
Abstract
The most lethal subtype of diffuse intrinsic pontine glioma (DIPG) is H3K27M. Although ACVR1 mutations have been implicated in the pathogenesis of this currently incurable disease, the impacts of bone morphogenetic protein (BMP) signaling on more than 60% of H3K27M DIPG carrying ACVR1 wild-type remain unknown. Here we show that BMP ligands exert potent tumor-suppressive effects against H3.3K27M and ACVR1 WT DIPG in a SMAD-dependent manner. Specifically, clinical data revealed that many DIPG tumors have exploited the capacity of CHRDL1 to hijack BMP ligands. We discovered that activation of BMP signaling promotes the exit of DIPG tumor cells from ‘prolonged stem-cell-like’ state to differentiation by epigenetically regulating CXXC5, which acts as a tumor suppressor and positive regulator of BMP signaling. Beyond showing how BMP signaling impacts DIPG, our study also identified the potent antitumor efficacy of Dacinostat for DIPG. Thus, our study delineates context-dependent features of the BMP signaling pathway in a DIPG subtype.
Similar content being viewed by others

Main
Diffuse intrinsic pontine glioma (DIPG) is a rare and incurable pediatric brain cancer with survival of less than 1 year1. Understanding the tumorigenesis mechanisms of DIPG and identifying potential therapeutic strategies are main research foci in the DIPG field. The most frequent mutation in DIPG is a lysine to methionine (K27M) mutation that occurs on H3F3A or HIST1H3B/C, encoding histone variants H3.3 and H3.1 (refs. 2,3,4,5,6). These mutations are present in around 80% of DIPG cases and are believed to be the drivers of DIPG tumorigenesis7,8. Single-cell RNA-sequencing (scRNA-seq) analysis of thousands of DIPG tumor cells containing H3K27M mutations showed that most of these tumor cells resemble oligodendrocyte precursor cells (OPC-like)—a cell type that exhibits obviously enhanced proliferation and tumor-propagating potential compared with other cell subsets in tumor samples9. Thus, targeting the prolonged stem-cell-like state of DIPG cells to differentiation has been proposed as a potential strategy for DIPG treatment.
Studies have demonstrated that H3K27M DIPG exists in one of two main disease states, distinguished by H3.3K27M or H3.1K27M10,11. H3.3K27M DIPG is associated with the poorest outcomes, whereas H3.1K27M DIPG cases tend to have longer survival12,13. Hence, it seems reasonable that research and potential therapeutic strategies for these two subtypes of DIPG should be tailored specifically, focused on the now-known differential impacts of the H3.1K27M versus H3.3K27M mutations on cancer epigenomes. About 20% of DIPG patients carry mutations in ACVR1 co-occurring with H3.1K27M2,3,4,5,10,14. ACVR1 encodes a BMP type I receptor. Most ACVR1 mutations in DIPG confer ligand-independent activation of BMP signaling15. Mouse studies have suggested that Acvr1G328V cooperates with Hist1h3bK27M and Pik3caH1047R to generate high-grade diffuse gliomas, specifically by arresting the differentiation of oligodendroglial lineage cells16. Studies have therefore proposed that blocking ACVR1 kinase activity may represent a useful therapeutic strategy for the DIPG subgroup carrying ACVR1 mutations14,17.
The research attention given to the role of BMP signaling in DIPG has so far strongly emphasized the ACVR1 mutation DIPG subtype. Yet, it is conspicuous that no studies have considered the possible involvement of BMP signaling in the etiology of the remaining DIPG cases (that is, fully 60% of DIPG cases occur in patients carrying wild-type (WT) ACVR1 and H3K27M). BMPs are members of the transforming growth factor β (TGF-β) superfamily that were originally discovered based on their functions in promoting bone formation18; we now understand that these proteins function in multiple biological processes—including in the central nervous system (CNS)—and act in diverse and highly context-dependent roles19.
Here, we used a set of treatment-naïve patient-derived ACVR1 WT DIPG cells containing the H3.3K27M mutation20 to investigate any functional impact(s) of BMP signaling in this DIPG subtype. We show that BMPs can exert potent tumor-suppressive effects against this subtype of DIPG tumors in a SMAD-dependent manner, mainly by forcing DIPG tumors to exit a prolonged stem-cell-like state and to differentiate. Moreover, we identified several drugs (including the histone deacetylase (HDAC) inhibitor Dacinostat) with antitumor efficacy against DIPG, all of which upregulate BMP signaling.
Results
BMP signaling is downregulated in H3.3K27M and ACVR1 WT DIPG subtype
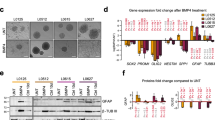
We set out to examine the signaling pathways impacted by several drugs with previously reported antitumor efficacy against DIPG21,22,23,24. Specifically, gene set enrichment analysis (GSEA) analyses indicated BMP signaling upregulation upon treatment of SU-DIPG6 and SU-DIPG13 cells with the BRD4 inhibitor JQ1 (ref. 21), the multi-HDAC inhibitor Panobinostat22, the CDK7 inhibitor THZ1 (ref. 23) and the LSD1-HDAC inhibitor Corin24; however, none of these drugs obviously affected TGF‐β, WNT, Notch or Hedgehog signaling with a consistent pattern (Fig. 1a and Extended Data Fig. 1a).

a, GSEA analysis using the ‘BMP signaling signature’ gene set to compare the control group and the drugs (JQ1, Panobinostat and THZ1) treatment group in SU-DIPG6 and SU-DIPG13, respectively (GEO: GSE94259). b, Immunoblotting analysis of the indicated proteins in PPCs and a group of H3K27M and ACVR1 WT DIPG cells. c, Left, GSEA analysis using the BMP signaling signature gene set to compare PPCs and the H3.3K27M and ACVR1 WT DIPG cells (TT150714 and TT150630). Right, GSEA analysis using the BMP signaling signature gene set to compare H3.3K27M HGG tumor or cell lines and H3.3K27WT tumor or cell lines (GEO: GSE128745). d, qPCR analysis of CHRDL1 expression in PPC and a group of H3.3K27M DIPG cell lines. All P values were generated by comparing with PPC; n = 3 independent experiments. e, Kaplan–Meier survival curves for ACVR1 WT DIPG patients in the cBioPortal DIPG patient cohort52. Log-rank test was performed; n = 14 patients in high expression of CHRDL1 group and n = 15 patients in low expression of CHRDL1 group. f, Immunoblotting analysis of p-SMAD1/5 and SMAD1 in indicated DIPG cell lines (Ctr or CHRDL1 KD DIPG cells) with or without BMP4 (25 ng ml–1) for 2 h. The relative intensity of the p-SMAD1/5 protein level compared with SMAD1 is indicated. g, qPCR analysis of mRNA expression of BMP signaling response genes in Ctr and CHRDL1 KD TT150630 DIPG cells with or without BMP4 (25 ng ml–1) treatment; n = 3 independent experiments. h, Viability of indicated cells (n = 3 independent experiments). i, Representative bioluminescence images from five mice in each group implanted with 5 × 105 Ctr or CHRDL1 KD of luciferase-GFP engineered-TT150630 cells in the pons at day 84. The heatmap superimposed over the mouse heads represents the degree of photon emission by DIPG cells expressing firefly luciferase. j, Kaplan–Meier analysis from animals implanted with TT150630 cells with (n = 5 mice) or without (n = 5 mice) CHRDL1 KD in the pons. Log-rank test was performed. The experiments in b and f were repeated three times with similar results. For d, g and h, data represents the mean ± s.d.; statistical significance was calculated by two-tailed unpaired Student’s t-test.
The consistent upregulation of BMP signaling by these drugs is surprising, so we wanted to know the roles of BMP signaling in DIPG. We further examined BMP signaling in patient-derived H3.3K27M and ACVR1 WT DIPG cells (SU-DIPG17, TT150714, TT150630 and TT150728), together with pons progenitor cells (PPCs) from the pons of a 9-week-old fetus as a normal cell control (Supplementary Table 1)20,23,25. These cells were cultured in serum-free medium, thereby eliminating the possibility of BMP signaling activation by serum factors. The phosphorylation levels of SMAD1/5 at Ser 463/465 (active SMAD1/5) were significantly lower in these DIPG cells compared with the PPCs (Fig. 1b). Moreover, the global level of H3K27me3 was decreased markedly in examined DIPG cell lines compared with PPCs (Fig. 1b).
We next performed RNA-seq analysis and then used the GSEA ‘BMP signaling’ set to compare DIPG cells (TT150630, TT150714, SU-DIPG6, SU-DIPG13 and SU-DIPG4) (NCBI GEO accession number (GEO:) GSE94259)23 with the PPCs. BMP signaling is significantly downregulated in four of the tested H3.3K27M and ACVR1 WT DIPG cell lines (Fig. 1c and Extended Data Fig. 1b). BMP signaling is also downregulated markedly in H3.3K27M HGG tumors and cell lines compared with H3.3 WT HGG (Fig. 1c) (NCBI GEO: GSE128745). No significant difference was detected in SU-DIPG4 cells that have ACVR1 mutation (Extended Data Fig. 1c; false discovery rate (FDR) q = 0.312). Consistently, we detected downregulated BMP signaling in publicly available data for H3.3K27M and ACVR1 WT DIPG tumor tissues (Extended Data Fig. 1d) (NCBI GEO: GSE50021)2. These results support the view that BMP signaling is downregulated in most of the examined H3.3K27M and ACVR1 WT DIPG.
High expression of CHRDL1 in DIPG
To explore potential factors that contribute to the downregulation of BMP signaling in H3.3K27M and ACVR1 WT DIPG cells, we examined the expression levels of 15 known BMP signaling components and 14 known regulators of BMP signaling, including antagonists from within the ‘BMP signaling’ GSEA dataset (Supplementary Table 2)26. CHRDL1 is consistently expressed at a significantly higher level in all the H3.3K27M and ACVR1 WT DIPG cells or tumor tissues compared with the PPCs or the pons tissues (Fig. 1d, Extended Data Fig. 1e,f and Supplementary Table 3).
We then examined the CHRDL1 expression from the recent scRNA-seq of six H3K27M DIPG patient samples9. These H3K27M-glioma contain primarily cells that resemble OPC-like cells, which all have high expression of OLIG2, ASCL1 and SOX2 (Extended Data Fig. 1g–j)9. Interestingly, most of the OPC-like cells have higher expression of CHRDL1 than their counterparts and normal cells (Extended Data Fig. 1k). Among the 422 CHRDL1-expressing cells, ACVR1 WT cells have significantly higher expression of CHRDL1 than ACVR1 mutant cells (Extended Data Fig. 1l). Further, cells with H3.3K27M tend to have higher expression of CHRDL1 than cells with H3.1K27M (Extended Data Fig. 1l). Moreover, we found that high CHRDL1 expression is associated with poor prognosis in ACVR1 WT DIPG and high-grade midline gliomas (including DIPG) (Fig. 1e and Extended Data Fig. 2a) (E-TABM-1107) (PedcBioPortal)27.
CHRDL1 contributes to the tumor progression in DIPG subtype
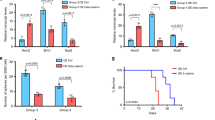
CHRDL1 is an antagonist of BMP signaling that prevents BMP ligand binding to receptors28. This observation of high CHRDL1 expression raised the possibility that CHRDL1 may be a factor driving the downregulation of BMP signaling29. Indeed, shRNA-mediated knockdown (KD) of CHRDL1 significantly increased p-SMAD1/5 levels and responses of main BMP signaling transcriptional target genes in DIPG cells upon BMP4 treatment (Fig. 1f,g and Extended Data Fig. 2b–d).
Further, CHRDL1 KD significantly reduced cell proliferation and sphere formation in this DIPG subtype (Fig. 1h and Extended Data Fig. 2e,f). Luciferase-engineered WT or CHRDL1 KD DIPG cells were established xenografts via orthotopic injection of 1 × 106 tumor cells into the pons of a cohort of B-NDG mice (B, Biocytytogen; N, NOD background; D, DNAK (Prkdc) null; G, IL2rg knockout). Consistently, xenograft models showed that CHRDL1 KD significantly inhibits the tumor growth (Fig. 1i and Extended Data Fig. 2g–i) and increases mouse survival (Fig. 1j and Extended Data Fig. 2j). In addition, immunofluorescence (IF) staining of tissue section from the xenograft mouse model of TT150630 showed that the CHRDL1 KD group has fewer tumor cells with lower OLIG2-positive percentage and higher glial fibrillary acidic protein (GFAP) levels than the control group (Extended Data Fig. 2k,l). These results indicate that CHRDL1 KD apparently reduces the stemness and induces differentiation of DIPG cells.
Together, these results strongly support the view that (1) high expression of CHRDL1 is correlated with DIPG development/pathogenesis and (2) CHRDL1 mediates downregulation of BMP signaling in DIPG. It is possible that CHRDL1 could be a vulnerable target for developing therapies to treat H3.3K27M and ACVR1 WT DIPG patients. Accordingly, active BMP signaling might exert tumor-suppressive functions and may be an indicator of better prognoses in the clinic.
BMPs exert tumor-suppressive functions in DIPG subtype
We treated DIPG cells and PPCs with a variety of TGF-β family ligands, including BMP2, BMP4, BMP7, Activin A and TGF-β1, as well as known inhibitors of the BMP pathway (LDN-193189). LDN-193189 at 200 nM is enough to block activation of BMP signaling (Extended Data Fig. 3a). The one-dose BMP4-treated cells were cultured in suspension for 10 days in neural sphere formation assays. Only BMP ligands including BMP2, BMP4 and BMP7 significantly disrupted sphere formation of DIPG cells (Fig. 2a,b and Extended Data Fig. 3b). Notably, none of the treatments affected sphere formation of PPC (Fig. 2a,b). Series concentration of BMP4 treatment showed inhibitory effects of sphere formation at higher doses such as 50, 25 and 10 ng ml–1 of BMP4, but not low doses of 100 and 10 pg ml–1 BMP4 (Extended Data Fig. 3c).

a, Neural sphere formation of the indicated cell lines treated with vehicle (Ctr), BMP4, BMP7, LDN-193189 (LDN), activin A (AC) or TGF-β1 (all for 10 days); n = 3 independent experiments. b, Neural sphere counts from a. c, Viability (metabolic capacity) of indicated cells (n = 3 independent experiments) (all BMP4 treatments 50 ng ml–1 unless specifically mentioned). d, Representative bioluminescence images from animals implanted with luciferase-GFP engineered-TT150630 cells (n = 6 mice) with or without BMP4 pretreatment (n = 6 mice) in the pons at day 94. e, Representative bioluminescence images from animals implanted with SU-DIPG17 cells with (n = 6 mice) or without (n = 6 mice) BMP4 treatment in the pons at day 150. f, Kaplan–Meier analysis from animals implanted with TT150630 cells with (n = 6 mice) or without (n = 6 mice) BMP4 treatment in the pons. Log-rank test was performed. g, Kaplan–Meier analysis from animals implanted with SU-DIPG17 cells with indicated treatment (n = 6 mice per group) in the pons. Log-rank test was performed. h, Representative images of pons from animals implanted with TT150630 cells with or without BMP4 treatment analyzed by H&E staining. Regions marked by the box are magnified below. Scale bars, 1,000 µm (top), 50 µm (middle) and 20 µm (bottom). i, IF of pons section from animals implanted with TT150630 cells with or without BMP4 treatment for anti-HNA. Scale bars, 1,000 µm (top), 50 µm (middle) and 20 µm (bottom). For b and c, data represent the mean ± s.d.; two-tailed unpaired Student’s t-test was performed. Experiments in h and i were repeated three times with similar results.
Moreover, we conducted cell viability assays for DIPG cells carrying ACVR1 WT or mutant (SU-DIPG4)23 with BMP4, LDN-193189 or vehicle and found that BMP4 treatment significantly inhibited the proliferation of H3.3K27M and ACVR1 WT DIPG cells; none of these treatments affected PPCs and only LDN-193189 has inhibitory effects on SU-DIPG4 cells, which is consistent with a previous report (Fig. 2c and Extended Data Fig. 3d,e)14.
We also investigated whether BMP signaling affects tumor growth in orthotopic xenograft mouse models. Luciferase-engineered DIPG cells were pretreated with either 50 ng ml–1 BMP4 or vehicle for 24 h, and then used to establish xenografts via orthotopic injection of tumor cells into the pons of a cohort of B-NDG mice (Extended Data Fig. 4a). The mice transplanted with BMP4-treated TT150630 and SU-DIPG17 cells all showed consistently significant weaker luciferase signals than the control (Fig. 2d,e and Extended Data Fig. 4b,c). Kaplan–Meier survival curves showed that the BMP4 pretreatment significantly prolonged the survival of the mice (Fig. 2f,g and Extended Data Fig. 4d).
Hematoxylin and eosin (H&E) staining of tumor regions revealed that BMP4 pretreated mice had markedly lower numbers of tumor cells compared with control mice (Fig. 2h and Extended Data Fig. 4e). Further, IF staining against the human nuclear antigen (HNA) showed that there were significantly fewer tumor cells in the BMP4 pretreated mice (Fig. 2i and Extended Data Fig. 4f). These results demonstrate that one-dose BMP4 pretreatment perturbs intracerebral tumor establishment and significantly inhibits tumor growth.
BMP4 inhibits DIPG subtype through SMAD1/5 and SMAD4
Many BMP functions are related to SMAD1/5/8- and SMAD4-dependent pathways, mainly through transcriptional regulation of BMP signaling target genes26. The above results motivated us to investigate the necessity of the canonical BMP pathway for the observed growth inhibition of DIPG cells by BMP4 treatment. The expression of SMAD8 is relatively lower compared with that of SMAD1 and SMAD5 in DIPG cells (Supplementary Table 2). We successfully established both SMAD1/5 KD and SMAD4 KD DIPG cell lines (TT150630 and TT150714), as validated by both immunoblotting analysis for proteins level and abolishment of BMP response of the main BMP signaling transcriptional target genes SMAD7 (Fig. 3a–c). Sphere formation assays showed no inhibitory effect of BMP4 treatment of SMAD1/5 KD or SMAD4 KD cells (Fig. 3d,e). Therefore, stimulation of BMP signaling activity inhibits DIPG cell growth through SMAD1/5 and SMAD4, which prominently impacts transcriptome profiles30.

a, SMAD1/5 double KD in DIPG TT150630 and TT150714 was validated by immunoblotting analysis using antibodies against SMAD1/5 and GAPDH. b, SMAD4 KD in DIPG TT150630 and TT150714 was validated by immunoblotting analysis using antibodies against SMAD4 and GAPDH. c, qPCR analysis of SMAD7 expression in indicated control and SMAD1/5 or SMAD4 KD cells. BMP4, (50 ng ml–1) for 2 h. n = 3 independent experiments. d,e, Neural sphere formation of control and SMAD1/5 KD TT150630 and TT150714 DIPG cells (d) or of control and SMAD4 KD TT150630 and TT150714 DIPG cells (e) treated with vehicle, BMP4 (50 ng ml–1) or LDN (200 nM) for 10 days; n = 3 independent experiments (left). Neural sphere counts (right). The experiments in a and b have been repeated three times with similar results. For c, d and e, data represents the mean ± s.d.; statistical significance was calculated by two-tailed unpaired Student’s t-test.
BMP4 forces DIPG subtype to exit from stemness state
We explored the molecular consequences of BMP4 treatment in H3.3K27M and ACVR1 WT DIPG cells. Specifically, TT150630 cells were cultured in suspension and treated with 50 ng ml–1 BMP4 for 2 h, 24 h or 10 days. BMP4 treatment significantly downregulated OPC-related genes such as OLIG2 and ASCL1, induced AC-related gene like GFAP and repressed 50 known cell-cycle-related genes (positive regulators)9 (Fig. 4a). BMP4 treatment at 24 h significantly induced BMP signaling direct regulated genes, such as SNAI1, ID1 and SMAD7 in TT150630 cells (Fig. 4b). Two apoptosis related genes BMF and GADD45B and cell-cycle inhibitor p21CIP1 are also markedly induced31,32 (Fig. 4b). Note that similar expression trend impacts for these genes were also observed at earlier timepoints for the BMP4-treated TT150714 cells, but not for the SU-DIPG4 cells (Extended Data Fig. 5a–d).

a, OPC-related genes, AC-related and cell-cycle-related genes heatmap from RNA-seq transcriptome analysis of TT150630 cells treated with BMP4 at the indicated timepoints (n = 2 independent experiments). b, Volcano plots showing differentially expressed genes from transcriptome datasets of TT150630 with or without BMP4 treatment for 24 h. Upregulated genes (n = 619; red dots, |LFC (log2 fold change)| ≥1.5; P < 0.05); blue dots, downregulated genes (n = 1,860; |LFC | ≥ 1.5-fold; P< 0.05). Individual genes of interest are depicted; P value was calculated by Cuffdiff. c, Immunoblotting analysis of phosphorylated SMAD1/5 (Ser 463/465), total SMAD1, OLIG2 and SOX2 proteins in TT150630, TT150714 and SU-DIPG17 cell lysates with or without BMP4 treatment (50 ng ml–1) at indicated timepoints; H3 was used as a loading control. d, IF staining for phosphorylated SMAD1/5 (Ser 463/465), OLIG2, GFAP and CD133 in 3D cultures of TT150630 using microscaffold. BMP4, 50 ng ml–1. Scale bars, 600 μm. e, IF staining for GFAP, OLIG2 and phosphorylated SMAD1/5 proteins in TT150630 cells with or without BMP4 treatment (50 ng ml–1) for 48 h. Scale bars, 50 μm. f, IF of pons section from animals implanted with TT150630 cells with or without BMP4 treatment for anti-HNA, OLIG2 and GFAP. Scale bars, 50 µm. This figure represents nine independent tissues. g, Quantification of OLIG2-positive cells in all tumor cells (HNA positive) from animals implanted with TT150630 cells with or without BMP4 treatment; n = 9 independent tissue samples, data represents the mean ± s.d., statistical significance was calculated by two-tailed unpaired Student’s t-test. h, GSEA analysis using the indicated E2F (left) and apoptosis pathway (right) signature genes to compare TT150630 control and BMP4-treated 10-day neural spheres. i, Immunoblotting analysis of cell-cycle-related proteins phosphorylated Rb (Ser 780), total Rb, p-SMAD1/5 and GAPDH in TT150630, TT150714 and SU-DIPG17 cell lines. BMP4, 50 ng ml–1. j, Immunoblotting analysis of PARP1, cleavage PARP1 and activated cleavage CASPASE3 and GAPDH in TT150630, TT150714 and SU-DIPG17 neural spheres with or without BMP4 treatment (50 ng ml–1) for 10 days. The experiments in c, d, e, f, i and j were repeated three times with similar results.
Consistently, the proteins level of stemness markers SOX2 and OLIG2 are decreased significantly in BMP4-treated DIPG cells (Fig. 4c); IF staining of both three-dimensional (3D) culture and regular culture of TT150630 cells showed BMP signaling significantly prevents oligodendrocyte-like cells proliferation indicated by decreased expression of CD133 (the marker for tumor initiating/propagating cells) and OLIG2 (the marker for oligodendrocyte progenitor cells). And induction of astrocyte marker GFAP suggests that BMP signaling promotes cell differentiation towards astrocyte (Fig. 4d,e and Extended Data Fig. 5e). Moreover, IF staining of tissue section from the xenograft mouse model of TT150630 showed that BMP4 pretreatment group has fewer tumor cells with lower OLIG2-positive percentage and higher GFAP levels than the control group (Fig. 4f,g and Extended Data Fig. 5f). These results indicate that activation of BMP signaling apparently reduces the proliferation of DIPG cells by forcing their exit from the recently described ‘prolonged stem-cell-like state9.’
BMP4 leads to cell-cycle arrest and apoptosis in DIPG subtype
We found that BMP4 treatment significantly downregulated E2F targets and clearly upregulated apoptosis markers in TT150630 cells (Fig. 4h). The phosphorylation of retinoblastoma protein (Rb) at Ser 780 was also downregulated upon BMP4 treatment (Fig. 4i). The cell cycle inhibitor p21CIP1 is significantly upregulated upon BMP4 treatment for 24 h (Extended Data Fig. 5g). The cell cycle arrest by BMP4 treatment was also confirmed by propidium iodide staining assay (Extended Data Fig. 5h,i).
The apoptosis impact was examined at the protein levels of cleaved PARP1 and cleaved-CASPASE3. Both the cleaved PARP1 and cleaved-CASPASE3 proteins were elevated by BMP4 treatment in DIPG cells (Fig. 4j). FACS analysis of annexin V revealed a time-dependent increased apoptosis upon BMP4 treatment of TT150630 cells (Extended Data Fig. 5j,k). Thus, beyond forcing the DIPG cells to exit the prolonged stem-cell-like state (possibly towards an astrocyte lineage), BMP4 treatment also induces both apoptosis and cell cycle arrest of DIPG cells.
BMP4 alters global epigenetic landscape in DIPG subtype
H3K27M mutation reprograms the cancer epigenome to lead to tumorigenesis in DIPG10. To explore the molecular mechanism of BMP signaling in this DIPG subtype, we also examined the epigenome impact of this subtype DIPG by activation of BMP signaling. We performed assay for transposase-accessible chromatin with high throughput sequencing (ATAC-seq) and chromatin immunoprecipitation sequencing (ChIP–seq) analyses of SMAD1, H3K27ac, H3K27me3 and H3K27M in TT150630 cells. ChIP–seq analysis showed that SMAD1 is highly enriched at promoter regions upon activation of BMP signaling (Ctr 3,490 versus BMP4 9,257) (Extended Data Fig. 6a). Among the top 4,000 SMAD1 peaks in the BMP4-treated cells, H3K27ac and H3K27M signals, but not H3K27me3, are also enriched at these sites in the BMP4-treated cells (Fig. 5a). This indicates that activated SMAD1/5 co-occupies with active histone H3K27ac. Consistent with another published study, H3K27M signals colocalized with H3K27ac21 (Fig. 5a).

a, Heatmaps of ChIP–seq signals of SMAD1, H3K27ac, H3K27me3 and H3K27M at top 4,000 SMAD1 ChIP–seq significant peaks in TT150630 DIPG cells with or without BMP4 treatment (50 ng ml–1) for 2 h (top). Average ChIP–seq signal for SMAD1, H3K27ac, H3K27me3 and H3K27M in the indicated conditions corresponding with their ChIP–seq heatmaps (bottom); n = 2 independent experiments. b, Metagene plots showing the average ChIP–seq signal for representative H3K27ac signals upregulated (SMADs) and downregulated (OLIG2) motifs in TT150630 DIPG cells treated with vehicle or BMP4. c, HOMER identified enriched motifs in the indicated ATAC-seq peaks (TT150630). d, SEs detected in TT150630 DIPG cells with vehicle or 2 h-BMP4 treatment. e, Differential SEs restricted to vehicle and BMP4 treatment for 2 h of TT150630 as detected by DiffBind in DESeq2 mode. x axis, rank of differential SEs; y axis, LFC of SEs; n = 2 independent experiments. f, Representative IGV tracks for RNA-seq and ATAC-seq in PPC, TT150630 DIPG cells with vehicle or BMP4 treatment at CXXC5, ID1 and UBE2D2 (negative control, in gray) gene loci and for SMAD1 and H3K27ac ChIP–seq in TT150630 DIPG cells with vehicle or BMP4 treatment at CXXC5, ID1 and UBE2D2 (negative control, in gray) gene loci.
Enrichment of H3K27ac, but not H3K27me3, modification is seen in the upregulated genes upon BMP4 treatment (Extended Data Fig. 6b). These upregulated genes also show enrichment of SMAD1 and H3K27M signals (Extended Data Fig. 6b). The genes promoters with stronger H3K27ac peaks in BMP4 treatment cells were primarily those of the upregulated genes identified in RNA-seq analysis, for example, ID1, SMAD7, NOG and CXXC5. (Extended Data Fig. 6c). Conversely, the promoters with weaker H3K27ac peaks corresponded to the downregulated genes in BMP4-treated cells, for example, ASCL1, TLR5, CBLN4 and SLA (Extended Data Fig. 6c).
Interestingly, the downregulated signals of H3K27ac were found at the oligodendrocyte progenitor marker motif OLIG2 and upregulated signals of H3K27ac were found at SMADs motif (Fig. 5b), indicating that this neuroprogenitor regulator might not be able to access its target genes upon BMP signaling, and consequently failed to maintain the stemness. Moreover, this is consistent with ATAC-seq analysis of TT150630. The chromatin regions that became less accessible following BMP4 treatment revealed binding elements of neuroprogenitor marker proteins including SOX2, OLIG2 and ASCL1 (Fig. 5c). The known SMAD4 and SMAD1 binding motifs in the chromatin regions became more accessible upon BMP4 treatment (Fig. 5c).
Previous studies have shown that, although DIPGs with the H3K27M mutation show global loss of H3K27me3, several loci, including HOXA and HOXB clusters, retain H3K27me3 (refs. 33,34). Interestingly, we also found enrichment binding motifs of HOX family transcription factors in BMP4-treated DIPG cells (Fig. 5c), which is consistent with the increased expression of HOX genes and decreased H3K27me3 levels in HOXA and HOXB cluster loci (Extended Data Fig. 6d,e). In addition, decreased H3K27me3 levels in HOXA and HOXB cluster loci were also observed in the CHRDL1 KD DIPG cells, which mimics what we observed following BMP4 treatment (Extended Data Fig. 6e).
Thus, stimulating BMP signaling triggers widespread changes in the epigenetic landscapes that encompass reduced chromatin accessibility for lineage master regulators like SOX2, OLIG2 and ASCL1, and increased chromatin accessibility for SMAD1/5 and SMAD4. These chromatin landscape changes lead to downregulation of stemness and likely exit to differentiation.
CXXC5 associates with super-enhancers upon BMP signaling in DIPG
Super-enhancers (SEs) are large clusters of enhancer elements that are considered as main regulatory hubs35,36. We set out to determine SE pattern changes between control and BMP4-treated TT150630 cells. Consistent with previous studies, CDK6, SOX2, OLIG2 and NESTIN, which control both DIPG cell identity and malignant state, are SE-associated genes in TT150630 (Fig. 5d)10,23. Interestingly, the rank of these genes SEs dropped significantly and expression decreased significantly in BMP4-treated TT150630 cells, suggesting that BMP signaling prevents DIPG cell growth through downregulating these SE-associated genes (Fig. 5e and Extended Data Fig. 6f). Consistently, these SE-associated genes are all ranked down in CHRDL1 KD cells (Extended Data Fig. 6g).
The top-ranking SE-associated genes in BMP4-treated cells could potentially be the candidate genes that have a tumor-suppressive effect. The top-ranking SE-associated genes in BMP4-treated cells are CXXC5, ID3 and SMAD7 from top to bottom (Fig. 5d). The CXXC5 promoter region is highly accessible in PPC compared with TT150630 and the expression level in PPC is much higher than TT150630 (Fig. 5f). BMP4 treatment also makes the promoter region more accessible, which correlates with increasing CXXC5 expression (Fig. 5f). The SMAD1 ChIP–seq reveals SMAD1 associates with the promoter of CXXC5 after BMP4 treatment (Fig. 5f). Therefore, CXXC5 is an SE-associated gene in H3.3K27M and ACVR1 WT DIPG upon BMP4 treatment.
CXXC5 functions as a tumor suppressor in DIPG subtype
We next validated that CXXC5 is a direct transcriptional target gene of BMP signaling: pretreating the four DIPG cell lines with the protein synthesis inhibitor cycloheximide (CHX) did not block CXXC5 mRNA upregulation upon BMP4 treatment (Extended Data Fig. 7a); the CXXC5 mRNA level increased after BMP4 treatment and we detected a significantly elevated protein level in these cell lines within 24 h (Extended Data Fig. 7b,c); we also found that SMAD1/5 KD and SMAD4 KD abolished the capacity of BMP signaling to induce CXXC5 transcription (Extended Data Fig. 7d–f). In contrast, no CXXC5 induction was observed in the SU-DIPG4 cells upon BMP4 treatment, although BMP4 treatment caused increased p-SMAD1/5 levels and increased expression of ID1 and SMAD7 in this cell line (Extended Data Fig. 7g,h).
CXXC5 has been demonstrated as a regulator that coordinates TGF‐β, BMP and Wnt signaling37,38, and CXXC5 acts as a tumor suppressor by inducing apoptosis in hepatocellular carcinoma39. Indeed, CXXC5 expression in the eight primary DIPG cell lines examined is reduced compared with PPC cells (Extended Data Fig. 7i). Moreover, CXXC5 KD blocks the growth inhibitory role of BMP4 in both TT150630 and TT150714 DIPG cells (Fig. 6a,b and Extended Data Fig. 8a,b). Interestingly, analysis of previous published dataset indicates that an elevated CXXC5 expression level is correlated with prolonged survival times for high-grade midline glioma patients (including DIPG)27 (Fig. 6c), suggesting that CXXC5 may function as a tumor suppressor in DIPG.

a, Neural sphere formation of control and CXXC5 KD TT150630 cells treated with vehicle, BMP4 (50 ng ml–1) for 2 h or LDN (200 nM) for 10 days; n = 3 independent experiments. b, Neural sphere counts in a. c, Kaplan–Meier survival curves for DIPG patients in the University of California Santa Cruz (UCSC) Xena cohort27. The patient cohort was separated into CXXC5 high (n = 26 patients) and low (n = 25 patients) expression groups. Log-rank test was performed. d, Viability of control and CXXC5 KD TT150630 and TT150714 cells; n = 3 independent experiments. e, GSEA analysis using the E2F targets and apoptosis gene sets to compare control and CXXC5 KD TT150714 DIPG cells. f, Heatmaps for cell-cycle inhibitor and cell-cycle positive regulator genes of RNA-seq transcriptome analysis in control and CXXC5 KD TT150714 DIPG cells (n = 2 independent experiments). g, Immunoblotting analysis for CXXC5 and TUBULIN proteins in the Dox-inducible overexpression CXXC5 TT150630 and TT150714 cells with or without Dox for 72 h. h, Viability of indicated cells treated with Dox or not for the number of days indicated (n = 3 independent experiments). i, GSEA analysis using the E2F target gene set to compare control and CXXC5-inducible overexpression TT150630 DIPG cells; n = 2 independent experiments. j, Schematic for the construction of a xenograft mouse model upon orthotopic injection of 1 × 106 luciferase-GFP engineered CXXC5-TET-ON TT150630 DIPG cells in the pons of B-NDG mice. Created with BioRender.com. k, The normalized bioluminescence activity was plotted and the statistical difference between control (–Dox) and Dox-fed (+Dox) groups was significant (n = 6 mice in each group). Boxplots define the interquartile range (IQR) split by the median, with whiskers extending to the most extreme values within 1.5 × IQR beyond the box; statistical significance was calculated by two-tailed unpaired Student’s t-test. l, Representative bioluminescence images from animals implanted with luciferase-GFP engineered-TT150630 cells containing inducible CXXC5 expression system in the pons at day 115 fed with (n = 6 mice) or without (n = 6 mice) Dox. For b, d and h, data represents the mean ± s.d.; two-tailed unpaired Student’s t-test was performed. The experiments in g were repeated three times with similar results.
Specifically, CXXC5 KD impairs cell cycle arrest by BMP4 treatment in DIPG (Extended Data Fig. 8c,d). Consistently, depletion of CXXC5 increases the proliferation rate of DIPG cells (Fig. 6d). GSEA analysis for CXXC5 KD DIPG cells showed significant upregulation of a set of cell-cycle positive regulator genes (Fig. 6e,f) that are downregulated upon BMP4 treatment in DIPG cells. Beyond these, we found that depletion of CXXC5 also decreased apoptosis (Fig. 6e). Moreover, we found that expression of CXXC5 in TT150630 cells induced apoptosis (Extended Data Fig. 8e,f). Consistently, we found that the inducible expression of CXXC5 in DIPG cells triggered both obvious cell growth inhibition and apoptosis (Fig. 6g–i and Extended Data Fig. 8g–i).
We next established xenografts via orthotopic injection of luciferase-engineered-TT150630 cells (enabling Dox-inducible expression of CXXC5) with or without pretreatment of Dox into the pons of B-NDG mice (Fig. 6j). The mice injected with Dox-treated DIPG cells were maintained on Dox for the duration of the study. TT150630 tumor growth was significantly slower in the Dox-fed mice group (Fig. 6k,l). Viewed together, these results suggest that CXXC5 function as a tumor suppressor in H3.3K27M and ACVR1 WT DIPG.
CXXC5 regulates cell-cycle-related genes expression in DIPG subtype
CXXC5 has been reported to bind to chromatin to regulate pluripotency network genes in mouse embryonic stem cells40,41. We then investigated the potential downstream target genes of CXXC5 in DIPG by RNA-seq and CXXC5 ChIP–seq analysis (Extended Data Fig. 9a,b). Consistently, there is markedly decreased expression of the cyclin-dependent kinase inhibitor p21CIP1 in CXXC5 KD DIPG. Interestingly, GFAP is almost abolished in CXXC5 KD DIPG (Extended Data Fig. 9b,c), indicating differentiation promoted by BMP signaling is also impaired by CXXC5 KD. The ChIP–seq data show that CXXC5 occupies the regulatory regions of known cell-cycle positive regulator genes loci such as p21CIP1 (Extended Data Fig. 9b).
Cell-cycle positive regulators were downregulated upon BMP4 treatment (Extended Data Fig. 9c) and CXXC5 binds at their promoters (Extended Data Fig. 9b), suggesting that CXXC5 negatively regulates the expression of these genes upon BMP4 treatment. Further, the fact that BMP4-signaling-triggered induction of p21CIP1 was totally inhibited in CXXC5 KD cells (Extended Data Fig. 9c) demonstrates that p21CIP1 is positively regulated by CXXC5. Accumulation of p21CIP1 is also significantly reduced in CXXC5 KD DIPG cells (Extended Data Fig. 9d) and the accumulation of p21CIP1 in DIPG cells is also dependent on SMAD1/5 and SMAD4 (Extended Data Fig. 9e,f). Moreover, overexpression of CXXC5 significantly reduced the expression of cell-cycle-related genes (Extended Data Fig. 9g). However, we observed no induction of p21CIP1 in BMP4-treated SU-DIPG4 cells (Extended Data Fig. 9h); accordingly, there was no BMP4-triggered induction of CXXC5 in SU-DIPG4 cells.
These results collectively support the view that CXXC5 functions as a tumor suppressor by binding the promoters and activating or repressing transcription of known cell cycle inhibitors p21CIP1 or cell-cycle-related genes, respectively, in DIPG subtype.
CXXC5 positively regulates BMP signaling
GSEA analysis for CXXC5 KD DIPG cells also revealed reduced BMP and TGF-β signaling activity (Fig. 7a), which is consistent with a previous report that CXXC5 coordinates TGF‐β and BMP signaling37. TGF‐β1 treatment of TT150630 and TT150714 cells did not induce CXXC5 expression (Fig. 7b), nor did TGF‐β1 treatment impact OLIG2 or p21CIP1 (Fig. 7b), which explained our earlier finding that TGF‐β1 treatment did not inhibit the growth of DIPG cells.

a, GSEA analysis using the ‘BMP signaling signature’ and ‘TGF-β signaling signature’ gene sets to compare control and CXXC5 KD TT150714 DIPG cells (n = 2 independent experiments). b, qPCR analysis of relative expression of the genes indicated in TT150630 and TT150714 cells treated with TGF-β1 (10 ng ml–1) at indicated timepoints; n = 3 independent experiments. c, Immunoblotting for phosphorylated SMAD1/5 (Ser 463/465), total SMAD1, CXXC5 and GAPDH in TET- ON inducible CXXC5 expression TT150630 (left) and TT150714 (right) cells treated with vehicle or BMP4 (50 ng ml–1) for 24 h. The relative intensity of the p-SMAD1/5 protein level compared with total SMAD1 is indicated. d, qPCR analysis of indicated genes relative expression in the indicated cells treated with BMP4 (50 ng ml–1) at indicated timepoints; n = 3 independent experiments. e, IGV tracks for CXXC5, SMAD1 and H3K27ac ChIP–seq in TT150630 cells treated with vehicle or BMP4 for 2 h (SMAD1 and H3K27ac) or 24 h (CXXC5) at indicated gene loci. The experiments in c were repeated three times with similar results. For b and d, data represents the mean ± s.d.; statistical significance was calculated by two-tailed unpaired Student’s t-test.
Moreover, we have two lines of direct evidence linking CXXC5 to regulate the expression of BMP signaling components ACVR1, BMPR2 and BMP2: these genes are all significantly downregulated in CXXC5 KD DIPG cells, and our ChIP–seq data showed CXXC5 occupancy at the promoters of these three loci (Extended Data Fig. 10a). In line with decreased expression of BMPR2, ACVR1 and BMP2 in the CXXC5-depleted DIPG cells, p-SMAD1/5 level is decreased markedly in CXXC5 KD cells (Extended Data Fig. 10b). Moreover, p-SMAD1/5 level is increased in the inducible overexpression CXXC5 DIPG cells suggesting that CXXC5 enhances the BMP signaling through upregulation of these components of the pathway (Fig. 7c).
Indirectly, we found BMP4 treatment of CXXC5 KD DIPG cells caused weaker or abolished induction of SMAD1 target genes compared with control DIPG cells (Fig. 7d). It was highly interesting to note in our ChIP–seq data that CXXC5 apparently regulates its own expression: the promoter of the CXXC5 locus was among the detected ChIP–seq peaks upon BMP4 treatment (Fig. 7e). The results showed that there is an enrichment of CXXC5 binding signal among the top 4,000 peaks from SMAD1 ChIP–seq in BMP4-treated TT150630 cells (Fig. 7e and Extended Data Fig. 10c). Consistently, there is an enrichment of CXXC5 binding signal among the upregulated 5,868 peaks from H3K27ac ChIP–seq in BMP4-treated TT150630 cells (Extended Data Fig. 10c). These results suggest that CXXC5 positively regulates BMP signaling by two mechanisms: CXXC5 regulates transcription of components of the BMP signaling pathway, such as, for example, ACVR1, BMP2 and BMPR2; CXXC5 is recruited to the SMAD1 binding sites of the BMP direct target genes and promotes the expression of these genes.
We then examined whether ACVR1 expression might be correlated positively with DIPG prognosis. We found that aberrantly elevated ACVR1 expression is correlated positively with longer survival time (Extended Data Fig. 10d) (E-TABM-1107)27. Coupled with our earlier finding that high CHRDL1 expression is an indicator for poor prognosis, it seems that selective modulation of BMP signaling may represent an attractive therapeutic strategy for treating this subtype of DIPG with H3.3K27M and ACVR1 WT.
HDAC inhibitors are potential therapeutic strategy for this subtype DIPG
It is well known that HDAC proteins are the inhibitory factors for transcriptional regulation of TGF‐β/BMP signaling and our earlier studies showed that the HDAC inhibitor Panobinostat can upregulate the BMP signaling. Thus, we performed a drug screen to find potential drugs that could inhibit DIPG growth using Panobinostat as a positive control. We found three efficient HDAC inhibitors (Dacinostat, Quisinostat and Panobinostat) that all significantly upregulated BMP signaling, as indicated by the increased p-SMAD1/5 signals and increased expression of BMP signaling genes upon drug treatments (Fig. 8a,b); DIPG growth in vitro showed marked inhibition (Fig. 8c,d and Supplementary Table 4). Consistently, these drugs also lead to an increase in both CXXC5 protein and mRNA levels (Fig. 8a,b). Notably, Panobinostat and Quisinostat have been reported to have antitumor efficacy in DIPG22,23,42. We found that both Panobinostat and Quisinostat have great inhibitory effects on PPC while Dacinostat has only a slight inhibitory effect on PPC (Fig. 8e), suggesting that Dacinostat might be a better choice for a therapeutic strategy. Thus, we provide a better option, Dacinostat, for potential treatment of DIPG.

a, Immunoblotting for phosphorylated SMAD1/5 (Ser 463/465), total SMAD1, CXXC5, H3K27ac and Histone H3 in TT150630 and TT150714 with or without Quisinostat, Dacinostat and Panobinostat for 48 h. Experiments were repeated at least three times with similar results. b, qPCR analysis of indicated genes relative expression in TT150630 and TT150714 cells treated with indicated drugs for 48 h. Data represents the mean ± s.d., statistical significance was calculated by two-tailed unpaired Student’s t-test; n = 3 independent experiments. c, TT150630, TT150714 and SU-DIPG17 were treated with Quisinostat, Dacinostat and Panobinostat with series of concentration for 72 h. Cell viabilities normalized to 0.1% DMSO control were plotted. Relative viability is calculated as T72-T0, and the value for DMSO-treated cells is set at 100%. The IC50 for individual drugs in three cell lines are listed in Supplementary Table 4. Data represents the mean ± s.d.; n = 3 independent experiments. d, Viability of indicated cells treated with Quisinostat, Dacinostat and Panobinostat for the number of days indicated (n = 3 independent experiments). e, Viability of PPCs treated with Quisinostat, Dacinostat and Panobinostat at 100 nM for the number of days indicated (n = 3 independent experiments). f, Viability of indicated cells treated with Quisinostat, Dacinostat or Panobinostat at 100 nM for the number of days indicated (n = 3 independent experiments). g, In H3.3K27M and ACVR1 WT DIPG, elevated CHRDL1 expression level contributes to reduced BMP signaling activity. BMP signaling has a significant growth inhibitory effect on this subgroup of DIPG tumors and cells in a SMAD-dependent manner. The growth inhibition effects of BMP signaling are mediated largely through transcriptional activation of CXXC5 gene expression. CXXC5 is a multifunctional tumor suppressor: CXXC5 induces the transcription of cell cycle inhibitors (for example, p21CIP1) and also positively regulates BMP signaling, specifically by inducing transcription of both BMP target genes and BMP pathway component genes (BMPR2, BMP2 and ACVR1) expression, forming a positive feedback loop. Indicated HDAC inhibitor (HDACi) drugs block DIPG growth and boost BMP signaling. For d–f, data represent the mean ± s.d.; statistical significance was calculated by two-tailed unpaired Student’s t-test.
More importantly, our findings suggest that upregulation of BMP signaling by HDAC inhibitors could be the general function for this type of drug in DIPG. Interestingly, HDAC inhibitors were identified by Connectivity Map analysis (L1000) for their similar impacts on the transcriptome of DIPG cells treated with BMP4 (ref. 43). The viability assays demonstrated that SMAD1/5 KD can partially block the inhibitory function of HDAC inhibitors on this subtype DIPGs (Fig. 8f), suggesting that HDAC inhibitors function as growth inhibitors partially through activation of BMP signaling in DIPG. Thus, designing better HDAC inhibitors that can be delivered through the blood–brain barrier and boost BMP signaling should be pursued in future DIPG therapeutic strategies.
Discussion
Here, we elucidated the context-dependent role of BMP signaling in H3.3K27M and ACVR1 WT DIPG. We detected reduced BMP signaling activity, and implicated elevated CHRDL1 levels in the observed reduction of BMP signaling in this DIPG subtype. We subsequently demonstrated that BMP signaling has a significant growth inhibitory effect on this DIPG subtype. Moreover, we showed that the growth inhibition effects of BMP signaling are mediated largely through activation of CXXC5 gene expression. In addition, we found that CXXC5 is a multifunctional tumor suppressor: CXXC5 induces the transcription of cell cycle inhibitors and also positively regulates BMP signaling, specifically by inducing transcription of both BMP target genes and BMP pathway component gene expression. Lastly, we showed that several epigenetic regulator inhibitors, particularly HDAC inhibitors, that have antitumor efficacy on DIPG all upregulate BMP signaling (Fig. 8g).
In agreement with BMP signaling tumor-suppressive effects on H3.3K27M and ACVR1 WT DIPGs, patients with DIPG who have high expression of CXXC5 or ACVR1 tend to have a relatively good prognosis; better prognosis is also associated with low CHRDL1 expression. These trends suggest at least three opportunities for developing therapies to treat this deadly cancer. First, considering that CHRDL1 is an antagonist of the BMP pathway and is correlated with poor prognosis of DIPG, promoting BMP activity could be achieved by blocking CHRDL1 activity; perhaps a neutralizing antibody against CHRDL1 would be suitable for this secreted protein. Second, given that activation of BMP signaling inhibits tumor growth, degrading or blocking FPKBP12—a negative regulator of BMP receptors—for example, using PRO-TAC technology or FK506 (ref. 44) could be another option for anti-cancer therapy by inducing BMP signaling activation. Third, enhancing CXXC5 activity would be expected to suppress tumor growth by positively regulating BMP signaling. Given that multiple HDAC inhibitor drugs, including our finding of Dacinostat having antitumor efficacy for DIPG, are capable of positively regulating BMP signaling, combining these drugs with our suggested strategy could lead to better therapeutic outcomes.
Beyond these biomedical considerations, our findings raise multiple scientific questions that could help deepen our understanding of DIPG oncogenesis. As elevated CHRDL1 expression is correlated with poor prognosis, it will be interesting to investigate the mechanism(s) underlying the high expression of CHRDL1 in H3K27M DIPG cells. It should be informative to test whether the H3K27M mutation promotes CHRDL1 expression in DIPG cells. Investigating the BMP-dependence of the oncogenic impacts of CHRDL1 will also probably expand our understanding of the apparently diverse tumor-related functions of this protein. Mirroring findings from previous studies of other DIPG subtypes, our study extended understanding about how arrested differentiation and a prolonged stem-cell-like state drive the etiopathology of DIPG. Recalling that ID1/2/3 each function in maintaining stemness45, and given that BMP signaling induces the expression of ID1/2/3, it will be fascinating to determine whether any of these proteins specifically contribute to the oncogenesis of this DIPG subtype. It is reasonable to expect that these and related investigations will further delineate the roles of BMP signaling in DIPG and will provide clues enabling the development of precision therapeutics.
Methods
Our research complies with all relevant ethical regulations of the Tsinghua University. The animal experiments conducted as part of this research were completed in accordance with the guidelines provided by the Tsinghua University Animal Care and Use Committee. Mice were monitored weekly for signs of ill health or overt tumors; once mice displayed signs of hydrocephalus (domed head) or neurological duress, they were killed humanely as defined by IACUC (17-XQR1, PI: Qiaoran Xi). IACUC guidelines recommend limiting solid tumors to 10% of the host’s body weight. This criterion was not exceeded in this study. All human cell cultures were generated with informed consent and in compliance with Institutional Review Board (IRB)-approved protocols (KY 2018-042-02).
Reagents
Recombinant BMP4 (catalog no. 120-05ET), BMP7 (catalog no. 120-03p) and TGF-β 1 (catalog no. 100-21c) were purchased from PeproTech; Activin A (catalog no. 338-AC-010) is from R&D Systems; LDN-193189 (catalog no. SML0559) is from Sigma; SB431542 (catalog no. S1067) is from Selleck; CHX (catalog no. 94271) is from amresco; Doxycycline hyclate (catalog no. D8960) is from Solarbio.
Antibodies
Phosphorylated SMAD1/5/8 (catalog no. 9511L), SMAD1 (catalog no. 9743), cleaved-CASPASE3 (catalog no. 9664), CXXC5 (catalog no. 84546 S), cleaved PARP1 (catalog no. 5625S), H3K27ac (catalog no. 8173S) and H3K27M (catalog no. 74829S) are from Cell Signaling Technology; SMAD1/5 antibody (catalog no. ab75273), CD133 (catalog no. ab19898), phosphorylated Rb S780 (catalog no. ab47763) and RB (catalog no. ab181616) are from Abcam; SMAD4 (catalog no. sc-7966) is from Santa Cruz Biotechnology; OLIG2 (catalog no. AB9610) and HNA (catalog no. 4383) are from Millipore; GFAP (catalog no. z0334) is from Dako; p21CIP1 (catalog no. 556431) is from BD Biosciences; SOX2 (catalog no. 3579) and H3K27me3 (catalog no. 61017) are from Active Motif; histone H3 (catalog no. BE3015) is from Easybio. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (catalog no. ta-08) is from ZSGB-BIO; β-tubulin (catalog no. Abm59005-37B-PU) is from Beijing protein innovation; horseradish peroxidase (HRP) anti-mouse IgG (catalog no. 115-035-003) and HRP anti-rabbit IgG (catalog no. 111-035-003) are from Jackson ImmunoResearch Labs; Alexa Fluor 594 secondary antibody (catalog no. A-21207) is from Invitrogen. For detailed antibody information, please refer to the Reporting Summary.
Cell lines and cell culture
The patient-derived DIPG cell lines were maintained following a previously described method20. Briefly, DIPG cells were cultured in plates coated with matrigel (catalog no. 356243, Corning) (1%, 4–12 h at 37 °C) and containing a serum-free medium with the following composition: DMEM (C11995500BT, Invitrogen), B27 (catalog no. 17504044, Gibco), N2 (catalog no. 17502048, Gibco), bFGF (20 ng ml–1; 100-18B, PeproTech), EGF (20 ng ml; AF-100-15, PeproTech), PDGF-AB (20 ng ml; 100-00AB, PeproTech) and 1% penicillin/streptomycin (03-033-1B, Biological Industries). PPC were cultured in the above medium without PDGF-AB. HEK293T cells were grown in a DMEM medium supplemented with 10% FBS (FCS500, ExCell Bio) and 1% penicillin/streptomycin. SU-DIPG17 cells and SU-DIPG4 were kindly provided by Y. Tang23. These two cells were maintained in Tumor Stem Medium (TSM) (1:1 mixture of Neurobasal-A medium (catalog no. 10888-022, Thermo Fisher) and DMEM/F-12 (catalog no. 11330-032, Thermo Fisher)) supplemented with 10 mM HEPES (catalog no. 15630-080, Thermo Fisher), 1 mM sodium pyruvate (catalog no. 11360-070, Thermo Fisher), 0.1 mM MEM nonessential amino acids (catalog no. 11140-050, Thermo Fisher), 1× GlutaMAX-I supplement (catalog no. 35050-061, Thermo Fisher), 1% penicillin/streptomycin (03-033-1B, Biological Industries), 1× B27 supplement without vitamin A (catalog no. 12587001, Gibco), EGF (20 ng ml–1), bFGF, PDGF-AA (10 ng ml–1; 100-13A, PeproTech), PDGF-BB (10 ng ml–1; AF-100-14B-100, PeproTech) and heparin (2 μg ml–1; catalog no. 07980, StemCell Technologies). The cells were digested with TrypLE (catalog no. 12604013, Gibco) and subsequently transferred to new plates every 2–3 days. We confirmed the authenticity of all cells by analyzing short tandem repeats. Importantly, all cells tested negative for the presence of mycoplasma.
Immunocompromised mice
The present xenograft animal study used 4-week-old female NOD-Prkdcscid1l2rgtm1/Bcgen mice (B-NDG mice) (Biocytogen). Animals were housed at 20–22 °C with 12 h:12 h light:dark cycles at 50–60% humidity.
Intracranial xenotransplantation
All experiments were performed using orthotopic cell xenograft models that were generated by injecting luciferase-engineered DIPG cells into the pons of 4-week-old female B-NDG mice. Specifically, luciferase-engineered DIPG cells were divided into two cohorts and pretreated with either 50 ng ml–1 BMP4 for 24 h or with a control vehicle instead. We resuspended 1 × 105 DIPG cells in 5 μl PBS and implanted them into the brainstem of immunodeficient mice under the control of Nanoject III Programmable Nanoliter Injector (Drummond Scientific Company). At 2 weeks after implantation, we measured the tumor burden once a week by bioluminescence imaging using the IVIS Spectrum imaging system (PerkinElmer). For Tet-On CXXC5 in vivo studies, Tet-On CXXC5 cells were injected into the brainstem of B-NDG mice and half of the mice were maintained on dox (200 mg kg–1 fodder and 2 mg ml–1 water) throughout the duration of the study.
Viral production and generation of DIPG stable cell lines
We established DIPG cell lines containing luciferase-green fluorescent protein (GFP) by infecting the DIPG cells with pLEX-based lentivirus carrying luciferase-GFP. The lentivirus plasmid was kindly provided by H. Zheng. To generate plasmids containing short hairpins against SMAD1, SMAD5, CXXC5 and CHRDL1, we digested the pLKO.1 vector with EcoRI/AgeI enzymes and then ligated it with the annealed oligos. J. Massagué provided the SMAD4 shRNA lentivirus vector. We provide a list of the shRNA oligonucleotide pairs sequences in Supplementary Table 5. The construct containing inducible overexpression of CXXC5 was generated by cloning the cDNA of the CXXC5 gene into the DOX-inducible pLVX-Tight-Puro vector using ClonExpress MultiS One Step Cloning Kit (catalog no. C113, Vazyme Biotech).
Target plasmids RRE, REV and VSVG were cotransfected into HEK293T cells for lentivirus packaging using Lipofectamine 2000. The supernatants containing lentivirus particles were collected 48 h after transfection. Cells were infected with lentiviruses in the presence of 6–8 μg ml–1 Polybrene (Sigma-Aldrich). The infected cells were selected with puromycin (catalog no. p8833, Sigma) (pLKO.1, pLVX vector) or sorted by GFP IF (pLEX vector).
Cell viability assay
We performed cell viability assays using the CellTiter-Blue Luminescent Cell Viability Assay (catalog no. PR-G8081, Promega) according to the specifications available from the manufacturer. The cells were plated in 96-well plates (seeding density of 2,000 cells per well). In the growth assays, the cells were incubated with CellTiter-Blue reagent and luminosity intensity was measured every 2 days until days 6–8. As for viability analyses, the cells were treated with various concentrations of specific drugs after cell seed 24 h. To ensure statistical robustness, we performed three independent triplicates of each condition and inferred significance using two-tailed unpaired Student’s t-test.
Sphere formation assay
The sphere formation assay was performed by seeding 2,000 DIPG cells in each of the ultralow attachment 96 wells (Corning) present in each plate; cells were treated with either a vehicle or designated reagents in triplicate. A total of three replicates were performed for each condition. The cells were cultured for a total of 10 days, with an extra 20–30 μl medium being added to each well every other day. We used a stereoscope to obtain the Panoramic images for each condition.
Cell cycle analysis
Cell cycle analysis was performed using propidium iodide (Beyotime) staining. A total of 106 DIPG cells were washed with ice-cold PBS buffer twice and incubated in 70% ethanol at 4 °C overnight. The cell pellets were collected and washed with cold PBS twice. The cell pellets were then resuspended and incubated in staining buffer containing RNase A (100 μg ml–1; Transgen biotech) and propidium iodide (50 μg ml–1; Beyotime) at 37 °C for 30 min. After centrifugation, discard the staining buffer and resuspend the pellet in cold PBS, then the cell cycle was analyzed by flow cytometry (BD FACS Calibur) and the data were analyzed using FlowJo v.10 (FlowJo, LLC).
Apoptosis assay
We harvested the cells and then stained them using the Annexin V/PI Apoptosis Detection Kit, following the instructions of the manufacturer (catalog no. 556547, BD). We then performed FACS analysis to count the proportion of cells that underwent apoptosis. The original data were analyzed using FlowJo v.10 (FlowJo, LLC).
Bioluminescence imaging
Mice were anesthetized with isoflurane before bulbus oculi injection. Imaging started 1 min after the injection of d-luciferin. The bioluminescence signal was measured using the region of interest tool available in the Living Image v.4.4 software.
Histopathology and immunohistochemistry
Histopathological evaluation of mouse pontine was performed on H&E-stained paraffin sections. For H&E, we used xylene to deparaffinize 5 μm-thick sections twice and for 3 min each time. The slides were then gradually and consecutively immersed for 3 min in 100%, 95%, 70% and 50% ethanol. The slides were counterstained with H&E and dehydrated before adding the mounting medium. To perform immunohistochemistry staining, sections were deparaffinized with xylene, rehydrated and finally subjected to antigen retrieval in a citrate-based buffer (Origene ZLI-9064) in a microwave oven for 15 min. The slides were incubated with 3% H2O2 for 10 min at room temperature and blocked with a solution containing PBS and 5% bovine serum albumin (V900933) 1 h before overnight incubation with OLIG2 (1:200) or GFAP (1:500) antibody. The slides were then incubated with secondary antibody conjugated with ImmPACT, then incubated with ImmPACT DAB EqV Substrate (catalog no. ZLI-9019, ZSGB-BIO). Finally, the slides were fixed in the mounting medium (Solarbio).
Western blotting
Cell lysates were prepared using RIPA lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.1% SDS and 1% NP-40) together with phosphatase (Roche) and proteinase (Sigma) inhibitor cocktails. Extracted proteins were boiled at 100 °C for 10 min and then subjected to electrophoresis through 8–15% SDS–polyacrylamide gel electrophoresis. For detailed antibody information, please refer to the Reporting Summary.
ChIP–seq
ChIP–seq was performed according to the protocol implemented in a previous study, with some minor modifications46,47. Specifically, approximately 1 × 107 cells were crosslinked for 10 min at 37 °C in 1% formaldehyde PBS (Thermo Scientific) followed by quenching with 0.125 M glycine for 5 min. The cells were then washed twice in ice-cold PBS, and the cell pellets were either frozen and stored at −80 °C, or instead lysed with SDS lysis buffer (1% SDS) on ice. The chromatin was sheared using a sonicator (Nanjing Xinchen Biotechnology) with the following settings: ten cycles of 30 s each, and off for 30 s, at an amplitude of approximately 30%. The lysates were then centrifuged for 10 min at 4 °C, after which the supernatants were collected and diluted with dilution buffer to make it possible to reach a final concentration of 0.1% SDS. The sonicated lysates were precleared with Protein G Agarose Resin (catalog no. 36405ES08, Smart-Lifesciences) for 1 h at 4 °C, and then incubated overnight at 4 °C with antibody-conjugated Protein G Agarose beads. The beads were washed once with a low-salt buffer composed of 0.1% SDS, 1% Triton X-100, 20 mM Tris-HCl (pH 8.1), 2 mM EDTA, 150 mM NaCl and 1× complete protease inhibitor. After this, the beads were washed again once with a high-salt buffer composed of 0.1% SDS, 1% Triton X-100, 20 mM Tris-HCl (pH 8.1), 2 mM EDTA, 500 mM NaCl and 1× complete protease inhibitor. Finally, the beads were washed twice with an LiCl buffer composed of 10 mM Tris-HCl (pH 8.1), 1% IGEPAL-CA630, 1% sodium deoxycholate, 1 mM EDTA, 250 mM LiCl and 1× complete protease inhibitor and a Tris-EDTA buffer. The DNA was then eluted from beads twice on the rotator with an elution buffer (0.1 M NaHCO3, 1% SDS) for 15 min at room temperature. High-speed centrifugation was performed to pellet the beads and collect the supernatants. The crosslinking was reversed with a final concentration of 0.2 M NaCl overnight at a temperature of 65 °C. RNA and proteins were digested using RNase A and proteinase K, respectively. We purified the DNA by performing phenol chloroform extraction and ethanol precipitation. Purified ChIP DNA was used to prepare Illumina multiplexed sequencing libraries, which were run on the Illumina HiSeq X platform (Novogen).
ATAC-seq library generation and sequencing
The ATAC-seq libraries were prepared as per described in the Omni-ATAC protocol, with inhouse assembled transposon. Briefly, 50,000 cells were washed with PBS and then centrifuged at 500g for 5 min in a prechilled (4 °C) fixed-angle centrifuge. After centrifugation, we discarded the supernatant and resuspended the cell pellets on 50 μl of a lysis buffer composed of 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.01% Tween 20 and 0.01% digitonin. The cells were then incubated for 3 min on ice. After lysis, we added 1 ml of lysis buffer that did not contain NP-40 and digitonin, and pelleted the nuclei at 500g for 10 min. Nuclei pellets were then suspended in 50 μl of a transposition mix composed of 10 μl 5× LM buffer, 16.5 μl PBS, 4 μl 2 mM transposon (Robust Tn5 Transposase, Robustnique), 0.5 μl 1% digitonin, 0.5 μl 10% Tween 20 and 18.5 μl water. This was achieved by pipetting up and down six times. The transposition reactions were incubated in a thermomixer at a temperature of 37 °C for 30 min, with shaking at 800g. The reactions were cleaned up with Zymo DNA Clean and Concentrator-5 columns. We amplified the eluted DNA with the NEBNext 2× MasterMix, and implemented the following thermocycler conditions: 72 °C for 5 min; 98 °C for 30 s; ten cycles of 98 °C for 10 s, 63 °C for 30 s and 72 °C for 1 min; and a final hold temperature of 4 °C. We implemented a final fragment size selection using the upper cutoff of 0.5× Ampure XP beads, followed by the lower cutoff of 1.3× Ampure XP beads and elution in 20 μl water. The sequencing libraries were qualified with Qubit and Agilent Bioanalyzer, and sequenced on an Illumina Hiseq ×10 to obtain 30 M 2 × 150 base pair (bp) paired-end reads for each library.
RNA-seq and qRT–PCR
Total RNA was extracted using a Total RNA Purification Kit according to protocol made available by the manufacturer (catalog no. 8034111, DAKEWE), and resuspended in 30 μl nuclease-free water. RNA libraries were prepared for sequencing using the Illumina TruSeq Stranded Total RNA Library Prep Kit. The libraries were sequenced on the Illumina HiSeq X platform (Novogen). A total of 1 μg purified RNA was reverse transcribed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) following the manufacturer’s specifications, and quantitative PCR was performed using SYBR Green (A311-10, GenStar) on a Viia7 Real-Time PCR system (Thermo Fisher Scientific). The primers are listed in Supplementary Table 6. The experiments were performed in biological triplicates unless otherwise stated, and normalized to GAPDH as an internal control.
3D microtumor culture and IF staining
We performed 3D culture of DIPG cells using a 3D PlaTrix (CytoNiche). Specifically, 3 μl cell suspension with a density of 6.67 × 106 cells per milliliter was seeded directly on the dry microscaffold etched at the bottom of each well to ensure that all cells were automatically absorbed into the pores of the microscaffold. After this, we added 80 μl of culture medium to each of the reservoir wells. The whole device was maintained on a humidified chamber at a temperature of 37 °C to form the microtumorl 50 ng ml–1 BMP4 was added overnight to the microplates after tumor cell inoculation.
After treatment with BMP4 for either 24 h or 48 h, the microscaffold was removed and fixed in 4% paraformaldehyde for 20 min, followed by rinsing with PBS and blocking with 5% bovine serum albumin (Sigma). We then incubated the 3D microtumors overnight at 4 °C with primary antibodies, including p-Smad1/5/8 (1:200), Olig2 (1:200), CD133 (1:200) and GFAP (1:500). This was followed by 1 h incubation with the Alexa Fluor 594 secondary antibody (1:1,000) at room temperature. We used 4,6-diamidino-2-phenylindole (DAPI) (1:10,000; Invitrogen) as a nuclear stainer, and observed the stained 3D microtumors using a Lightsheet microscope (Zeiss).
Gene expression analyses
We mapped the RNA-seq data to the human reference genome (hg38) using HISAT2 (v.2.1.0). The levels of gene expression were calculated by Cufflinks (v.2.2.1) based on hg38 annotations. Differential testing and log2 fold change calculations were performed using Cuffdiff (v.2.2.1), with the implementation of two biological replicates. Gene ontology analysis was performed using DAVID (https://david.ncifcrf.gov/). We generated Volcano plots using ggplot2 (v.3.2.1) showing the log transformed P values in the y axis and the log2 fold change in the x axis. GSEA of DIPG cells was performed by GSEA software (v.4.0.0)48 (www.broadinstitute.org/software/gsea). We calculated the normalized enrichment score (NES) and the false discovery rate (FDR) Q value by permuting the gene set types. We assigned a cutoff of FDR ≤ 0.25 to identify significantly enriched gene sets.
ChIP–seq analysis
We performed ChIP–seq experiments to determine the changes in chromatin modification and SMAD1 and CXXC5 signals as a response to the two treatments (dimethylsulfoxide (DMSO) and BMP4). These experiments were implemented in duplicate for each of the analyzed histone marks (H3K27ac, H3K27M and H3K27me3), SMAD1 and CXXC5 and sequenced on an Illumina platform. The resulting FASTQ files were trimmed using TrimGalore (v.0.6.1) and then aligned with hg38 using Bowtie2 (v.2.3.3)49. PCR duplicates were removed using PicardTools (v.2.26.10). We chose MACS2 (v.2.1.4) to call the peaks using the ‘broad peaks’ setting for H3K27me3 and H3K27M (FDR < 0.1); we used the narrow peak of H3K27ac in the analysis. ChIP–seq peaks that were significantly increased or decreased in the presence of BMP4 in both replicates were then identified using the DiffBind (v.3.0.10) package in R (v.4.0). We subsequently generated bigwig files from the bam files using the Coverage function in deepTools (v.3.4.4)50. For visualization purposes, we normalized the data to 1× genome coverage (hg38), ignoring PCR duplicates. Representative track diagrams were generated using the Integrated Genomics Viewer (IGV) software (v.2.5.3)51.
We identified SEs using ROSE (v.1.2.0)36, and joined together enhancers within 12.5 kb of each other. All enhancers were ranked according to read density in H3K27ac ChIP–seq compared with reads from input.
Transcription factor binding motifs in different regulatory regions were identified and analyzed using the HOMER (v.4.11) suite of tools available on the findMotifsGenome.pl script. From the set of identified motifs, we filtered those exhibiting a Q value <0.001 and a fold enrichment of more than two. The frequency of the indicated motif was plotted over the distance from the center of the peak (±500 bp).
ATAC-seq analysis
Reads were aligned to hg38 using bowtie2 with the parameters –X 2000 and –m 1. We removed duplicates for all data files using PicardTools. For downstream analysis, we normalized the read counts by computing the number of reads per kilobase of bin per million of reads sequenced. To visualize the ATAC-seq signal in the IGV software, we extended each read by 250 bp and counted the sequencing coverage for each base. The ATAC-seq peaks were called using MACS2 with the parameters –nolambda –nomodel.
Statistics and reproducibility
The statistical analyses employed in each plot are either described in the figure legends or in the corresponding Methods section. Briefly, the grouped data are presented as the mean ± s.d. unless stated otherwise. Statistical significance for pairwise comparisons was determined using two-tailed unpaired Student’s t-tests. Data distribution was assumed to be normal but this was not formally tested. Survival analysis was performed using the Kaplan–Meier method, and differences between the groups were calculated by the log-rank test. As above, all quantitative analyses are expressed as means ± s.d. of three biological replicates. RNA-seq, ChIP–seq and ATAC-seq data were obtained from at least two independent experiments. For mouse studies, a minimum of five mice were used. For IHC or IF experiments, staining was performed on the entire cohort (minimum of n = 3 biological samples) at the same time. Image analysis was performed at the same time for each experiment. The experiments in Figs. 1b,f, 3a,b, 4c,i–j, 6g, 7c and 8a and Extended Data Figs. 2c, 3a, 5g, 7c,f,h–i, 8a,d, 9d and 10b have been repeated three times with similar results. No statistical methods were used to predetermine the sample size in the different experiments, but our sample sizes are similar to those reported in previous publications23. Unless stated otherwise, the experiments were not randomized and the investigators were not blinded to allocation during the experimental procedures and the assessment of the outcomes. No data were excluded from the analyses.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The raw RNA-seq, ChIP–seq and ATAC-seq data that support the findings of this study have been deposited with the Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences, https://bigd.big.ac.cn/gsa-human, under the accession number: HRA000612. Previously published data that were reanalyzed here are available under accession numbers GSE50021, GSE126319, GSE94259, GSE128745 and GSE1105722. Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Change history
05 September 2022
In the version of this article initially published, the three columns at the right of the SU-DIPG17 panel of Fig. 2b were obscured by a white box, which has been removed from the HTML and PDF versions of the article.
References
Hargrave, D., Bartels, U. & Bouffet, E. Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol. 7, 241–248 (2006).
Buczkowicz, P. et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 46, 451–456 (2014).
Fontebasso, A. M. et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 46, 462–466 (2014).
Taylor, K. R. et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 46, 457–461 (2014).
Wu, G. et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 46, 444–450 (2014).
Mackay, A. et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537.e5 (2017).
Funato, K., Major, T., Lewis, P. W., Allis, C. D. & Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533 (2014).
Pathania, M. et al. H3.3K27M cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell 32, 684–700.e9 (2017).
Filbin, M. G. et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018).
Nagaraja, S. et al. Histone variant and cell context determine H3K27M reprogramming of the enhancer landscape and oncogenic state. Mol. Cell 76, 965–980.e12 (2019).
Castel, D. et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun. 6, 117 (2018).
Khuong-Quang, D. A. et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 124, 439–447 (2012).
Castel, D. et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 130, 815–827 (2015).
Carvalho, D. et al. ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun. Biol. 2, 156 (2019).
Mucha, B. E., Hashiguchi, M., Zinski, J., Shore, E. M. & Mullins, M. C. Variant BMP receptor mutations causing fibrodysplasia ossificans progressiva (FOP) in humans show BMP ligand-independent receptor activation in zebrafish. Bone 109, 225–231 (2018).
Fortin, J. et al. Mutant ACVR1 arrests glial cell differentiation to drive tumorigenesis in pediatric gliomas. Cancer Cell 37, 308–323.e12 (2020).
Chaikuad, A. & Bullock, A. N. Structural basis of intracellular TGF-β signaling: receptors and Smads. Cold Spring Harb. Perspect. Biol. 8, a022111 (2016).
Urist, M. R. Bone: formation by autoinduction. Science 150, 893–899 (1965).
Sanchez-Duffhues, G., Williams, E., Goumans, M. J., Heldin, C. H. & Ten Dijke, P. Bone morphogenetic protein receptors: structure, function and targeting by selective small molecule kinase inhibitors. Bone 138, 115472 (2020).
Sun, Y. et al. Potent anti-tumor efficacy of palbociclib in treatment-naive H3.3K27M-mutant diffuse intrinsic pontine glioma. Ebiomedicine 43, 171–179 (2019).
Piunti, A. et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 23, 493–500 (2017).
Grasso, C. S. et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 21, 555–559 (2015).
Nagaraja, S. et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652.e6 (2017).
Anastas, J. N. Re-programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell 36, 528–544.e10 (2019).
Xu, C. et al. Patient-derived DIPG cells preserve stem-like characteristics and generate orthotopic tumors. Oncotarget 8, 76644–76655 (2017).
Ouahoud, S., Hardwick, J. C. H. & Hawinkels, L. Extracellular BMP antagonists, multifaceted orchestrators in the tumor and Its microenvironment. Int. J. Mol. Sci. 21, 3888 (2020).
Puget, S. et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS ONE 7, e30313 (2012).
Walsh, D. W., Godson, C., Brazil, D. P. & Martin, F. Extracellular BMP-antagonist regulation in development and disease: tied up in knots. Trends Cell Biol. 20, 244–256 (2010).
Cyr-Depauw, C. et al. Chordin-Like 1 suppresses bone morphogenetic protein 4-induced breast cancer cell migration and invasion. Mol. Cell. Biol. 36, 1509–1525 (2016).
Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 (2012).
VanBrocklin, M. W., Verhaegen, M., Soengas, M. S. & Holmen, S. L. Mitogen-activated protein kinase inhibition induces translocation of Bmf to promote apoptosis in melanoma. Cancer Res. 69, 1985–1994 (2009).
Yoo, J. et al. Transforming growth factor-β-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J. Biol. Chem. 278, 43001–43007 (2003).
Mohammad, F. et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 23, 483–492 (2017).
Harutyunyan, A. S. et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 10, 1262 (2019).
Whyte, WarrenA. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Loven, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Xiong, X., Tu, S., Wang, J., Luo, S. & Yan, X. CXXC5: a novel regulator and coordinator of TGF-beta, BMP and Wnt signaling. J. Cell. Mol. Med. 23, 740–749 (2019).
Andersson, T. et al. CXXC5 is a novel BMP4-regulated modulator of Wnt signaling in neural stem cells. J. Biol. Chem. 284, 3672–3681 (2009).
Yan, X. et al. CXXC5 suppresses hepatocellular carcinoma by promoting TGF-beta-induced cell cycle arrest and apoptosis. J. Mol. Cell. Biol. 10, 48–59 (2018).
Voo, K. S., Carlone, D. L., Jacobsen, B. M., Flodin, A. & Skalnik, D. G. Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shares a CXXC domain with DNA methyltransferase, human trithorax, and methyl-CpG binding domain protein 1. Mol. Cell Biol. 20, 2108–2121 (2000).
Ravichandran, M. et al. Rinf regulates pluripotency network genes and Tet enzymes in embryonic stem cells. Cell Rep 28, 1993–2003.e5 (2019).
Vitanza, N. A. et al. Optimal therapeutic targeting by HDAC inhibition in biopsy-derived treatment-naive diffuse midline glioma models. Neuro Oncol. 23, 376–386 (2021).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452.e17 (2017).
Sun, X. et al. A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov. 5, 10 (2019).
Sachdeva, R. et al. ID1 is critical for tumorigenesis and regulates chemoresistance in glioblastoma. Cancer Res. 79, 4057–4071 (2019).
Xi, Q. et al. A poised chromatin platform for TGF-beta access to master regulators. Cell 147, 1511–1524 (2011).
Wang, Q. et al. The p53 family coordinates Wnt and Nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell 20, 70–86 (2017).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Thorvaldsdóttir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192 (2013).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013).
Acknowledgements
We thank all the patients and families for donating tissues for this research. We are grateful to H. Zheng for the luciferase-GFP plasmid; to Y. Lu from the Laboratory Animal Research Center (Tsinghua University) for technical assistance with histological analyses and immunohistochemistry experiments; to G. Wang (Tsinghua University) for the cleaved-CASPASE3 antibody and helpful discussion and to J. Massagué (Memorial Sloan Kettering Cancer Center), Y.-G. Chen, C. David, H. Zheng, W. Wu, Y. Rao (Tsinghua University) and Y. Sun (Tiantan Hospital) for valuable discussions; and to staff of the Laboratory Animal Research Center for animal experiments during the COVID-19 pandemic. Finally, we thank Z. Zhou (Tsinghua University) for the graphic design. Q.X. is supported by Ministry of Science and Technology of China (2018YFA0107702), National Natural Science Foundation of China (31471229, 31771622 and 91540108) and Tsinghua-Peking Center for Life Sciences. Y.T. is supported by The Recruitment Program of Global Experts of China, National Natural Science Foundation of China (81772655), and Innovative Research Team of High-Level Local Universities in Shanghai (SSMU-ZDCX20180800). L.Z. is supported by Beijing municipal administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201608), Beijing Municipal Natural Science Foundation (7161004) and National Natural Science Foundation of China (81872048).
Author information
Authors and Affiliations
Contributions
Y. Sun and K.Y. performed most of the experiments. Y.W., C.X., S.S. and L. Z. provided the biopsy samples and clinical data of DIPG patients from Tiantan hospital (Beijing). Y.H. and Y.T. provided SU-DIPG4 and SU-DIPG17 cell samples. D.W. performed 3D culture of DIPG cells. S.G. helped with western blot assay. Y. Sun and K.Y. performed RNA-seq and ChIP–seq experiments. L.T. performed ATAC-seq experiments. Q.C.Z. supervised the ATAC-seq experiments. K.Y. performed RNA-seq, ChIP–seq, and ATAC-seq analyses. K.Y. and W.Z. performed scRNA-seq analysis; Y. Shao provided bioinformatic analysis tools. Q.X., Y. Sun and K.Y. designed the experiments. All authors contributed ideas to the project. Q.X. supervised the project. Q.X., Y. Sun and K.Y. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks Rosemary Akhurst, Joan Seoane and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 BMP signaling is downregulated and CHRDL1 is highly expressed in H3.3K27M and ACVR1 WT DIPG subtype.
(a) Heatmap to show single sample GSEA (ssGSEA) scores of hallmark gene sets in control group and the drug (JQ1, Panobinostat, THZ1, or Corin) treatments group in SU-DIPG6 or SU-DIPG13 cells (GEO: GSE94259, GSE1105722). Each column represents a sample with or without drug. Each row represents the specific gene signature of the pathway and the normalized z-score of the ssGSEA score corresponding to the sample is displayed in the heatmap where the colors correspond to the normalized z-score. (b) GSEA analysis using the “BMP signaling signature” gene set to compare PPCs and the H3.3K27M and ACVR1 WT DIPG cells (SU-DIPG6 and SU-DIPG13). (c, d) GSEA analysis using the “BMP signaling signature” gene set to compare PPCs and the H3.1K27M and ACVR1 mutant DIPG cell (SU-DIPG4)23 (c); or to compare 10 normal tissues and 18 H3.3K27M and ACVR1 WT DIPG tissues (d). (e) FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) values of CHRDL1 in RNA-seq data of TT150630, TT150714, and PPCs (n = 2 independent experiments). (f) FPKM values of CHRDL1 in RNA-seq data of pons and a group of H3K27M DIPG tissues. Genotype of H3 and ACVR1 in each sample is marked10. (g) Unsupervised clustering of single-cell H3K27M DIPG RNA-seq data using the most variable genes9. (h-k) Gene expression of OLIG2, ASCL1, SOX2 and CHRDL1 in (g). Expression of each gene was scaled to [0, 2] for visualization. (l) Among the CHRDL1-expressing cells in (g), boxplots representing CHRDL1 gene expression differences between ACVR1 mutant (MUT, blue, n = 286 tumor single cells) and wild-type (WT, red, n = 136 tumor single cells) (left). Boxplots representing CHRDL1 gene expression differences between H3.3K27M (red, n = 406 tumor single cells) and H3.1K27M (blue, n = 16 tumor single cells) (right). Boxplots define the interquartile range (IQR) split by the median, with whiskers extending to the most extreme values within 1.5 × IQR beyond the box, statistical significance was calculated by two-tailed unpaired Student’s t-test.
Extended Data Fig. 2 High expression of CHRDL1 contributes to the low activity of BMP signaling and tumor progression in this DIPG subtype.
(a) Kaplan–Meier survival curves for DIPG patients in the UCSC xena patient cohort27, separated into CHRDL1 high(n = 8 patients) and CHRDL1 low(n = 9 patients) survival groups. Log-rank test was performed. (b) qPCR analysis of CHRDL1 expression in indicated Ctr or CHRDL1 KD cells. n = 3 independent experiments. (c) Immunoblotting analysis of p-SMAD1/5 and SMAD1 in TT150714 and SU-DIPG17 (Ctr or CHRDL1 KD DIPG cells) with or without BMP4 (25 ng/mL) for 2 hr. (d) qPCR analysis of BMP signaling response genes mRNA expression in Ctr and CHRDL1 KD TT150714 and SU-DIPG17 cells with or without BMP4 (25 ng/mL) treatment. Data represents the mean ± S.D., statistical significance was calculated by two-tailed unpaired Student’s t-test, n = 3 independent experiments. (e) Neural sphere formation of indicated cell lines (Ctr or CHRDL1 KD DIPG cells) for 10 days (n = 3 independent experiments). (f) Neural sphere counts from (e). (g) The normalized bioluminescence activity was plotted and the statistical difference between TT150630 Ctr and CHRDL1 KD groups was significant (n = 5 mice in Ctr and n = 5 mice in CHRDL1 KD). (h) Representative bioluminescence images from animals implanted with 5 ×105 Ctr (n = 5 mice) or CHRDL1 KD(n = 5 mice) of luciferase-GFP engineered-SU-DIPG17 cells in the pons at day 238. The heatmap superimposed over the mouse heads represents the degree of photon emission by DIPG cells expressing firefly luciferase. (i) The normalized bioluminescence activity was plotted and the statistical difference between SU-DIPG17 Ctr and CHRDL1 KD groups was significant (n = 5 mice in Ctr and n = 5 mice in CHRDL1 KD). (j) Kaplan–Meier analysis from animals implanted with SU-DIPG17 cells with (n = 5 mice) or without (n = 5 mice) CHRDL1 KD in the pons. Log-rank test was performed. (k) Immunofluorescence of pons section from animals implanted with TT150630 cells with or without CHRDL1 KD for anti-human nuclear antigen (HNA), OLIG2 and GFAP. Scale bars, 50 µm. This figure represents 9 independent tissues. (l) Quantification of OLIG2-positive cells in all tumor cells (HNA positive) from animals implanted with TT150630 cells with or without CHRDL1 KD. n = 9 independent tissue samples. For g and i, boxplots define the interquartile range (IQR) split by the median, with whiskers extending to the most extreme values within 1.5 × IQR beyond the box, statistical significance was calculated by two-tailed unpaired Student’s t-test. The experiments in c have been repeated three times with similar results. For b, f and i, data represents the mean ± S.D., statistical significance was calculated by two-tailed unpaired Student’s t-test.
Extended Data Fig. 3 BMPs inhibit ACVR1 WT DIPG subtype cells sphere formation and growth.
(a) Immunoblotting analysis of p-SMAD1/5 and SMAD1 levels in TT150630 and TT150714 treated with LDN-193189 (LDN) (200 nM), BMP4 (50 ng/mL), or BMP4 (50 ng/mL) plus LDN-193189 (200 nM). (b) top: Neural sphere formation of the indicated cell lines treated with vehicle, 50 ng/mL BMP2 or 25 ng/mL BMP2 for 10 days, n = 3 independent experiments; bottom: Neural sphere counts from top panel. All p value were generated by comparing to control group. (c) top: Neural sphere formation of the indicated cell lines treated with vehicle or indicated concentration of BMP4 for 10 days, n = 3 independent experiments; bottom: Neural sphere counts from top panel. All p value were generated by comparing to control group.(d) Viability (metabolic capacity) of indicated cells (n = 3 independent experiments) treated with vehicle or indicated concentration of BMP4.(e) Viability (metabolic capacity) of SU-DIPG4 cells (n = 3 independent experiments) treated with vehicle or indicated concentration of BMP4 or LDN. For b-e, data represents the mean ± S.D., statistical significance was calculated by two-tailed unpaired Student’s t-test. The experiments in a have been repeated three times with similar results.
Extended Data Fig. 4 BMP4 prolongs the survival of DIPG xenograft model mice.
(a) Schematic for the construction of a xenograft mouse model upon orthotopic injection of 1 ×105 BMP4-pretreated (50 ng/mL) 24 hr or untreated DIPG cells. Created with BioRender.com. (b) The normalized bioluminescence activity in TT150630 mouse model was plotted and the statistical difference between BMP4 pre-treated and vehicle treated groups was significant (n = 6 mice in each group). (c) The normalized bioluminescence activity in SU-DIPG17 mouse model was plotted and the statistical difference between BMP4 pre-treated and vehicle treated groups was significant (n = 6 mice in each group).(d) Kaplan–Meier analysis from animals implanted with TT150630 cells with indicated treatment (n = 6 mice in Ctr group vs n = 5 mice in BMP4 pretreatment group) in the pons. Log-rank test was performed. (e) Representative images of pons from animals implanted with SU-DIPG17 cells with or without BMP4 treatment analyzed by H&E staining. Regions marked by the box are magnified below. Scale bars, 1,000 µm (top), 50 µm (middle) and 20 µm (bottom). (f) Immunofluorescence of pons section from animals implanted with SU-DIPG17 cells with or without BMP4 treatment for anti-human nuclear antigen (HNA). Scale bars, 1,000 µm (top), 50 µm (middle) and 20 µm (bottom). For b and c, boxplots define the interquartile range (IQR) split by the median, with whiskers extending to the most extreme values within 1.5 × IQR beyond the box, statistical significance was calculated by two-tailed unpaired Student’s t-test. The experiments in e and f have been repeated three times with similar results.
Extended Data Fig. 5 BMP4 forces DIPG subtype cells to exit from a prolonged stem-cell-like state.
(a) The heatmap of OPC, AC and cell-cycle-related genes in RNA-seq transcriptome analysis of TT150714 cells treated with BMP4 (50 ng/mL) at indicated timepoints (n = 2 independent experiments). (b) Volcano plots showing differentially expressed genes from transcriptome datasets of TT150714 with or without BMP4 (50 ng/mL) treatment for 24 hr. Upregulated genes (n = 323; |LFC | ≥ 1.5-fold; p < 0.05) are red dots; downregulated genes (n = 193; |LFC | ≥ 1.5-fold; p < 0.05) are in blue. Individual genes of interest are depicted. (c) The heatmap of OPC, AC and cell-cycle-related genes in RNA-seq transcriptome analysis of SU-DIPG4 cells treated with BMP4 (50 ng/mL) at indicated timepoints (n = 2 independent experiments). (d) Volcano plots showing differentially expressed genes from transcriptome datasets of SU-DIPG4 with or without BMP4 treatment for 24 hr. Upregulated genes (n = 31; |LFC | ≥ 1.5-fold; p < 0.05) are red dots; downregulated genes (n = 55; |LFC | ≥ 1.5-fold; p < 0.05) are in blue. Individual genes of interest are depicted. (e) Immunofluorescence staining for GFAP, OLIG2 and phosphorylated SMAD1/5 proteins in TT150714 cells with or without BMP4 (50 ng/mL) treatment for 48 hr. Scale bars, 100 μm. (f) Representative immunohistochemistry of OLIG2 and GFAP in pons from animals implanted with TT150630 cells with or without BMP4 treatment. Scale bars, 50 μm.(g) Immunoblotting for p-SMAD1/5, SMAD1, p21CIP1 and GAPDH proteins in TT150630 and TT150714 DIPG cells with BMP4 treatment (50 ng/mL) at indicated timepoints. (h) Cell cycle analysis of indicated cells with or without BMP4 treatment (50 ng/mL) for 24 hr by propidium Iodide staining. n = 3 independent experiments. (i) Quantification of cells (%) in each cell cycle phase of cells analyzed by flow cytometry in (h), n = 3 independent experiments, data represents the mean ± S.D, statistical significance was calculated by two-tailed unpaired Student’s t-test. (j) Plots of annexin V (AV) and Propidium Iodide (PI) FACS analyses in TT150630 DIPG cells treated with indicated concentration at indicated conditions. n = 3 independent experiments (k) Bar plots show early apoptotic (AV + DAPI − ) or late apoptotic (AV + DAPI + ) cell population percentage from each condition. n = 3 independent experiments, two-tailed unpaired Student’s t-tests was performed. For b and d, p value was calculated by Cuffdiff. The experiments in e, f and g have been repeated three times with similar results.
Extended Data Fig. 6 Global alteration in H3K27ac, H3K27me3, and H3K27M-modified chromatin in DIPG subtype cells treated with BMP4.
(a) Pie charts show the percentage of genomic distribution of SMAD1 ChIP–seq peaks in the vehicle or BMP4 (50 ng/mL) treated TT150630 cells. The hg38 reference genome distribution was set up for control. (b) Metagene plots showing the average ChIP–seq signal for SMAD1, H3K27ac, H3K27me3 and H3K27M for all of the BMP4 up- (top) and downregulated (down) genes in TT150630 cells treated with vehicle or BMP4 for 2 hr. (c) Identification of subgroup-specific genes with concordant changes in both expression and SEs in TT150630 DIPG cells. x axis: LFC of gene expression between vehicle and BMP4 treatment for 2 hr in TT150630 DIPG cells. y axis: LFC of SEs associated with the gene. Red: significantly upregulated genes. Blue: significantly downregulated genes. Gray: no significant changes in expression. (d) The HOXA family genes heatmap from RNA-seq transcriptome analysis of TT150630 cells treated with BMP4 at the indicated timepoints (n = 2 independent experiments). (e) IGV tracks for H3K27me3 ChIP–seq (with vehicle or BMP4 treated for 24 hr or CHRDL1 KD) in TT150630 cells at indicated genes loci. (f) Identification of subgroup-specific genes with concordant changes in both expression and H3K27ac signals in TT150630 cells. X axis: LFC of gene expression between control (n = 2 independent experiments) and BMP4 treatment (n = 2 independent experiments). Y axis: LFC of H3K27ac signals at a promoter or enhancer associated with the gene. Genes with significantly differentially acetylated regulatory regions between control and BMP4 treatment are shown. Red: significantly upregulated genes. Blue: significantly downregulated genes. Gray: no significant changes in expression. (g) Super enhancers (SEs) detected in TT150630 DIPG CHRDL1 KD cells.
Extended Data Fig. 7 CXXC5 is the primary target gene of BMP signaling in DIPG subtype cells.
(a) qPCR analysis of CXXC5 mRNA expression in control or 2 hr-BMP4 (50 ng/mL) -treated indicated DIPG cells with or without pretreatment with 10 µg/mL cycloheximide (CHX) for 1 hr. n = 3 independent experiments. (b) FPKM values of CXXC5 in RNA-seq data of TT150630 DIPG cells treated with BMP4 at indicated timepoints (n = 2 independent experiments). (c) Immunoblotting analysis of CXXC5 and TUBULIN in indicated DIPG cell lines treated with BMP4 (50 ng/mL) at the indicated timepoints. The relative intensity of the CXXC5 protein level compared to TUBULIN is indicated. (d, e) qPCR analysis of CXXC5 relative expression in control and SMAD4 KD (d) or control, SMAD1 KD, and SMAD1/5 KD (e) DIPG cells treated with BMP4 (50 ng/mL) at indicated timepoints. n = 3 independent experiments. (f) Immunoblotting analysis for phosphorylated SMAD1/5, total SMAD1, CXXC5 and GAPDH proteins in the indicated cell lines. (g) qPCR analysis of ID1, SMAD7 and CXXC5 expression in SU-DIPG4 cells treated with BMP4 (50 ng/mL) at the indicated timepoints. n = 3 independent experiments. (h) Immunoblotting analysis for phosphorylated SMAD1/5, total SMAD1, CXXC5 and GAPDH proteins in SU-DIPG4 cell lines treated with BMP4 (50 ng/mL) at the indicated timepoints. (i) Immunoblotting analysis of CXXC5 and TUBULIN in indicated DIPG cell lines and PPC. SU-DIPG17 (1), TT150714 (2), TT150630 (3), TT160728 (4), TT150603 (5), TT160310 (6), TT151201 (7) and TT160518 (8). The relative intensity of the CXXC5 protein level is indicated. The experiments in c, f, h and i have been repeated three times with similar results. For a, d, e and g, data represents the mean ± S.D., statistical significance was calculated by two-tailed unpaired Student’s t-test.
Extended Data Fig. 8 CXXC5 functions as a tumor suppressor in DIPG subtype.
(a) CXXC5 KD verification in TT150630 and TT150714 cells. Cells were validated by immunoblotting analysis using antibodies against CXXC5 and TUBULIN. (b) Neural sphere formation of control and CXXC5 KD TT150714 cells (n = 3 independent experiments) and neural sphere counts. (c) Cell cycle analysis of indicated cells by propidium Iodide staining. (d) Quantification of cells (%) in each cell cycle phase of cells analyzed by flow cytometry in (c), n = 3 independent experiments samples, data represents the mean ± S.D, statistical significance was calculated by two-tailed unpaired Student’s t-test. (e) Immunoblotting for PARP1, cleaved PARP1, CXXC5 and GAPDH in the indicated TT150630 DIPG cell line.(f) Overlapping plots of Annexin V (AV) and Propidium Iodide (PI) FACS analyses in CXXC5 instantaneous overexpression TT150630 cells. (g) Overlapping plots of Annexin V (AV) and Propidium Iodide (PI) FACS analyses in Dox-inducible CXXC5 overexpression TT150630 DIPG cells at indicated timepoints. (h) Neural sphere formation of indicated cells treated with Dox (750 ng/mL) or none for 10 days. (n = 3 independent experiments). (i) Neural sphere counts in (h). For b and i, data represents the mean ± S.D., statistical significance was calculated by two-tailed unpaired Student’s t-test. The experiments in a and e have been repeated three times with similar results.
Extended Data Fig. 9 Depletion of CXXC5 alters transcription of cell-cycle-related genes.
(a) Pie charts show the percentage of genomic distribution of CXXC5 ChIP–seq peaks in the vehicle or BMP4 treated TT150630 cells. The hg38 reference genome distribution was set up for control. (b) IGV tracks for RNA-seq and CXXC5 ChIP–seq (with vehicle or BMP4 treated for 24 hr) in TT150630 cells at indicated genes loci. (c) qPCR analysis of indicated genes expression in control and CXXC5 KD DIPG cells treated with BMP4 (50 ng/mL) at indicated timepoints. n = 3 independent experiments.(d) Immunoblotting for CXXC5, p21CIP1 and GAPDH proteins in TT150630 and TT150714 control and CXXC5 KD cells with or without BMP4 treatment (50 ng/mL) for 24 hr. (e) qPCR analysis of SMAD1, SMAD5 and p21CIP1 relative expression in TT150630 control, SMAD1 KD and SMAD1/5 double KD cells treated with BMP4 (50 ng/mL) at indicated timepoints. n = 3 independent experiments. (f) qPCR analysis of SMAD4 and p21CIP1 relative expression in TT150630 control and SMAD4 KD cells treated with BMP4 (50 ng/mL) at indicated timepoints. n = 3 independent experiments. (g) GSEA analysis using the G2M checkpoint gene set to compare control and CXXC5-inducible overexpression TT150630 DIPG cells. (h) qPCR analysis of p21CIP1 relative expression in SU-DIPG4 cells treated with BMP4 (50 ng/mL) at indicated timepoints. Data represents the mean ± S.D., statistical significance was calculated by two-tailed unpaired Student’s t-test, n = 3 independent experiments. The experiments in d have been repeated three times with similar results. For c, e and f, data represents the mean ± S.D., statistical significance was calculated by two-tailed unpaired Student’s t-test.
Extended Data Fig. 10 CXXC5 positively regulates BMP signaling.
(a) IGV tracks for RNA-seq and CXXC5 ChIP–seq (treated with vehicle or BMP4 for 2 hr) in TT150630 DIPG cells at BMP2, BMPR2 and ACVR1 gene loci. (b) Immunoblotting for phosphorylated SMAD1/5 (Ser 463/465), total SMAD1, CXXC5 and GAPDH in control and CXXC5 KD TT150714 DIPG cells. The experiments have been repeated three times with similar results. (c) Heatmaps of ChIP–seq signals of CXXC5 at top 4000 SMAD1 significant peaks or 5868 H3K27ac upregulated peaks respectively in TT150630 DIPG cells with or without BMP4 treatment for 2 hr (top). Average CXXC5 ChIP–seq signals corresponding with their ChIP–seq profiles in the indicated conditions (bottom). (d) Kaplan–Meier survival curves for DIPG patients in the UCSC xena patient cohort27, separated into ACVR1 high (n = 26 patients) and ACVR1 low (n = 25 patients) survival groups. Log-rank test was performed.
Supplementary information
Supplementary Information
Supplementary Tables 1–6.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 6
Unprocessed western blots.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 7
Unprocessed western blots.
Source Data Fig. 8
Statistical source data.
Source Data Fig. 8
Unprocessed western blots.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 2
Unprocessed western blots.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 3
Unprocessed western blots.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 7
Unprocessed western blots.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 8
Unprocessed western blots.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 9
Unprocessed western blots.
Source Data Extended Data Fig. 10
Unprocessed western blots.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sun, Y., Yan, K., Wang, Y. et al. Context-dependent tumor-suppressive BMP signaling in diffuse intrinsic pontine glioma regulates stemness through epigenetic regulation of CXXC5. Nat Cancer 3, 1105–1122 (2022). https://doi.org/10.1038/s43018-022-00408-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-022-00408-8
This article is cited by
-
BMP signaling in cancer stemness and differentiation
Cell Regeneration (2023)
-
The moving target of cancer cell plasticity
Nature Cancer (2022)
