
Abstract
Bispecific antibodies targeting GPRC5D demonstrated promising efficacy in multiple myeloma, but acquired resistance usually occurs within a few months. Using a single-nucleus multi-omic strategy in three patients from the MYRACLE cohort (ClinicalTrials.gov registration: NCT03807128), we identified two resistance mechanisms, by bi-allelic genetic inactivation of GPRC5D or by long-range epigenetic silencing of its promoter and enhancer regions. Molecular profiling of target genes may help to guide the choice of immunotherapy and early detection of resistance in multiple myeloma.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Data availability
WGS, bulk RNA-seq and single-cell Multiome data generated for this study have been deposited to the European Genome Archive (accession no. EGAS00001007014). These data contain identifiable genetic variants and are thus accessible under controlled access for patient privacy concerns, by contacting the data access committee. Source data for Figs. 1 and 2 and Extended Data Figs. 1–10 have been provided as Source Data files. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
All bioinformatic tools used in this study are indicated in the relevant sections of the Methods, with versions and modified parameters. Custom codes developed for this project have been deposited on Zenodo (https://doi.org/10.5281/zenodo.8183942) and are available under an open source Creative Commons Attribution 4.0 International license.
Change history
08 September 2023
A Correction to this paper has been published: https://doi.org/10.1038/s43018-023-00646-4
References
Moreau, P. & Touzeau, C. T-cell-redirecting bispecific antibodies in multiple myeloma: a revolution? Blood 139, 3681–3687 (2022).
Moreau, P. et al. Teclistamab in relapsed or refractory multiple myeloma. New Engl. J. Med. 387, 495–505 (2022).
Chari, A. et al. Talquetamab, a T-cell–redirecting GPRC5D bispecific antibody for multiple myeloma. New Engl. J. Med. 387, 2232–2244 (2022).
Benaniba, L. et al. The MYRACLE protocol study: a multicentric observational prospective cohort study of patients with multiple myeloma. BMC Cancer 19, 855 (2019).
Moore, J. E. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Mifsud, B. et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 47, 598–606 (2015).
Fishilevich, S. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database 2017, bax028 (2017).
Da Vià, M. C. et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 27, 616–619 (2021).
Samur, M. K. et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 12, 868 (2021).
Maclachlan, K. H. et al. Copy number signatures predict chromothripsis and clinical outcomes in newly diagnosed multiple myeloma. Nat. Commun. 12, 5172 (2021).
Hon, G. C. et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 22, 246–258 (2012).
Kumar, S. et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 17, e328–e346 (2016).
Shinde, J. et al. Palimpsest: an R package for studying mutational and structural variant signatures along clonal evolution in cancer. Bioinformatics 34, 3380–3381 (2018).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Letouzé, E. et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat. Commun. 8, 1315 (2017).
Walker, B. A. et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 132, 587–597 (2018).
Barwick, B. G. et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat. Commun. 10, 1911 (2019).
Zhang, H., Meltzer, P. & Davis, S. RCircos: an R package for Circos 2D track plots. BMC Bioinform. 14, 244 (2013).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Satpathy, A. T. et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Zhang, Y. et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 (2008).
Prashant, N. M. et al. SCReadCounts: estimation of cell-level SNVs expression from scRNA-seq data. BMC Genom. 22, 689 (2021).
Gao, T. et al. Haplotype-aware analysis of somatic copy number variations from single-cell transcriptomes. Nat. Biotechnol. 41, 417–426 (2023).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Miller, C. A. et al. Visualizing tumor evolution with the fishplot package for R. BMC Genom. 17, 880 (2016).
Acknowledgements
This study was supported by the Région Pays de la Loire (Connect Talent program, RPH2114NNA), Nantes Métropole (RPH2114NNA), CHU Nantes, the I-SITE NexT (ANR-16-IDEX-0007), HéMA-NExT research cluster and the SIRIC ILIAD (INCa-DGOS-INSERM-ITMO Cancer_18011). We thank the Biological Resource Centre for Biobanking (CHU Nantes, Hôtel Dieu, Tumorothèque, Nantes, F-44093, France). The authors acknowledge the Cytocell - Flow Cytometry and FACS core facility (SFR Bonamy, BioCore, Inserm UMS 016, CNRS UAR 3556, Nantes, France) for its technical expertise and help, member of the Scientific Interest Group Biogenouest and the Labex IGO program supported by the French National Research Agency (no. ANR-11-LABX-0016-01). We warmly thank the patients who participated in this study.
Author information
Authors and Affiliations
Contributions
E.L. conceived and directed the research. J.D., S.G., C.T. and E.L. designed the study and wrote the manuscript. E.D. and M.D. performed the experiments. J.D., S.G., F.M., S.M. and E.L. analyzed and interpreted the data. J.D., S.G., A.F., N.G., M.C., V.G., R.B. and E.L. performed bioinformatics and statistical analysis. P.M. and C.T. provided essential biological resources and collected clinical data. All authors approved the final manuscript and contributed critical revisions to its intellectual context.
Corresponding author
Ethics declarations
Competing interests
P.M. participates in advisory boards and receives honoraria from pharmaceutical companies producing MM treatments: Janssen, Celgene, Abbvie, Pfizer, Amgen, Sanofi and Takeda. C.T. participates in advisory boards and receives honoraria from Janssen, the company that produces talquetamab. All other authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks P. Leif Bergsagel and Bruno Paiva for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
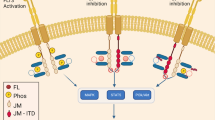
Extended Data Fig. 1 Chromosome 12p deletion and structural variant affecting GPRC5D in MM-01-0288.
a, Copy-number profile along chromosome 12 in MM-01-0288 t1 sample, showing a deletion on the 12p arm encompassing CDKN1B and GPRC5D. Upper panel: coverage log-ratio between the tumor and normal samples. Middle: B Allele Frequency of common polymorphisms. Bottom: Absolute copy-number estimated by FACETs algorithm. The location of CDKN1B and GPRC5D genes is shown with dashed lines. b, Focal deletion encompassing the transcription start site of GPRC5D, identified in 7.9% of cells of MM-01-0288-t2.
Extended Data Fig. 2 Mutational signature analysis of tumors MM-0288-t1 and t2.
Proportion of the 96 mutation categories in MM-01-0288-t1 (a) and t2 (b). We can distinguish a high prevalence of C > T mutations in TCA and TCT contexts, characteristic of APOBEC mutagenesis. c, Deconvolution of COSMIC mutational signatures in MM-01-0288 t1 and t2 samples.
Extended Data Fig. 3 Single-cell GPRC5D mRNA expression in MM-01-0288.
a, Projection of GPRC5D mRNA expression onto the single-nucleus RNA-seq UMAP of patient MM-01-0288. b, Violin plot showing the distribution of single-nucleus GPRC5D expression in cells from t1 and t2 time points. c, Violin plot showing the distribution of single-nucleus GPRC5D expression in the 5 copy-number subclones.
Extended Data Fig. 4 Flow cytometry gating.
The gating strategy is shown on top and the results for each sample below.
Extended Data Fig. 5 Cell surface protein expression of GPRC5D analyzed by flow cytometry.
GPRC5D cell-surface protein expression was analyzed in 2 MM cell lines (a), sorted tumor cells from 4 multiple myelomas (b) and sorted tumor cells from multiple myelomas with genetic (MM-01-0288) or epigenetic (MM-01-0221) inactivation of GPRC5D (c). Celles were labeled using an anti-GPRC5D antibody (red line; 571961 clone; R&D Systems) or an isotype control antibody (gray line; IC0041P clone; R&D Systems) for 60 minutes at 4 °C and analyzed by flow cytometry.
Extended Data Fig. 6
Integration of WGS and single-cell data reveals the clonal evolution of MM-01-0288 a, Clonality of somatic mutations identified by whole genome sequencing in MM-01-0288. Each point represents a mutation, with its cancer cell fraction (CCF) in t1 on the x-axis, and in t2 on the y-axis. Trunk mutations (CCF ~ 1 in both t1 and t2) are highlighted, together with two subclones: subclone A (dominant in t1 and minor in t2) and subclone B (minor in t1 and dominant in t2). Private mutations encountered in only one sample are located on the axes. In addition to these, 4,049 mutations were identified with low CCF in both samples. b, Uniform Manifold Approximation and Projection (UMAP) of the 14,849 cells analyzed from MM-01-0288 t1 and t2 samples. Each point represents a cell. They are grouped according to the similarity of their transcriptomic profiles, and colored by sample of origin. c, Projection of somatic mutations identified by WGS onto single-cell RNA-seq UMAPs. For each set of mutations, the total number of mutated reads per cell is represented with a color code. Trunk mutations are detected in all cells. Subclone A mutations are detected in the major subclone of t1 and 2 minor subclones of t2. Subclone B mutations are detected in the minor subclone of t1 and 3 subclones of t2. The proportion of cells harboring each set of mutations is consistent with their CCF estimated from WGS data in each tumor.
Extended Data Fig. 7 Clonal and subclonal copy-number changes identified by whole genome sequencing in MM-01-0288.
Pangenomic copy-number profiles of MM-01-0288 before (a) and after (b) talquetamab treatment. The two samples display 10 common clonal copy-number changes: +1q, +3q, −4p, −4q, +6p, −6q, −7p, −12p, −20p and −X. One aberration (−1p) is absent in t1 and subclonal in t2, and 3 aberrations (−14q, −14qtel and +17q) are subclonal with different cancer cell fractions (CCF) in each sample. Subclonal copy-number number losses and gains are highlighted in blue and red, respectively. For each sample, the upper panel shows the coverage log-ratio between the tumor and normal samples, the middle panel shows the B Allele Frequency of common polymorphisms, and the bottom panel shows the absolute copy-number estimated by FACETs algorithm. c, Detailed characterization of subclonal copy-number changes, including the detailed coordinates and the cancer cell fraction (CCF, for example proportion of tumor cells harboring the alteration) at t1 and t2. We indicated a CCF ‘<20%‘ for alterations visible on the profile but below FACETs detection threshold.
Extended Data Fig. 8 Identification of 5 copy-number subclones in MM-01-0288.
a, Unsupervised classification by hierarchical clustering as implemented in InferCNV (Ward.D method) reveals 9 clusters. b, Proportion of cells within each cluster carrying each of the 6 reliable subclonal CNAs identified in the patient. We used InferCNV HMM predictions to determine the presence of aberrations in each cell, except for the -14q deletion that was idenfied using Numbat tool. c, Numbat identifies in single-cell data a subclone carrying the same 14q deletion identified by FACETs in WGS data. The upper track shows the log(fold-change) of expression in cells of the subclone vs. reference cells, and the bottom track shows the allele frequencies of polymorphisms. d, Assignment of final copy-number subclones. InferCNV clusters with identical copy-number profiles were merged: InferCNV 1 and 7 into cn4, InferCNV 3-6 into cn5.
Extended Data Fig. 9 Proportion of cells carrying each GPRC5D alteration identified by WGS and single-cell sequencing data.
The type of alteration is highlighted by a color code. The p-value and correlation score were obtained using Pearson’s correlation test.
Extended Data Fig. 10 Genomic landscape and regulation of GPRC5D in the two post-talquetamab relapses with epigenetic silencing.
a, CIRCOS plot showing (from outer to inner layer) the driver mutations, copy-number changes and structural variants identified by whole genome sequencing in the post-talquetamab sample of patient MM-01-0221. b, UMAPs showing the classification of 65,700 cells from 11 samples (2 normal bone marrow samples and 9 multiple myelomas) based on their snRNA-seq profiles. Cells are colored according to their sample of origin (left) or their level of GPRC5D expression (right). c, Chromatin landscape at GPRC5D locus. ATAC-seq signals are showing two normal bone marrow samples (in green), the 2 GPRC5D-silenced post-talquetamab relapses MM-01-0221-t2 and MM-01-0302-t2 (in red) and 5 other MM samples (in blue), together with H3K27Ac (enhancer mark) and H3K4Me3 (promoter mark) ChIP-seq signals in 2 MM samples from BLUEPRINT project. All samples display a major peak of accessibility at GPRC5D promoter, and up to 5 minor peaks in the gene body. All loci are closed in MM-01-0221-t2 and MM-01-0302-t2, as demonstrated by the lack of ATAC-seq reads. The ATAC-seq peak overlapping GPRC5D promoter has both H3K27Ac and H3K4Me3 marks in BLUEPRINT samples, consistent with an active promoter role. Other ATAC-seq peaks match H3K27Ac peaks, indicating their putative enhancer role.
Supplementary information
Supplementary Table
Supplementary Tables 1–7.
Source data
Source Data Fig. 1
Genomic alterations and processed single-cell data from MM-01-0288 annotated by copy-number subclone and GPRC5D mutation.
Source Data Fig. 2
GPRC5D expression data and normalized ATAC-seq tracks.
Source Data Extended Data Fig. 1
Coverage log-ratio data.
Source Data Extended Data Fig. 2
Number and proportion of mutations per category and signature.
Source Data Extended Data Fig. 3
SnRNA-seq data from MM-01-0288 annotated by GPRC5D expression.
Source Data Extended Data Fig. 4
Raw immunofluorescence signals for all cells.
Source Data Extended Data Fig. 5
Raw immunofluorescence signals for plasma cells.
Source Data Extended Data Fig. 6
Mutation cancer cell fractions and detection in single-cell data.
Source Data Extended Data Fig. 7
FACETs segments.
Source Data Extended Data Fig. 8
SnRNA-seq data from MM-01-0288 annotated by InferCNV cluster.
Source Data Extended Data Fig. 9
Proportions of mutated cells estimated from WGS and single-cell data.
Source Data Extended Data Fig. 10
Genomic alterations and snRNA-seq data from all samples annotated by GPRC5D expression.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Derrien, J., Gastineau, S., Frigout, A. et al. Acquired resistance to a GPRC5D-directed T-cell engager in multiple myeloma is mediated by genetic or epigenetic target inactivation. Nat Cancer 4, 1536–1543 (2023). https://doi.org/10.1038/s43018-023-00625-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-023-00625-9
This article is cited by
-
GPRC5D as a novel target for the treatment of multiple myeloma: a narrative review
Blood Cancer Journal (2024)
-
Myeloma evades T cell-engaging therapies
Nature Cancer (2023)
