
Abstract
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure but rather determined from the equilibrium distribution of structures. Conventional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. Here we introduce a deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG uses deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system such as a chemical graph or a protein sequence. This framework enables the efficient generation of diverse conformations and provides estimations of state densities, orders of magnitude faster than conventional methods. We demonstrate applications of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst–adsorbate sampling and property-guided structure generation. DiG presents a substantial advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in the molecular sciences.
Similar content being viewed by others
Main
Deep learning methods excel at predicting molecular structures with high efficiency. For example, AlphaFold predicts protein structures with atomic accuracy1, enabling new structural biology applications2,3,4; neural network-based docking methods predict ligand binding structures5,6, supporting drug discovery virtual screening7,8; and deep learning models predict adsorbate structures on catalyst surfaces9,10,11,12. These developments demonstrate the potential of deep learning in modelling molecular structures and states.
However, predicting the most probable structure only reveals a fraction of the information about a molecular system in equilibrium. Molecules can be very flexible, and the equilibrium distribution is essential for the accurate calculation of macroscopic properties. For example, biomolecule functions can be inferred from structure probabilities to identify metastable states; and thermodynamic properties, such as entropy and free energies, can be computed from probabilistic densities in the structure space using statistical mechanics.
Figure 1a shows the difference between conventional structure prediction and distribution prediction of molecular systems. Adenylate kinase has two distinct functional conformations (open and closed states), both experimentally determined, but a predicted structure usually corresponds to a highly probable metastable state or an intermediate state (as shown in this figure). A method is desired to sample the equilibrium distribution of proteins with multiple functional states, such as adenylate kinase.

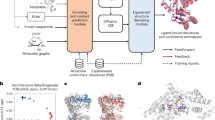
a, DiG takes the basic descriptor \({{{\mathcal{D}}}}\) of a target molecular system as input—for example, an amino acid sequence—to generate a probability distribution of structures that aims at approximating the equilibrium distribution and sampling different metastable or intermediate states. In contrast, static structure prediction methods, such as AlphaFold1, aim at predicting one single high-probability structure of a molecule. b, The DiG framework for predicting distributions of molecular structures. A deep learning model (Graphormer10) is used as modules to predict a diffusion process (→) that gradually transforms a simple distribution towards the target distribution. The model is trained so that the derived distribution pi in each intermediate diffusion time step i matches the corresponding distribution qi in a predefined diffusion process (←) that is set to transform the target distribution to the simple distribution. Supervision can be obtained from both samples (workflow in the top row) and a molecular energy function (workflow shown in the bottom row).
Unlike single structure prediction, equilibrium distribution research still depends on classical and costly simulation methods, while deep learning methods are underdeveloped. Commonly, equilibrium distributions are sampled with molecular dynamics (MD) simulations, which are expensive or infeasible13. Enhanced sampling simulations14,15 and Markov state modelling16 can accelerate rare event sampling but need system-specific collective variables and are not easily generalized. Another approach is coarse-grained MD17,18, where deep learning approaches have been proposed19,20. These deep learning coarse-grained methods have worked well for individual molecular systems but have not yet demonstrated generalization. Boltzmann generators21 are a deep learning approach to generate equilibrium distributions by creating a probability flow from a simple reference state, but this also hard to generalize to different molecules. Generalization has been demonstrated for flows generating simulations with longer time steps for small peptides but has not yet been scaled to large proteins22.
In this Article, we develop DiG, a deep learning approach to approximately predict the equilibrium distribution and efficiently sample diverse and function-relevant structures of molecular systems. We show that DiG can generalize across molecular systems and propose diverse structures that resemble observations in experiments. DiG draws inspiration from simulated annealing23,24,25,26, which transforms a uniform distribution to a complex one through a simulated annealing process. DiG simulates a diffusion process that gradually transforms a simple distribution to the target one, approximating the equilibrium distribution of the given molecular system27,28 (Fig. 1b, right arrow symbol). As the simple distribution is chosen to enable independent sampling and have a closed-form density function, DiG enables independent sampling of the equilibrium distribution and also provides a density function for the distribution by tracking the process. The diffusion process can also be biased towards a desired property for inverse design and allows interpolation between structures that passes through high-probability regions. This diffusion process is implemented by a deep learning model based upon the Graphormer architecture10 (Fig. 1b), conditioned on a descriptor of the target molecule, such as a chemical graph or a protein sequence. DiG can be trained with structure data from experiments and MD simulations. For data-scarce cases, we develop a physics-informed diffusion pre-training (PIDP) method to train DiG with energy functions (such as force fields) of the systems. In both data-based or energy-supervised modes, the model gets a training signal in each diffusion step independently (Fig. 1b, left arrow symbol), enabling efficient training that avoids long-chain back-propagation.
We evaluate DiG on three predictive tasks: protein structure distribution, the ligand conformation distribution in binding pockets and the molecular adsorption distribution on catalyst surfaces. DiG generates realistic and diverse molecular structures in these tasks. For the proteins in this Article, DiG efficiently generated structures resembling major functional states. We further demonstrate that DiG can facilitate the inverse design of molecular structures by applying biased distributions that favour structures with desired properties. This capability can expand molecular design for properties that lack enough data. These results indicate that DiG advances deep learning for molecules from predicting a single structure towards predicting structure distributions, paving the way for efficient prediction of the thermodynamic properties of molecules.
Results
Here, we demonstrate that DiG can be applied to study protein conformations, protein–ligand interactions and molecule adsorption on catalyst surfaces. In addition, we investigate the inverse design capability of DiG through its application to carbon allotrope generation for desired electronic band gaps.
Protein conformation sampling
At physiological conditions, most protein molecules exhibit multiple functional states that are linked via dynamical processes. Sampling of these conformations is crucial for the understanding of protein properties and their interactions with other molecules. Recently, it was reported that AlphaFold1 can generate alternative conformations for certain proteins by manipulating input information such as multiple sequence alignments (MSAs)29. However, this approach is developed on the basis of varying the depth of MSAs, and it is hard to generalize to all proteins (especially those with a small number of homologous sequences). Therefore, it is highly desirable to develop advanced artificial intelligence (AI) models that can sample diverse structures consistent with the energy landscape in the conformational space29. Here, we show that DiG is capable of generating diverse and functionally relevant protein structures, which is a key capability for being able to efficiently sample equilibrium distributions.
Because the equilibrium distribution of protein conformations is difficult to obtain experimentally or computationally, there is a lack of high-quality data for training or benchmarking. To train this model, we collect experimental and simulated structures from public databases. To mitigate the data scarcity, we generated an MD simulation dataset and developed the PIDP training method (see Supplementary Information sections A.1.1 and D.1 for the training procedure and the dataset). The performance of DiG was assessed at two levels: (1) by comparing the conformational distributions against those obtained from extensive (millisecond timescale) atomistic MD simulations and (2) by validating on proteins with multiple conformations. As shown in Fig. 2a, the conformational distributions are obtained from MD simulations for two proteins from the SARS-CoV-2 virus30 (the receptor-binding domain (RBD) of the spike protein and the main protease, also known as 3CL protease; see Supplementary Information section A.7 for details on the MD simulation data). These two proteins are the crucial components of the SARS-CoV-2 and key targets for drug development in treating COVID-1931,32. The millisecond-timescale MD simulations extensively sample conformation space, and we therefore regard the resulting distribution as a proxy for the equilibrium distribution.

a, Structures generated by DiG resemble the diverse conformations of millisecond MD simulations. MD-simulated structures are projected onto the reduced space spanned by two time-lagged independent component analysis (TICA) coordinates (that is, independent component (IC) 1 and 2), and the probability densities are depicted using contour lines. Left: for the RBD protein, MD simulation reveals four highly populated regions in the 2D space spanned by TICA coordinates. DiG-generated structures are mapped to this 2D space (shown as orange dots), with a distribution reflected by the colour intensity. Under the distribution plot, structures generated by DiG (thin ribbons) are superposed on representative structures. AlphaFold-predicted structures (stars) are shown in the plot. Right: the results for the SARS-CoV-2 main protease, compared with MD simulation and AlphaFold prediction results. The contour map reveals three clusters, DiG-generated structures overlap with clusters II and III, whereas structures in cluster I are underrepresented. b, The performance of DiG on generating multiple conformations of proteins. Structures generated by DiG (thin ribbons) are compared with the experimentally determined structures (each structure is labelled by its PDB ID, except DEER-AF, which is an AlphaFold predicted model, shown as cylindrical cartoons). For the four proteins (adenylate kinase, LmrP membrane protein, human BRAF kinase and D-ribose binding protein), structures in two functional states (distinguished by cyan and brown) are well reproduced by DiG (ribbons).
Taking protein sequences as the descriptor inputs for DiG, structures were generated and compared with simulation data. Although simulation data of RBD and the main protease were not used for DiG training, generated structures resemble the conformational distributions (Fig. 2a). In the two-dimensional (2D) projection space of RBD conformations, MD simulations populate four regions, which are all sampled by DiG (Fig. 2a, left). Four representative structures are well reproduced by DiG. Similarly, three representative structures from main protease simulations are predicted by DiG (Fig. 2a). We noticed that conformations in cluster I are not well recovered by DiG, indicating room for improvement. In terms of conformational coverage, we compared the regions sampled by DiG with those from simulations in the 2D manifold (Fig. 2a), observing that about 70% of the RBD conformations sampled by simulations can be covered with just 10,000 DiG-generated structures (Supplementary Fig. 1).
Atomistic MD simulations are computationally expensive, therefore millisecond-timescale simulations of proteins are rarely executed, except for simulations on special-purpose hardware such as the Anton supercomputer13 or extensive distributed simulations combined in Markov state models16. To obtain an additional assessment on the diverse structures generated by DiG, we turn to proteins with multiple structures that have been experimentally determined. In Fig. 2b, we show the capability of DiG in generating multiple conformations for four proteins. Experimental structures are shown in cylinder cartoons, each aligned with two structures generated by DiG (thin ribbons). For example, DiG generated structures similar to either open or closed states of the adenylate kinase protein (for example, backbone root mean square difference (r.m.s.d.) < 1.0 Å to the closed state, 1ake). Similarly, for the drug transport protein LmrP, DiG generated structures covering both states (r.m.s.d. < 2.0 ): one structure is experimentally determined, and the other (denoted as DEER-AF) is the AlphaFold prediction29 supported by double electron electron resonance (DEER) experiments33. For human BRAF kinase, the overall structural difference between the two states is less pronounced. The major difference is in the A-loop region and a nearby helix (the αC-helix, indicated in the figure)34. Structures generated by DiG accurately capture such regional structural differences. For D-ribose binding protein, the packing of two domains is the major source of structural difference. DiG correctly generates structures corresponding to both the straight-up conformation (cylinder cartoon) and the twisted or tilted conformation. If we align one domain of D-ribose binding protein, the other domain only partially matches the twisted conformation as an ‘intermediate’ state. Furthermore, DiG can generate plausible conformation transition pathways by latent space interpolations (see demonstration cases in Supplementary Videos 1 and 2). In summary, beyond static structure prediction for proteins, DiG generates diverse structures corresponding to different functional states.
Ligand structure sampling around binding sites
An immediate extension of protein conformational sampling is to predict ligand structures in druggable pockets. To model the interactions between proteins and ligands, we conducted MD simulations for 1,500 complexes to train the DiG model (see Supplementary Information section D.1 for the dataset). We evaluated the performance of DiG with 409 protein–ligand systems35,36 that are not in the training dataset. The inputs of DiG include protein pocket information (atomic type and position) and the ligand descriptor (a SMILES string). We pad the input node and pair representations with zeros to handle the different number of atoms surrounding a pocket and the different length of SMILES strings. The predicted results are the atomic coordinate distributions of both the ligand and the protein pocket. For protein pockets, changes in atomic positions are up to 1.0 Å in terms of r.m.s.d. compared with the input values, reflecting pocket flexibility during ligand structure generation. For the ligand structures, the deviation comes from two sources: (1) the conformational difference between generated and experimental structures, and (2) the difference in the binding pose due to ligand translation and rotation. Among all the tested cases, the conformational differences are small, with an r.m.s.d. value of 1.74 Å on average, indicating that generated structures are highly similar to the ligands resolved in crystal structures (Fig. 3a). When including the binding pose deviations, larger discrepancies are observed. Yet, the DiG predicts structures that are very similar to the experimental structure for each system. The best matched structure among 50 generated structures for each system is within 2.0 Å r.m.s.d. compared with the experimental data for nearly all 409 testing systems (see Fig. 3a for the r.m.s.d. distribution, with more cases shown in Supplementary Fig. 3). The accuracy of generated structures for ligand is related to the characteristics of the binding pocket. For example, in the case of the TYK2 kinase protein, the ligand shown in Fig. 3b (top) deviated from the crystal structure by 0.91 Å (r.m.s.d.) on average. For target P38, the ligand exhibited more diverse binding poses, probably owing to the relatively shallow binding pocket, making the most stable binding pose less dominant compared with other poses (Fig. 3b, bottom). MD simulations reveal similar trends as DiG-generated structures, with ligand binding to TYK2 more tightly than in the case of P38 (Supplementary Fig. 2). Overall, we observed that the generated structures resemble experimentally observed poses.

a, The results of DiG on poses of ligands bound to protein pockets. DiG generates ligand structures and binding poses with good accuracy compared with the crystal structures (reflected by the r.m.s.d. statistics shown in the red histogram for the best matching cases and the green histogram for the median r.m.s.d. statistics). When considering all 50 predicted structures for each system, diversity is observed, as reflected in the r.m.s.d. histogram (yellow colour, normalized). All r.m.s.d. values are calculated for ligands with respect to their coordinates in complex structures. b, Representative systems show diversity in ligand structures, and such predicted diversity is related to the properties of the binding pocket. For a deep and narrow binding pocket such as for the TYK2 protein (shown in the surface representation, top panel), DiG predicts highly similar binding poses for the ligand (in atom bond representations, top panel). For the P38 protein, the binding pocket is relatively flat and shallow and predicted ligand poses are more diverse and have large conformational flexibility (bottom panel, following the same representations as in the TYK2 case). The average r.m.s.d. values and the associated standard deviations are indicated next to the complex structures.
Catalyst–adsorbate sampling
Identifying active adsorption sites is a central task in heterogeneous catalysis. Owing to the complex surface–molecule interactions, such tasks rely heavily on a combination of quantum chemistry methods such as density functional theory (DFT) and sampling techniques such as MD simulations and grid search. These lead to large and sometimes intractable computational costs. We evaluate the capability of DiG for this task by training it on the MD trajectories of catalyst–adsorbate systems from the Open Catalyst Project and carrying out further evaluations on random combinations of adsorbates and surfaces that are not included in the training set9. DiG takes the atomic types, initial positions of atoms in substrate, and the lattice vectors of the substrate, with an initial structure of the molecular adsorbate, as joint inputs. Besides, we use a cross-attention sub-layer to handle the periodic boundary conditions, as detailed in Supplementary Information section B.5. On feeding the model with a substrate and a molecular adsorbate, DiG can predict adsorption sites and stable adsorbate configurations, along with the probability for each configuration (see Supplementary Information sections A.4 and A.7 for training and evaluation details). Figure 4a,b shows the adsorption configurations of an acyl group on a stepped TiIr alloy surface. Multiple adsorption sites are predicted by DiG. To test the plausibility of these predicted configurations and evaluate the coverage of the predictions, we carry out a grid search using DFT methods. The results confirm that DiG predicts all stable sites found by the grid search and that the adsorption configurations are in close agreement, with an r.m.s.d. of 0.5–0.8 Å (Fig. 4b). It should be noted that the combinations of substrate and adsorbate shown in Fig. 4b are not included in the training dataset. Therefore, the result demonstrates the cross-system generalization capability of DiG in catalyst adsorption predictions. Here we show only the top view. Supplementary Fig. 4 in addition shows the front view of the adsorption configurations.

a, The problem setting: the prediction of the adsorption configuration distribution of an adsorbate on a catalyst surface. b, The adsorption sites and corresponding configurations of the adsorbate found by DiG (in colour) compared with DFT results (in white). DiG finds all adsorption sites, with adsorbate structures close to the DFT calculation results (see Supplementary Information for details of the adsorption sites and configurations). c–f, Adsorption prediction results of single N or O atoms on TiN (c), RhTcHf (d), AlHf (e) and TaPd (f) catalyst surfaces compared with DFT calculation results. Top: the catalyst surface. Middle: the probability distribution of adsorbate molecules on the corresponding catalyst surfaces on log scale. Bottom: the interaction energies between the adsorbate molecule and the catalyst calculated using DFT methods. The adsorption sites and predicted probabilities are highly consistent with the energy landscape obtained by DFT.
DiG not only predicts the adsorption sites with correct configurations but also provides a probability estimate for each adsorption configuration. This capability is illustrated in the systems with single-atom adsorbates (including H, N and O atoms) on ten randomly chosen metallic surfaces. For each combination of adsorbate and catalyst, DiG predicts the adsorption sites and the probability distributions. To validate the results, for the same systems, grid search DFT calculations are carried out to find adsorption sites and corresponding energies. Taking the adsorption sites identified by grid search as references, DiG achieved 81% site coverage for single-atom adsorbates on the ten metallic catalyst surfaces. Figure 4c–f shows closer examinations on adsorption predictions for four systems, namely single N or O atoms on TiN, RhTcHf, AlHf and TaPd metallic surfaces (top panels). The predicted adsorption probabilities projected on the plane in parallel with the catalyst surface are shown in the middle panels. The probabilities show excellent accordance with the adsorption energies calculated using DFT methods (bottom panels). It is worth noting that the speed of DiG is much faster compared with DFT; that is, it takes about 1 min to sample all adsorption sites for a catalyst–adsorbate system for DiG on a single modern graphics processing unit (GPU), but at least 2 hours for a single DFT relaxation with VASP, a number that will be further multiplied by a factor of >100 depending on the resolution of the searching grid37. Such fast and accurate prediction of adsorption sites and the corresponding distributional features can be useful in identifying catalytic mechanisms and guiding research on new catalysts.
Property-guided structure generation
While DiG by default generates structures following the learned training data distribution, the output distribution can be purposely biased to steer the structure generation to meet particular requirements. Here, we leverage this capability by using DiG for inverse design (described in ‘Property-guided structure generation with DiG’ section). As a proof of concept, we search for carbon allotropes with desired electronic band gaps. Similar tasks are critical to the design of novel photovoltaic and semi-conductive materials38. To train this model, we prepared a dataset composed of carbon materials by carrying out structure search based on energy profiles obtained from DFT calculations (L.Z., manuscript in preparation). The structures corresponding to energy minima form the dataset used to train DiG, which in turn are applied to generate carbon structures. We use a neural network model based on the M3GNet architecture11 as the property predictor for the electronic band gap, which is fed to the property-guided structure generation of carbon structures.
Figure 5 shows the distributions of band gaps calculated from generated carbon structures. In the original training dataset, most structures have a band gap of around 0 eV (Fig. 5a). When the target band gaps are supplied to DiG as an additional condition, carbon structures are generated with desired band gaps. Under the guidance of a band gap model in conditional generation, the distribution is biased towards the targets, showing pronounced peaks around the target band gaps. Representative structures are shown in Fig. 5. For conditional generation with a target band gap of 4 eV, DiG generates stable carbon structures similar to diamond, which has large band gaps. In the case of the 0 eV band gap, we obtain graphite-like structures with small band gaps. In Fig. 5a, we show some structures obtained by unconditional generation. To evaluate the quality of carbon structures generated by DiG, we calculate the percentage of generated structures that match relaxed structures in the dataset by using the ‘StructureMatcher’ in the PyMatgen package39. For unconditional generation, the match rate is 99.87%, and the average matched normalized r.m.s.d. computed from fractional coordinates over all sampled structures is 0.16. For conditional generation, the match rate is 99.99%, but with a higher average normalized r.m.s.d. of 0.22. While increasing the possibility of generating structures with the target band gap, conditional generation can influence the quality of the structures (see Supplementary Information section F.1 for more discussion). This proof-of-concept study shows that DiG not only captures the probability distributions with complex features in a large configurational space but also can be applied for inverse design of materials, when combined with a property quantifier, such as a machine learning (ML) predictor. Since the property prediction model (for example, the M3GNet model for band gap prediction) and the diffusion model of DiG are fully decoupled, our approach can be readily extended to inverse design of materials targeting for other properties.

a, The electronic band gaps of generated structures from the trained DiG with no specification on the band gap. Generated structures do not show any obvious preference on band gaps, closely resembling the distribution of the training dataset. b, Structures generated for three band gaps (0, 2 and 4 eV). The distributions of band gaps for generated structures peak at the desired values. In particular, DiG generates graphite-like structures when the desired band gap is 0 eV, while for the 4 eV band gap, the generated structures are mostly similar to diamonds. The vertical dashed lines represent the band gaps of generated structures near to 0, 2 and 4 eV. Inset: representative structures.
Discussion
Predicting the equilibrium distribution of molecular states is a formidable challenge in molecular sciences, with broad impacts for understanding structure–function relations, computing macroscopic properties and designing molecules and materials. Existing methods need numerous measurements or simulated samples of single molecules to characterize the equilibrium distribution. We introduce DiG, a deep generative framework towards predicting equilibrium probability distributions, enabling efficient sampling of diverse conformations and state densities across molecular systems. Inspired by the annealing process, DiG uses a sequence of deep neural networks to gradually transform state distributions from a simple form to the target ones. DiG can be trained to approximate the equilibrium distribution with suitable data.
We applied DiG to several molecular tasks, including protein conformation sampling, protein–ligand binding structure generation, molecular adsorption on catalyst surfaces and property-guided structure generation. DiG generates chemically realistic and diverse structures, and distributions that resemble MD simulations in low-dimensional projections in some cases. By leveraging advanced deep learning architectures, DiG learns the representation of molecular conformations from molecular descriptors such as sequences for proteins or formulas for compound molecules. Moreover, its capacity to model complex, multimodal distributions using diffusion models enables it to capture equilibrium distributions in high-dimensional space.
Consequently, the framework opens the door to a multitude of research opportunities and applications in molecular science. DiG can provide statistical understanding of molecules, enabling computation of macroscopic properties such as free energies and thermodynamic stability. These insights are critical for investigating physical and chemical phenomena of molecular systems.
Finally, with its ability to generate independent and identically distributed (i.i.d.) conformations from equilibrium distributions, DiG offers a substantial advantage over traditional sampling or simulation approaches, such as Markov chain Monte Carlo (MCMC) or MD simulations, which need rare events to cross energy barriers. DiG covers similar conformation space as millisecond-timescale MD simulations in the two tested protein cases. On the basis of the OpenMM performance benchmark, it would require about 7–10 GPU-years on NVIDIA A100s to simulate 1.8 ms for RBD of the spike protein, while generating 50k structures with DiG takes about 10 days on a single A100 GPU without inference acceleration (Supplementary Information section A.6). Similar or even better speed-up has been achieved for predicting the adsorbate distribution on a catalyst surface, as shown in Results. Combined with high-accuracy probability distributions, such order-of-magnitude speed-up will be transformative for molecular simulation and design.
Although the quantitative prediction of equilibrium distributions at given states will hinge upon data availability, the capacity of DiG to explore vast and diverse conformational spaces contributes to the discovery of novel and functional molecular structures, including protein structures, ligand conformers and adsorbate configurations. DiG can help to connect microscopic descriptors and macroscopic observations of molecular systems, with potential effect on various areas of molecular sciences, including but not limited to life sciences, drug design, catalysis research and materials sciences.
Methods
Deep neural networks have been demonstrated to predict accurate molecular structures from descriptors \({{{\mathcal{D}}}}\) for many molecular systems1,5,6,9,10,11,12. Here, DiG aims to take one step further to predict not only the most probable structure but also diverse structures with probabilities under the equilibrium distribution. To tackle this challenge, inspired by the heating–annealing paradigm, we break down the difficulty of this problem into a series of simpler problems. The heating–annealing paradigm can be viewed as a pair of reciprocal stochastic processes on the structure space that simulate the transformation between the system-specific equilibrium distribution and a system-independent simple distribution psimple. Following this idea, we use an explicit diffusion process (forward process; Fig. 1b, orange arrows) that gradually transforms the target distribution of the molecule \({q}_{{{{\mathcal{D}}}},0}\), as the initial distribution, towards psimple through a time period τ. The corresponding reverse diffusion process then transforms psimple back to the target distribution \({q}_{{{{\mathcal{D}}}},0}\). This is the generation process of DiG (Fig. 1b, blue arrows). The reverse process is performed by incorporating updates predicted by deep neural networks from the given \({{{\mathcal{D}}}}\), which are trained to match the forward process. The descriptor \({{{\mathcal{D}}}}\) is processed into node representations \({{{\mathcal{V}}}}\) describing the feature of each system-specific individual element and a pair representation \({{{\mathcal{P}}}}\) describing inter-node features. The \(\{{{{\mathcal{V}}}},{{{\mathcal{P}}}}\}\) representation is the direct input from the descriptor part to the Graphormer model10, together with the geometric structure input R to produce a physically finer structure (Supplementary Information sections B.1 and B.3). Specifically, we choose \({p}_{{{\mbox{simple}}}}:= {{{\mathcal{N}}}}({{{\bf{0}}}},{{{\bf{I}}}})\) as the standard Gaussian distribution in the state space, and the forward diffusion process as the Langevin diffusion process targeting this psimple (Ornstein–Uhlenbeck process)40,41,42. A time dilation scheme βt (ref. 43) is introduced for approximate convergence to psimple after a finite time τ. The result is written as the following stochastic differential equation (SDE):
where Bt is the standard Brownian motion (a.k.a. Wiener process). Choosing this forward process leads to a psimple that is more concentrated than a heated distribution, hence it is easier to draw high-density samples, and the form of the process enables efficient training and sampling.
Following stochastic process theory (see, for example, ref. 44), the reverse process is also a stochastic process, written as the following SDE:
where \(\bar{t}:= \tau -t\) is the reversed time, \({q}_{{{{\mathcal{D}}}},\bar{t}}:= {q}_{{{{\mathcal{D}}}},t = \tau -\bar{t}}\) is the forward process distribution at the corresponding time and \({{{{\bf{B}}}}}_{\bar{t}}\) is the Brownian motion in reversed time. Note that the forward and corresponding reverse processes, equations (1) and (2), are inspired from but not exactly the heating and annealing processes. In particular, there is no concept of temperature in the two processes. The temperature T mentioned in the PIDP loss below is the temperature of the real target system but is not related to the diffusion processes.
From equation (2), the only obstacle that impedes the simulation of the reverse process for recovering \({q}_{{{{\mathcal{D}}}},0}\) from psimple is the unknown \(\nabla \log {q}_{{{{\mathcal{D}}}},\bar{t}}({{{{\bf{R}}}}}_{\bar{t}})\). Deep neural networks are then used to construct a score model \({{{{\bf{s}}}}}_{{{{\mathcal{D}}}},t}^{\theta }({{{\bf{R}}}})\), which is trained to predict the true score function \(\nabla \log {q}_{{{{\mathcal{D}}}},t}({{{\bf{R}}}})\) of each instantaneous distribution \({q}_{{{{\mathcal{D}}}},t}\) from the forward process. This formulation is called a diffusion-based generative model and has been demonstrated to be able to generate high-quality samples of images and other content27,28,45,46,47. As our score model is defined in molecular conformational space, we use our previously developed Graphormer model10 as the neural network architecture backbone of DiG, to leverage its capabilities in modelling molecular structures and to generalize to a range of molecular systems. Note that the score model aims to approximate a gradient, which is a set of vectors. As these are equivariant with respect to the input coordinates, we designed an equivariant vector output head for the Graphormer model (Supplementary Information section B.4).
With the \({{{{\bf{s}}}}}_{{{{\mathcal{D}}}},t}^{\theta }({{{\bf{R}}}})\) model, drawing a sample R0 from the equilibrium distribution of a system \({{{\mathcal{D}}}}\) can be done by simulating the reverse process in equation (2) on N + 1 steps that uniformly discretize [0, τ] with step size h = τ/N (Fig. 1b, blue arrows), thus
where the discrete step index i corresponds to time t = ih, and βi := hβt=ih. Supplementary Information section A.1 provides the derivation. Note that the reverse process does not need to be ergodic. The way that DiG models the equilibrium distribution is to use the instantaneous distribution at the instant t = 0 (or \(\bar{t}=\tau\)) on the reverse process, but not using a time average. As RN samples can be drawn independently, DiG can generate statistically independent R0 samples for the equilibrium distribution. In contrast to MD or MCMC simulations, the generation of DiG samples does not suffer from rare events that link different states and can thus be far more computationally efficient.
PIDP
DiG can be trained by using conformation data sampled over a range of molecular systems. However, collecting sufficient experimental or simulation data to characterize the equilibrium distribution for various systems is extremely costly. To address this data scarcity issue, we propose a pre-training algorithm, called PIDP, which effectively optimizes DiG on an initial set of candidate structures that need not be sampled from the equilibrium distribution. The supervision comes from the energy function \({E}_{{{{\mathcal{D}}}}}\) of each system \({{{\mathcal{D}}}}\), which defines the equilibrium distribution \({q}_{{{{\mathcal{D}}}},0}({{{\bf{R}}}})\propto \exp (-\frac{{E}_{{{{\mathcal{D}}}}}({{{\bf{R}}}})}{{k}_{{{{\rm{B}}}}}T})\) at the target temperature T.
The key idea is that the true score function \(\nabla \log {q}_{{{{\mathcal{D}}}},t}\) from the forward process in equation (1) obeys a partial differential equation, known as the Fokker–Planck equation (see, for example, ref. 48). We then pre-train the score model \({{{{\bf{s}}}}}_{{{{\mathcal{D}}}},t}^{\theta }\) by minimizing the following loss function that enforces the equation to hold:
Here, the second term, weighted by λ1, matches the score model at the final generation step to the score from the energy function, and the first term implicitly propagates the energy function supervision to intermediate time steps (Fig. 1b, upper row). The structures \({\{{{{{\bf{R}}}}}_{{{{\mathcal{D}}}},i}^{(m)}\}}_{m = 1}^{M}\) are points on a grid spanning the structure space. Since these structures are only used to evaluate the loss function on discretized points, they do not have to obey the equilibrium distribution (as is required by structures in the training dataset), therefore the cost of preparing these structures can be much lower. As structure spaces of molecular systems are often very high dimensional (for example, thousands for proteins), a regular grid would have intractably many points. Fortunately, the space of actual interest is only a low-dimensional manifold of physically reasonable structures (structures with low energy) relevant to the problem. This allows us to effectively train the model only on these relevant structures as R0 samples. Ri samples are produced by passing R0 samples through the forward process. See Supplementary Information section C.1 for an example on acquiring relevant structures for protein systems.
We also leverage stochastic estimators, including Hutchinson’s estimator49,50, to reduce the complexity in calculating derivatives of high order and for high-dimensional vector-valued functions. Note that, for each step i, the corresponding model \({{{{\bf{s}}}}}_{{{{\mathcal{D}}}},i}^{\theta }\) receives a training loss independent of other steps and can be directly back-propagated. In this way, the supervision on each step can improve the optimizing efficiency.
Training DiG with data
In addition to using the energy function for information on the probability distribution of the molecular system, DiG can also be trained with molecular structure samples that can be obtained from experiments, MD or other simulation methods. See Supplementary Information section C for data collection details. Even when the simulation data are limited, they still provide information about the regions of interest and about the local shape of the distribution in these regions; hence, they are helpful to improve a pre-trained DiG. To train DiG on data, the score model \({{{{\bf{s}}}}}_{{{{\mathcal{D}}}},i}^{\theta }({{{{\bf{R}}}}}_{i})\) is matched to the corresponding score function \(\nabla \log {q}_{{{{\mathcal{D}}}},i}\) demonstrated by data samples. This can be done by minimizing \({{\mathbb{E}}}_{{q}_{{{{\mathcal{D}}}},i}({{{{\bf{R}}}}}_{i})}{\parallel {{{{\bf{s}}}}}_{{{{\mathcal{D}}}},i}^{\theta }({{{{\bf{R}}}}}_{i})-\nabla \log {q}_{{{{\mathcal{D}}}},i}({{{{\bf{R}}}}}_{i})\parallel }^{2}\) for each diffusion time step i. Although a precise calculation of \(\nabla \log {q}_{{{{\mathcal{D}}}},i}\) is impractical, the loss function can be equivalently reformulated into a denoising score-matching form51,52
where \({\alpha }_{i}:= \mathop{\prod }\nolimits_{j = 1}^{i}\sqrt{1-{\beta }_{j}}\), \({\sigma }_{i}:= \sqrt{1-{\alpha }_{i}^{2}}\) and p(ϵi) is the standard Gaussian distribution. The expectation under \({q}_{{{{\mathcal{D}}}},0}\) can be estimated using the simulation dataset.
We remark that this score-predicting formulation is equivalent (Supplementary Information section A.1.2) to the noise-predicting formulation28 in the diffusion model literature. Note that this function allows direct loss estimation and back-propagation for each i in constant (with respect to i) cost, recovering the efficient step-specific supervision again (Fig. 1b, bottom).
Density estimation by DiG
The computation of many thermodynamic properties of a molecular system (for example, free energy or entropy) also requires the density function of the equilibrium distribution, which is another aspect of the distribution besides a sampling method. DiG allows for this by tracking the distribution change along the diffusion process45:
where D is the dimension of the state space and \({{{{\bf{R}}}}}_{{{{\mathcal{D}}}},t}^{\theta }({{{{\bf{R}}}}}_{0})\) is the solution to the ordinary differential equation (ODE)
with initial condition R0, which can be solved using standard ODE solvers or more efficient specific solvers (Supplementary Information section A.6).
Property-guided structure generation with DiG
There is a growing demand for the design of materials and molecules that possess desired properties, such as intrinsic electronic band gaps, elastic modulus and ionic conductivity, without going through a forward searching process. DiG provides a feature to enable such property-guided structure generation, by directly predicting the conditional structural distribution given a value c of a microscopic property.
To achieve this goal, regarding the data-generating process in equation (2), we only need to adapt the score function from \(\nabla \log {q}_{{{{\mathcal{D}}}},t}({{{\bf{R}}}})\) to \({\nabla }_{{{{\bf{R}}}}}\log {q}_{{{{\mathcal{D}}}},t}({{{\bf{R}}}}| c)\). Using Bayes’ rule, the latter can be reformulated as \({\nabla }_{{{{\bf{R}}}}}\log {q}_{{{{\mathcal{D}}}},t}({{{\bf{R}}}}| c)=\nabla \log {q}_{{{{\mathcal{D}}}},t}({{{\bf{R}}}})+{\nabla }_{{{{\bf{R}}}}}\log {q}_{{{{\mathcal{D}}}}}(c| {{{\bf{R}}}})\), where the first term can be approximated by the learned (unconditioned) score model; that is, the new score model is
Hence, only a \({q}_{{{{\mathcal{D}}}}}(c| {{{\bf{R}}}})\) model is additionally needed45,46, which is a property predictor or classifier that is much easier to train than a generative model.
In a normal workflow for ML inverse design, a dataset must be generated to meet the conditional distribution, then an ML model will be trained on this dataset for structure distribution predictions. The ability to generate structures for conditional distribution without requiring a conditional dataset places DiG in an advantageous position when compared with normal workflows in terms of both efficiency and computational cost.
Interpolation between states
Given two states, DiG can approximate a reaction path that corresponds to reaction coordinates or collective variables, and find intermediate states along the path. This is achieved through the fact that the distribution transformation process described in equation (1) is equivalent to the process in equation (3) if \({{{{\bf{s}}}}}_{{{{\mathcal{D}}}},i}^{\theta }\) is well learned, which is deterministic and invertible, hence establishing a correspondence between the structure and latent space. We can then uniquely map the two given states in the structure space to the latent space, approximate the path in the latent space by linear interpolation and then map the path back to the structure space. Since the distribution in the latent space is Gaussian, which has a convex contour, the linearly interpolated path goes through high-probability or low-energy regions, so it gives an intuitive guess of the real reaction path.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Structures from the Protein Data Bank (PDB) were used for training and as templates (https://www.wwpdb.org/ftp/pdb-ftp-sites; for the associated sequence data and 100% sequence clustering see also https://ftp.wwpdb.org/pub/pdb/derived_data/and https://cdn.rcsb.org/resources/sequence/clusters/clusters-by-entity-100.txt). Training used a version of the PDB downloaded on 25 December 2020. The template search also used the PDB70 database, downloaded 13 May 2020 (https://wwwuser.gwdg.de/~compbiol/data/hhsuite/databases/hhsuite_dbs/). For MSA lookup at both the training and prediction time, we used Uniclust30 v.2018_08 (https://wwwuser.gwdg.de/~compbiol/uniclust/2018_08/). The milisecond MD simulation trajectories for the RBD and main protease of SARS-CoV-2 are downloaded from the coronavirus disease 2019 simulation database (https://covid.molssi.org/simulations/). We collect 238 simulation trajectories from the GPCRmd dataset (https://www.gpcrmd.org/dynadb/datasets/). Protein–ligand docked complexes are collected from CrossDocked2020 dataset v1.3 (https://github.com/gnina/models/tree/master/data/CrossDocked2020). The MD simulation trajectories for 1,500 protein–ligand complexes and the generated carbon structures are available upon request from the corresponding authors (S.Z., C.L., H.L. or T.Y.-L.) owing to Microsoft’s data release policy.
The OC20 dataset used for catalyst–adsorption generation modelling is publicly available (https://github.com/Open-Catalyst-Project/ocp/blob/main/DATASET.md). Specifically, we use the IS2RS part and MD part. The carbon polymorphs dataset is generated using random structure search where random initial structures are relaxed together with the lattice using density functional theory with conjugated gradient. The generated carbon structures are available upon request from the corresponding authors (S.Z., C.L., H.L. or T.-Y.L.) owing to Microsoft’s data release policy.
Code availability
Source code for the Distributional Graphormer model, inference scripts, and model weights are available via Zenodo at https://doi.org/10.5281/zenodo.10911143 (ref. 53). An online demo page is available at https://DistributionalGraphormer.github.io.
The DiG models are primarily developed using Python, PyTorch, Numpy, fairseq, torch-geometric and rdkit. We used HHBlits and HHSearch from the hh-suite for MSA and PDB70 template searches, and Gromacs for MD simulations. OpenMM, pdbfixer and the amber14 force field were utilized for energy function training. DFT calculations for the carbon polymorphs dataset were performed with VASP. Both the carbon polymorphs and OC20 datasets were converted to PyG graphs using torch-geometric and stored in lmdb databases. For more detailed information, please refer to the code repository.
Data analysis for proteins and ligands was conducted using Python, PyTorch, Numpy, Matplotlib, MDTraj, seaborn, SciPy, scikit-learn, pandas and Biopython. Visualization and rendering were done with ChimeraX and Pymol. Analysis and visualization of catalyst–adsorption systems and carbon structures were performed with Python, PyTorch, Numpy, Matplotlib, Pandas and VESTA. Adsorption configurations were searched using density functional theory computations with VASP.
References
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589 (2021).
Cramer, P. Alphafold2 and the future of structural biology. Nat. Struct. Mol. Biol. 28, 704–705 (2021).
Akdel, M. et al. A structural biology community assessment of AlphaFold2 applications. Nat. Struct. Mol. Biol. 29, 1056–1067 (2022).
Pereira, J. et al. High-accuracy protein structure prediction in casp14. Proteins Struct. Funct. Bioinf. 89, 1687–1699 (2021).
Stärk, H., Ganea, O., Pattanaik, L., Barzilay, R. & Jaakkola, T. Equibind: geometric deep learning for drug binding structure prediction. In Proc. International Conference on Machine Learning 20503–20521 (PMLR, 2022).
Corso, G., Stärk, H., Jing, B., Barzilay, R. & Jaakkola, T. DiffDock: diffusion steps, twists, and turns for molecular docking. In Proc. International Conference on Learning Representations (2023).
Diaz-Rovira, A. M. et al. Are deep learning structural models sufficiently accurate for virtual screening? application of docking algorithms to AlphaFold2 predicted structures. J. Chem. Inf. Model. 63, 1668–1674 (2023).
Scardino, V., Di Filippo, J. I. & Cavasotto, C. N. How good are AlphaFold models for docking-based virtual screening? iScience 26, 105920 (2022).
Chanussot, L. et al. Open catalyst 2020 (OC20) dataset and community challenges. ACS Catal. 11, 6059–6072 (2021).
Ying, C. et al. Do transformers really perform badly for graph representation? Adv. Neural Inf. Process. Syst. 34, 28877–28888 (2021).
Chen, C. & Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2, 718–728 (2022).
Schaarschmidt, M. et al. Learned force fields are ready for ground state catalyst discovery. Preprint at https://arxiv.org/abs/2209.12466 (2022).
Lindorff-Larsen, K., Piana, S., Dror, R. O. & Shaw, D. E. How fast-folding proteins fold. Science 334, 517–520 (2011).
Barducci, A., Bonomi, M. & Parrinello, M. Metadynamics. Wiley Interdisc. Rev. Comput. Mol. Sci. 1, 826–843 (2011).
Kästner, J. Umbrella sampling. Wiley Interdisc. Rev. Comput. Mol. Sci. 1, 932–942 (2011).
Chodera, J. D. & Noé, F. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 25, 135–144 (2014).
Monticelli, L. et al. The Martini coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 4, 819–834 (2008).
Clementi, C. Coarse-grained models of protein folding: toy models or predictive tools? Curr. Opin. Struct. Biol. 18, 10–15 (2008).
Wang, J. et al. Machine learning of coarse-grained molecular dynamics force fields. ACS Cent. Sci. 5, 755–767 (2019).
Arts, M. et al. Two for one: diffusion models and force fields for coarse-grained molecular dynamics. J. Chem. Theory Comput. 19, 6151–6159 (2023).
Noé, F., Olsson, S., Köhler, J. & Wu, H. Boltzmann generators: sampling equilibrium states of many-body systems with deep learning. Science 365, 1147 (2019).
Klein, L. et al. Timewarp: transferable acceleration of molecular dynamics by learning time-coarsened dynamics. In Advances Neural Information Processing Systems Vol 36 (2024).
Kirkpatrick, S., Gelatt Jr, C. D. & Vecchi, M. P. Optimization by simulated annealing. Science 220, 671–680 (1983).
Neal, R. M. Annealed importance sampling. Stat. Comput. 11, 125–139 (2001).
Del Moral, P., Doucet, A. & Jasra, A. Sequential Monte Carlo samplers. J. R. Stat. Soc. B 68, 411–436 (2006).
Doucet, A., Grathwohl, W.S., Matthews, A.G.d.G. & Strathmann, H. Annealed importance sampling meets score matching. In Proc. ICLR Workshop on Deep Generative Models for Highly Structured Data (2022).
Sohl-Dickstein, J., Weiss, E., Maheswaranathan, N. & Ganguli, S. Deep unsupervised learning using nonequilibrium thermodynamics. In Proc. International Conference on Machine Learning 2256–2265 (PMLR, 2015).
Ho, J., Jain, A. & Abbeel, P. Denoising diffusion probabilistic models. Adv. Neural Inf. Process. Syst. 33, 6840–6851 (2020).
Del Alamo, D., Sala, D., Mchaourab, H. S. & Meiler, J. Sampling alternative conformational states of transporters and receptors with alphafold2. eLife 11, 75751 (2022).
Zimmerman, M. I. et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 13, 651–659 (2021).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368, 409–412 (2020).
Tai, W. et al. Characterization of the receptor-binding domain (rbd) of 2019 novel coronavirus: implication for development of rbd protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 17, 613–620 (2020).
Masureel, M. et al. Protonation drives the conformational switch in the multidrug transporter LmrP. Nat. Chem. Biol. 10, 149–155 (2014).
Nussinov, R., Zhang, M., Liu, Y. & Jang, H. Alphafold, artificial intelligence (AI), and allostery. J. Phys. Chem. B 126, 6372–6383 (2022).
Schindler, C. E. et al. Large-scale assessment of binding free energy calculations in active drug discovery projects. J. Chem. Inf. Model. 60, 5457–5474 (2020).
Wang, L. et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc. 137, 2695–2703 (2015).
Hafner, J. Ab-initio simulations of materials using VASP: density-functional theory and beyond. J. Comput. Chem. 29, 2044–2078 (2008).
Lu, Z. Computational discovery of energy materials in the era of big data and machine learning: a critical review. Mater. Rep. Energy 1, 100047 (2021).
Ong, S. P. et al. Python materials genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Langevin, P. Sur la théorie du mouvement brownien. Compt. Rendus 146, 530–533 (1908).
Uhlenbeck, G. E. & Ornstein, L. S. On the theory of the Brownian motion. Phys. Rev. 36, 823–841 (1930).
Roberts, G. O. et al. Exponential convergence of Langevin distributions and their discrete approximations. Bernoulli 2, 341–363 (1996).
Wibisono, A., Wilson, A. C. & Jordan, M. I. A variational perspective on accelerated methods in optimization. Proc. Natl Acad. Sci. USA 113, 7351–7358 (2016).
Anderson, B. D. Reverse-time diffusion equation models. Stoch. Process. Their Appl. 12, 313–326 (1982).
Song, Y. et al. Score-based generative modeling through stochastic differential equations. In Proc. International Conference on Learning Representations (2021).
Dhariwal, P. & Nichol, A. Diffusion models beat GANs on image synthesis. Adv. Neural Inf. Process. Syst. 34, 8780–8794 (2021).
Ramesh, A., Dhariwal, P., Nichol, A., Chu, C. & Chen, M. Hierarchical text-conditional image generation with clip latents. Preprint at https://arxiv.org/abs/2204.06125 (2022).
Risken, H. Fokker–Planck Equation (Springer, 1996).
Hutchinson, M. F. A stochastic estimator of the trace of the influence matrix for Laplacian smoothing splines. Commun. Stats. Simul. Comput. 18, 1059–1076 (1989).
Grathwohl, W., Chen, R.T., Bettencourt, J., Sutskever, I. & Duvenaud, D. FFJORD: free-form continuous dynamics for scalable reversible generative models. In Proc. International Conference on Learning Representations (2019).
Vincent, P. A connection between score matching and denoising autoencoders. Neural Comput. 23, 1661–1674 (2011).
Alain, G. & Bengio, Y. What regularized auto-encoders learn from the data-generating distribution. J. Mach. Learn. Res. 15, 3563–3593 (2014).
Zheng, L. et al. Towards predicting equilibrium distributions for molecular systems with deep learning. Zenodo https://doi.org/10.5281/zenodo.10911143 (2024).
Acknowledgements
This work has been supported by the Joint Funds of the National Natural Science Foundation of China (grant no. U20B2047). We thank N. A. Baker, L. Sun, B. Veeling, V. García Satorras, A. Foong and C. Lu for insightful discussions; S. Luo for helping with dataset preparations; J. Su for managing the project; J. Bai for helping with figure design; G. Guo for helping with cover design; and colleagues at Microsoft for their encouragement and support.
Author information
Authors and Affiliations
Contributions
S.Z. and T.-Y.L. led the research. S.Z., J.H., C.L., Z.L. and H.L. conceived the project. J.H., C.L., Y.S., W.F., F.J. and J.Wang developed the diffusion model and training pipeline. J.H., Y.S., Z.L., J.Z., F.J., H.Z. and H.L. developed data and analytics systems. H.L., Y.S., Z.L., Y.M. and S.T. conducted simulations. H.H., P.J., C.C., and F.N. contributed technical advice and ideas. S.Z., J.H., C.L., Y.S., Z.L., F.N., H.Z. and H.L. wrote the paper with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
S.Z., C.L., Y.S., H.L. and T.-Y.L. are inventors of a pending patent application in the name of Microsoft Technology Licensing LLC concerning machine learning for predicting molecular systems as related to this paper. The other authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Tiago Rodrigues, Dacheng Tao, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary discussion, Figs. 1–9 and Tables 1–6.
Supplementary Video 1
DiG can generate plausible transition pathways that connect the two structures of adenylate kinase, corresponding to open and closed conformations. Using an interpolation approach, DiG generates pathways connecting open and closed states. The open and closed structures are shown as semi-transparent ribbons, and the structures along the predicted pathways are shown as cartoons (each secondary structure component is coloured differently for better visualization).
Supplementary Video 2
DiG can generate plausible transition pathways that connect the two structures of LmrP membrane protein, corresponding to open and closed conformations. Using an interpolation approach, DiG generates pathways connecting open and closed states. The open and closed structures are shown as semi-transparent ribbons, and the structures along the predicted pathways are shown as cartoons (each secondary structure component is coloured differently for better visualization).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, S., He, J., Liu, C. et al. Predicting equilibrium distributions for molecular systems with deep learning. Nat Mach Intell (2024). https://doi.org/10.1038/s42256-024-00837-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42256-024-00837-3
