in vivo stable isotope labelling of central carbon metabolism and related pathways with analysis by LC–MS/MS. Nat. Protoc. 14, 313–330 (2019).
DeBerardinis, R. J. & Chandel, N. S. We need to talk about the Warburg effect. Nat. Metab. 2, 127–129 (2020).
Kinnaird, A., Zhao, S., Wellen, K. E. & Michelakis, E. D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 16, 694–707 (2016).
Peng, M. et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484 (2016).
Sivanand, S., Viney, I. & Wellen, K. E. Spatiotemporal control of acetyl-CoA metabolism in chromatin regulation. Trends Biochem. Sci. 43, 61–74 (2018).
Wellen, K. E. et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009).
Campbell, S. L. & Wellen, K. E. Metabolic signaling to the nucleus in cancer. Mol. Cell 71, 398–408 (2018).
Cluntun, A. A. et al. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab. 3, 10 (2015).
Li, J. J. et al. 2-hydroxy-N-arylbenzenesulfonamides as ATP-citrate lyase inhibitors. Bioorg. Med. Chem. Lett. 17, 3208–3211 (2007).
Volker-Albert, M. C., Schmidt, A., Forne, I. & Imhof, A. Analysis of histone modifications by mass spectrometry. Curr. Protoc. Protein Sci. 92, e54 (2018).
Icard, P. et al. ATP citrate lyase: a central metabolic enzyme in cancer. Cancer Lett. 471, 125–134 (2020).
Lee, J. V. et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306–319 (2014).
Martinez Calejman, C. et al. mTORC2–AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat. Commun. 11, 575 (2020).
Covarrubias, A. J. et al. Akt–mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 5, e11612 (2016).
Migita, T. et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 68, 8547–8554 (2008).
Potapova, I. A., El-Maghrabi, M. R., Doronin, S. V. & Benjamin, W. B. Phosphorylation of recombinant human ATP:citrate lyase by cAMP-dependent protein kinase abolishes homotropic allosteric regulation of the enzyme by citrate and increases the enzyme activity. Allosteric activation of ATP:citrate lyase by phosphorylated sugars. Biochemistry 39, 1169–1179 (2000).
Vlahos, C. J., Matter, W. F., Hui, K. Y. & Brown, R. F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269, 5241–5248 (1994).
Mardis, E. R. ChIP–seq: welcome to the new frontier. Nat. Methods 4, 613–614 (2007).
Mizzen, C. A. & Allis, C. D. Linking histone acetylation to transcriptional regulation. Cell. Mol. Life Sci. 54, 6–20 (1998).
Kurdistani, S. K., Tavazoie, S. & Grunstein, M. Mapping global histone acetylation patterns to gene expression. Cell 117, 721–733 (2004).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Li, Z. et al. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 20, 45 (2019).
Fornes, O. et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92 (2020).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Welch, R. P. et al. ChIP-Enrich: gene set enrichment testing for ChIP–seq data. Nucleic Acids Res. 42, e105 (2014).
Lachmann, A. et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 (2010).
Consortium, E. P. The ENCODE (Encyclopedia of DNA Elements) Project. Science 306, 636–640 (2004).
Rouillard, A. D. et al. The Harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, baw100 (2016).
Davis, R. J. Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 (2000).
Sasaki, T. et al. Spatiotemporal regulation of c-Fos by ERK5 and the E3 ubiquitin ligase UBR1, and its biological role. Mol. Cell 24, 63–75 (2006).
Ferrari, S., Bandi, H. R., Hofsteenge, J., Bussian, B. M. & Thomas, G. Mitogen-activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. J. Biol. Chem. 266, 22770–22775 (1991).
Yamada, K., Saito, M., Matsuoka, H. & Inagaki, N. A real-time method of imaging glucose uptake in single, living mammalian cells. Nat. Protoc. 2, 753–762 (2007).
Kim, S. J. et al. A phase II study of everolimus (RAD001), an mTOR inhibitor plus CHOP for newly diagnosed peripheral T-cell lymphomas. Ann. Oncol. 27, 712–718 (2016).
Yukawa, M. et al. AP-1 activity induced by co-stimulation is required for chromatin opening during T cell activation. J. Exp. Med. 217, e20182009 (2020).
Lynn, R. C. et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019).
Qu, K. et al. Chromatin accessibility landscape of cutaneous T cell lymphoma and dynamic response to HDAC inhibitors. Cancer Cell 32, 27–41 (2017).
Parekh, S., Ziegenhain, C., Vieth, B., Enard, W. & Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci. Rep. 6, 25533 (2016).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Mootha, V. K. et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Shechter, D., Dormann, H. L., Allis, C. D. & Hake, S. B. Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 (2007).
Yuan, M., Breitkopf, S. B., Yang, X. & Asara, J. M. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 7, 872–881 (2012).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Lauterbach, M. A. et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-citrate lyase. Immunity 51, 997–1011 (2019).
Cernilogar, F. M. et al. Pre-marked chromatin and transcription factor co-binding shape the pioneering activity of Foxa2. Nucleic Acids Res. 47, 9069–9086 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Feng, J., Liu, T., Qin, B., Zhang, Y. & Liu, X. S. Identifying ChIP–seq enrichment using MACS. Nat. Protoc. 7, 1728–1740 (2012).
Choi, J. et al. Genomic landscape of cutaneous T cell lymphoma. Nat. Genet. 47, 1011–1019 (2015).
Daniels, J. et al. Cellular origins and genetic landscape of cutaneous γ δ T cell lymphomas. Nat. Commun. 11, 1806 (2020).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Bolotin, D. A. et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381 (2015).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Acknowledgements
We thank K. Burmeister, A. Doddajjappanavar, V. Höfl, N. Prause, M. Arens, K. Pechloff, W. Lee, Z. Kurgyis, P. Gaulard and A. Wahida for providing excellent technical assistance or helpful discussions and the Core Facility Cell Analysis at TranslaTUM at Klinikum rechts der Isar of the Technical University Munich for their support. We also acknowledge core facilities at Northwestern including the flow cytometry core and the Skin Biology and Disease Resource Center. This work was supported by research grants from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (project ID 210592381, SFB 1054; project ID 360372040, SFB 1335; project ID 395357507, SFB 1371; project ID 369799452, TRR 237; project ID 452881907, TRR 338, RU 695/9-1, RU 695/12-1), The Leukemia & Lymphoma Society and the European Research Council under the European Union’s Horizon 2020 Research and Innovation Programme awarded grant agreement 834154 to J.R. and grant agreement 820374 to F.S. F.S. was further supported by the DFG (project ID 68647618, SFB 824). J.C. was supported by the National Institutes of Health (1DP2AI136599-01), the American Cancer Society (Research Scholar Grant RSG-20-050-01), the Bakewell Foundation and the Leukemia Lymphoma Society Scholar Award (1377-21). T.W. was supported by the Cancer Research Institute (CRI4029) and is currently funded by NIH–NCI (1K99CA277586-01). J.D. was supported by NIH–NCI grants F30 CA265107 and T32 CA009560.Work in G.S.’s laboratory was funded by the DFG (project ID 213249687, SFB 1064 TP3; project ID 329628492, SFB 1321 TP13). D.B. was supported by FNR CORE grants (C21/BM/15796788 and C18/BM/12691266). M.M. was supported by research grants from the National Institutes of Health (R35CA197628, R01CA157644, R01CA213138 and P01CA233412) and the Howard Hughes Medical Institute (HHMI-55108547). Work in the laboratory of A.I. was funded by research grants from the DFG (project ID 325871075-SFB1309) and the Bundesministerium für Bildung und Forschung (FKZ, FKZ161L0214F, ClinspectM).
Author information
Authors and Affiliations
Contributions
T.W., J.D., E.H., J.C. and J.R. conceived and designed the study. T.W. and A.J. performed mouse experiments, bioinformatic analyses and metabolomic assays. M.S. and E.M. performed mouse experiments and signaling studies. J.D., J.P., C.L., Y.L. and S.C. performed experiments with primary patient samples and bioinformatic analyses. E.H. and A.V.V. performed mouse experiments and metabolic assays. F.K. and J.V. Gabler performed mouse experiments. A.V.V., I.F. and A.I. performed LC–MS/MS analysis of histones. F.M.C. and G.S. generated ChIP–seq data. F.H.A.v.H., C.H. and F.S. performed FDG PET and MRI imaging. H.S. guided metabolic LC–MS/MS experiments. C.K. and R.R. performed ATAC-seq and RNA-seq of murine cells. H.K., M.M., S.J., D.M.W., N.O., J. Guitart, J Giustinani and D.B. provided intellectual input and critical reagents. T.W., E.H., M.S., A.V.V. and J.D. generated the figures. T.W., J.D., M.S., J.C. and J.R. wrote the paper. J.C. and J.R. secured funding. All authors discussed the results and contributed to the paper.
Corresponding authors
Ethics declarations
Competing interests
T.W., J.D., J.C. and J.R. are listed as inventors on a patent application related to this work. The other authors declare no competing interests. D.M.W. is an employee of MSD–Merck. He has equity interest in Travera, Ajax and Bantam.
Peer review
Peer review information
Nature Cancer thanks Ping-Chih Ho, Keisuke Kataoka and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Increased glucose uptake upon Pdcd1 loss.
a, Percentage of eGFP-expressing cells as determined by flow cytometric analysis of peripheral blood. A cross indicates death of the animal. Individual data points are shown (n = 3 mice per genotype). TAM, tamoxifen. b, Remaining glucose in cell culture supernatants. ITK-SYK-expressing T cells were sorted by FACS for eGFP+ from the spleens of acutely induced ITK-SYKCD4-creERT2 (‘Pdcd1+/+’) and ITK-SYKCD4-creERT2;Pdcd1−/− mice (‘Pdcd1−/−‘) and cultured in vitro for four hours. Next, the cell culture supernatants were harvested, and glucose concentrations were determined by colorimetric measurements. All animals received single-dose intraperitoneal injections of tamoxifen five days before the experiment (n = 6 and n = 4 mice per genotype). Data was normalized to viable cell number. P=two-sided Student’s t-test. Shown are the mean ± SD and individual data points. a, Representative results from 8 independent experiments with three biological replicates per group. b, Representative results from 2 independent experiments.
Extended Data Fig. 2 PD-1 inactivation enables oncogene-enforced glycolysis in vivo.
a, Schematic outline of metabolic imaging analysis experiment. b, Exemplary splenic T1-weighted sequence of an acutely induced ITK-SYKCD4-creERT2;Pdcd1−/− animals with indicated ROI used for 7T-MRI [1-13C]pyruvate and [1-13C]lactate measurements. ROI, region of interest. c, Percentage of viable lymphocytes in the peripheral blood of anti-PD-L1 or isotype control antibodies treated ITK-SYKCD4-creERT2 mice. All animals received a single dose of tamoxifen on day zero. Cross indicates death of the animal (n = 3 mice per genotype). d, Schematic outline of metabolic flux analysis experiment. e, ECAR metabolic flux analysis of ITK-SYK-expressing T cells FACS sorted for eGFP+ from the spleens of acutely induced ITK-SYKCD4-creERT2 mice. As indicated in (d), the mice received anti-PD-L1 or isotype control antibodies on days one, three and five via intraperitoneal injection (n = 3 biological replicates per group). Data was normalized using total cellular protein. ECAR, extracellular acidification rate. P=two-sided Student’s t-test. The data are shown as box plots, in which the middle line denotes the median, the top and bottom box edges denote the 0.25 and 0.75 quantiles, respectively, and the whiskers denote the minimum and maximum values. f, OCR metabolic flux analysis from the same experiment as in (e) OCR, oxygen consumption rate. P=two-sided Student’s t-test. b, Representative data from a single mouse within the experiment shown in Fig. 2c, d. c, Representative results from 5 independent experiments with three biological replicates per group. e,f, Representative data from two independent experiments with three biological replicates per group.
Extended Data Fig. 3 Transformed T cells with Pdcd1 deletion depend on the mTOR-HIF1α-glycolysis cascade.
a,b,c, Lactate concentrations in the cell culture supernatants of eGFP+ FACS sorted cells derived from the spleens of acutely induced ITK-SYKCD4-creERT2;Pdcd1−/− mice and incubated in vitro for 48 h in the presence of the mTOR inhibitor Torin-1 (a; 0.1 μM), HIF1α inhibitor PX-478 (b; iHIF1α, 5 μM) or glycolysis inhibitor 2-DG (c; 1 mM) or DMSO as a control (n = 3 biological replicates). Lactate concentrations were normalized to the viable cell numbers. P=paired two-sided Student’s t-test. d,e,f, Number of viable ITK-SYK+PD-1- cells cultured in vitro for 72 h with the mTOR inhibitor Torin-1 (d; 0.1 μM), HIF1α inhibitor PX-478 (e; iHIF1α, 5 μM) or glycolysis inhibitor 2-DG (f; 1 mM) or DMSO as a control (n = 3 biological replicates). eGFP+ T cells were sorted by FACS from acutely induced ITK-SYKCD4-creERT2;Pdcd1−/− animals (n = 3 biological replicates). P=paired two-sided Student’s t-test. g, Phosflow analysis of serine 240/244 of S6 ribosomal protein in ITK-SYK+PD-1- cells isolated from NSG mice treated with Torin-1 or vehicle. Cells were fixed and permeabilized immediately after isolation. h, eGFP percentage of viable lymphocytes in the peripheral blood of Torin-1 or vehicle-treated NSG mice, which were transplanted with 1,000 eGFP+ FACS-sorted ITK-SYK+PD-1- cells isolated from acutely induced ITK-SYKCD4-creERT2;Pdcd1−/− mice. Blood was taken seven days after initiation of treatment. a,b,c,d,e,f, Representative results from 2 independent experiments with three biological replicates per group. g, Representative results from two out of the six animals analyzed. h, Representative results from two out of the thirteen animals analyzed.
Extended Data Fig. 4 PD-1 controls ACLY-dependent epigenetic reprogramming.
a, H3K27ac in CD4 + GFP + T cells of ITK-SYKCD4-creERT2 or C57BL/6 mice on d5 after TAM +/− anti-PD-L1 antibody treatment. b, c, Non-labeled (b) and labeled (c) acetyl-CoA determined by targeted LC-MS/MS in ITK-SYK+ T cells +/− PD-1 cultured with uniformly labeled [U-13C]glucose (11 mM) for 4 h in vitro (n = 4 mice genotype). Normalization to viable cell numbers. P=two-sided Student’s t-test; mean ± SEM. d, [U-13C]glucose-derived 13C-acetyl groups on histones determined by orbitrap LC-MS/MS in ITK-SYK+ T cells +/− PD-1 cultured with uniformly labeled [U-13C]glucose (11 mM) for 4 h in vitro (n = 3 mice genotype). Normalization as in (b, c). P=two-sided Student’s t-test; mean ± SD. e, H3K27ac in ITK-SYK+ T cells +/− PD-1 after 3 h in vitro incubation with BMS-303141 (‘iACLY’, 15 μM) +/− acetate (100 μM; n = 3 biological replicates). P=paired two-sided Student’s t-test; mean ± SD. f, Viable ITK-SYK+PD-1- cells after 72 h in vitro culture with BMS-303141 (‘iACLY,’ 5 μM) or DMSO (n = 3 biological replicates). P=paired two-sided Student’s t-test. g, Viable ITK-SYK+ cells in the blood of sgACLY or sgMOCK-injected mice (n = 4 per group). P=two-sided Student’s t-test. h, Viable ITK-SYK+ cells +/− PD-1 after 3-day in vitro incubation with FT113 (1 μM) or DMSO (n = 3 biological replicates). P=two-sided Student’s t-test; mean ± SD. i, Viable ITK-SYK+ cells in presence of NEO2734 (50 nM). Experiment as in (e). P=two-sided Student’s t-test; mean ± SD. j, Percentage of H3K27ac ChIP-seq peak distribution over different genomic features as determined from the experiment in (d). k, Genome-wide average H3K27ac scores in a 3 Kb window around the TSS. ChIP-seq was performed as indicated in Fig. 4h. ChIP-seq, chromatin immunoprecipitation sequencing; TSS, transcription start site. a, Representative data from two independent experiments with n = 3 biological replicates per genotype. b, c, one experiment, n = 4 biological replicates. d, Data from one experiment. e, Representative data from one experiment with n = 3 biological replicates per genotype. f, Representative data from two independent experiments. g, Data from a single experiment. h,i, Representative data from three independent experiments with n = 3 biological replicates per genotype. j,k, Data from one experiment.
Extended Data Fig. 5 PD-1 tumor suppressor function.
a, Schematic model: Loss of PD-1 tumor suppressor function enables enhanced glucose uptake and glycolytic remodeling upon oncogenic T cell signaling. These events are critical for energy production in pre-malignant cells, for providing building blocks for rapid proliferation and preserving redox balance during T cell transformation and lymphoma progression. In addition, loss of PD-1 signaling unleashes ACLY activity and thereby triggers glucose-dependent production of extramitochondrial acetyl-CoA to mediate the de novo acetylation of histones to open chromatin and enhance the activity of oncogenic AP-1 family transcription factors. b, T-cell receptor sequence analysis of three CTCL patients with hyperprogressive disease. The nucleotide sequence, V-D-J-C segments, amino acid sequence, and percentage of T-cell receptor reads are shown for each sample before (‘pre’) and after (‘post’) hyperprogression. b, Data from one experiment.
Extended Data Fig. 6 PD-1 controls post-translational AP-1 activation and transcriptional activity.
a, Oncoplot depicting mutation in T cell receptor (TCR) signaling pathway or other pathways detected in CTCL. Samples are grouped by PD-1 status, either wild-type (‘wt’) or mutant (‘mut’). b,c, MFI of p-c-JunS73 (b, n = 4) and percentage of cells positive for AP-1 reporter (c, n = 3) in Jurkat cells co-cultured with PD-L1 overexpressing Raji cells, with or without anti-CD3/CD28 beads. Jurkat cells were transduced with a lentivirus encoding either PD-1 or an empty vector control. In b, cells were co-cultured for 1 hour prior to analysis. In c, cells were co-cultured for 24 hours prior to analysis. P = 2-way ANOVA followed by Sidak’s multiple comparison test. MFI, mean fluorescence intensity. The data are presented as the mean ± SEM and individual data points are shown. a, Data from one experiment. b,c, Representative data from 2 independent experiments.
Supplementary information
Supplementary Information
Supplementary Fig. 1.
Supplementary Tables
Supplementary Tables 1–14.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 1
Uncropped western blots.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Uncropped western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Uncropped western blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wartewig, T., Daniels, J., Schulz, M. et al. PD-1 instructs a tumor-suppressive metabolic program that restricts glycolysis and restrains AP-1 activity in T cell lymphoma. Nat Cancer 4, 1508–1525 (2023). https://doi.org/10.1038/s43018-023-00635-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-023-00635-7
This article is cited by
-
Integrative analysis of chromatin accessibility and transcriptome landscapes in the induction of peritoneal fibrosis by high glucose
Journal of Translational Medicine (2024)
