
Abstract
Prime editors (PEs) are promising genome-editing tools, but effective optimization of prime-editing guide RNA (pegRNA) design remains a challenge owing to the lack of accurate and broadly applicable approaches. Here we develop Optimized Prime Editing Design (OPED), an interpretable nucleotide language model that leverages transfer learning to improve its accuracy and generalizability for the efficiency prediction and design optimization of pegRNAs. Comprehensive validations on various published datasets demonstrate its broad applicability in efficiency prediction across diverse scenarios. Notably, pegRNAs with high OPED scores consistently show significantly increased editing efficiencies. Furthermore, the versatility and efficacy of OPED in design optimization are confirmed by efficiently installing various ClinVar pathogenic variants using optimized pegRNAs in the PE2, PE3/PE3b and ePE editing systems. OPED consistently outperforms existing state-of-the-art approaches. We construct the OPEDVar database of optimized designs from over two billion candidates for all pathogenic variants and provide a user-friendly web application of OPED for any desired edit.
Similar content being viewed by others


Main
Prime editing is a versatile and precise genome-editing technology, enabling the introduction of insertions, deletions and all 12 possible point mutations without DNA double-strand breaks or donor DNA templates1,2. The prime editor 2 (PE2), a basic PE version, comprises a Cas9 nickase–reverse transcriptase fusion protein, with the Cas9 nickase inducing DNA single-strand breaks, alongside a prime-editing guide RNA (pegRNA). The pegRNA contains three essential sub-sequences: a guide sequence (spacer), a primer binding site (PBS) and a reverse transcription template (RTT). The spacer guides pegRNA to the genomic target site (protospacer) and initiates an nCas9-mediated strand nick. The resulting 3′ end hybridizes to the PBS and primes the reverse transcription. This results in the formation of two redundant single-stranded DNA flaps. The equilibrium between the edited 3′ flap and the unedited 5′ flap facilitates the cleavage and ligation, along with DNA repair processes, leading to the incorporation of the desired edit encoded in the RTT into the genome1. In advanced PE versions such as PE3/PE3b and ePE, an additional single guide RNA (sgRNA) is employed to induce a nick on the opposite strand1,2,3,4. At present, the prime-editing technique is undergoing rapid optimization, encompassing modifications to the sequence and architecture of pegRNA5,6,7,8, regulation of the associated repair pathway9,10,11 and utilization of the paired prime-editing strategy12,13. Importantly, the prime-editing efficiency is notably influenced by the characteristics of pegRNA1,14,15, underscoring the necessity and criticality of pegRNA optimization before final application.
Customizing the pegRNA design for different types of edit and different edit positions presents a complex and time-intensive task. Recently, several rule-based tools such as pegFinder16, PrimeDesign17 and PE-Designer18 have emerged to offer recommendations for pegRNA designs based on expert-driven design guidelines. Nonetheless, their performance remains constrained by the expertise and knowledge of human experts. To address this limitation, three machine learning-based models (DeepPE4, Easy-Prime19 and PRIDICT20) have been developed for optimizing pegRNA designs. However, these models heavily rely on manual feature engineering, involving the calculation of numerous predefined pegRNA features such as GC count and minimum self-folding free energy. These models may overlook critical insights and result in limited accuracy and generalizability. Moreover, they inherently lack interpretability, resembling black boxes. Consequently, post hoc interpretable approaches such as SHAP21 are employed to provide feature importance.
Here we developed Optimized Prime Editing Design (OPED) for predicting pegRNA efficiency and optimizing pegRNA design. OPED automatically learns a comprehensive and interpretable representation of the target DNA and pegRNA pair, thereby improving its generalizability and accuracy without relying on predefined features. We demonstrated the versatility and accuracy of OPED in predicting pegRNA efficiencies across different edit types, edit positions, endogenous sites, laboratory settings, tevopreQ1 conditions, mismatch repair (MMR) inhibition conditions, cell lines in vitro and mouse hepatocytes in vivo. Remarkably, pegRNAs with a higher OPED editing score resulted in substantially higher editing efficiencies (2.2–82.9-fold) compared with pegRNAs with lower OPED scores across the aforementioned scenarios. Furthermore, we validated the broad applicability and superior performance of OPED by efficiently installing various ClinVar22 pathogenic variants using optimized PE designs (pegRNAs and sgRNAs) for the PE2, PE3/PE3b and ePE editing systems. Notably, the PE designs recommended by OPED showed substantially higher average editing efficiencies compared with those generated by existing PE design tools (PE2, 2.1–6.8-fold; PE3/PE3b, 7.9–24.6-fold; ePE, 3.4–47.5-fold). OPED consistently outperformed existing PE design tools in all comparisons. Moreover, we constructed OPEDVar, a comprehensive database comprising optimized PE designs targeting over 77,000 ClinVar pathogenic variants, and developed a user-friendly web application for OPED (http://bicdb.ncpsb.org.cn/OPED/).
Results
Design and development of OPED
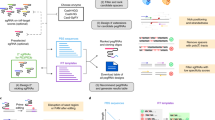
To enhance the accuracy and interpretability for pegRNA efficiency prediction, we devised OPED, a nucleotide language model inspired by the achievements of deep learning-based natural language models23,24,25 (Fig. 1, Extended Data Fig. 1 and Methods). OPED comprises three major modules: an input module, a feature learning module and a regression module. The upstream input module performs preprocessing, embedding and positional encoding24 of raw nucleotide sequences of the target DNA and pegRNA pair (Extended Data Fig. 1a). The core feature learning module utilizes specialized custom transformer and attention24 networks to automatically learn a comprehensive and interpretable representation of the DNA–pegRNA pair (Extended Data Fig. 1b). Specifically, a transformer encoder is employed to extract features from the target sequence, where self-attention mechanisms capture interdependencies between nucleotides within the target sequence. A custom transformer decoder is utilized to extract features from the PBS sequence, with self-attention mechanisms capturing interdependencies between nucleotides within the PBS sequence, and encoder–decoder attention capturing interdependencies between nucleotides of the PBS sequence and the target sequence. Similarly, another transformer decoder is customized to extract features from the RTT sequence. Subsequently, a tailored attention network is designed for the interpretation of nucleotide contributions and higher-level feature learning, generating a sequence description vector. These sequence description vectors are then concatenated into a comprehensive description vector that represents the DNA–pegRNA pair. The downstream regression module employs a multilayer fully connected feed-forward network to learn the underlying relationships between the DNA–pegRNA pair and the editing efficiency (Extended Data Fig. 1c). Collectively, OPED predicts a prime-editing score for each DNA–pegRNA pair.

a,b, Schematic representation of the OPED workflow designed for pegRNA efficiency prediction. OPED consists of three primary modules: an input module, a feature learning module and a regression module. a, Initially, OPED undergoes pre-training using a large specific dataset that focuses on pegRNAs inducing transversion point mutations from G to C at position +5 in HEK293T cells. b, Following pre-training, OPED is fine-tuned using a smaller general dataset that is randomly sampled from three training datasets encompassing pegRNAs associated with diverse edit types and positions in HEK293T cells. The details of the OPED modules can be found in Extended Data Fig. 1.
Furthermore, we adopted a pre-training and fine-tuning strategy of transfer learning to improve the accuracy and generalizability of OPED across diverse edits. Initially, we trained OPED from scratch using the published HT-training dataset4 in HEK293T (human embryonic kidney 293T) cells, which is a large specific PE2 dataset comprising 38,692 DNA–pegRNA pairs inducing G-to-C transversion point mutations at position +5 (Fig. 1a and Methods). The pre-trained intermediate model effectively captured common features in prime editing, allowing the feature learning module to be shared as a rough feature extractor for the DNA–pegRNA pair. Subsequently, we retrained the intermediate model using resampling technologies26,27 and transfer learning28,29 (Fig. 1b). Specifically, we randomly sampled an equivalent number of pegRNAs with replacement from the HT-training dataset and two additional smaller datasets (Type-training and Position-training4), which encompass pegRNAs associated with diverse edit types and positions. These three resampled sets were merged into a general dataset, upon which the intermediate model was fine-tuned. By leveraging these strategies, OPED aims to predict pegRNA editing efficiency with improved accuracy and generalizability, thus expanding its applicability.
Performance evaluation of OPED
We assessed the performance of OPED using three reserved datasets (HT-test, Type-test and Position-test4) that were never used for training. On the HT-test dataset, OPED achieved a Pearson correlation coefficient (r) of 0.769, a Spearman correlation coefficient (R) of 0.798, a mean absolute error (MAE) of 4.28% and a root mean squared error (RMSE) of 6.01% (Fig. 2a). Similarly, OPED attained r = 0.612, R = 0.624, MAE = 5.32% and RMSE = 6.91% and r = 0.628, R = 0.562, MAE = 4.14% and RMSE = 5.17% on the Type-test and Position-test datasets, respectively (Fig. 2b,c), demonstrating its accuracy and generalizability across various edit types and positions. Importantly, we observed that pegRNAs with higher predicted editing scores consistently exhibited substantially higher measured editing efficiency compared with those with lower editing scores (8.0-fold, 3.1-fold and 2.2-fold increase for the HT-test, Type-test and Position-test datasets, respectively; Fig. 2d), underlining the benefit of OPED for pegRNA designs. We compared OPED with three machine leaning-based models using the three test datasets. Remarkably, OPED consistently outperformed these models across all datasets (Fig. 2e). In addition, we developed and evaluated nine different machine learning approaches using the same datasets, further reaffirming the superior performance of OPED (Supplementary Table 1). When tested in 6 replicates of PE2 efficiencies at 33 endogenous sites4, OPED achieved r = 0.561–0.668, R = 0.663–0.756, MAE = 7.21–8.07% and RMSE = 9.69–11.05% (Fig. 2f and Extended Data Fig. 2a–e) and substantially increased the editing efficiency in all replicates (7.8–50.8-fold; Extended Data Fig. 2f). When assessed in two additional cell lines4, HCT116 (human colorectal carcinoma) and MDA-MB-231 (human breast adenocarcinoma), OPED consistently achieved good performance across biological and technical replicates in HCT116 cells (r = 0.569–0.611, R = 0.677–0.728, MAE = 8.48–8.99% and RMSE = 11.89–12.47%; Fig. 2g and Extended Data Fig. 2g–i) and MDA-MB-231 cells (r = 0.636–0.663, R = 0.730–0.766, MAE = 6.36–6.64% and RMSE = 9.05–9.56%; Fig. 2h and Extended Data Fig. 2j–l). Compared with pegRNAs with lower editing scores, pegRNAs with higher editing scores exhibited a substantial increase of editing efficiency in both cell lines (39.2–82.9-fold in HCT116 and 37.4–60.7-fold in MDA-MB-231; Extended Data Fig. 2m,n).

a–c, Performance evaluations using independent HT-test (a, n = 4,457), Type-test (b, n = 403) and Position-test (c, n = 200) datasets in HEK293T cells from ref. 4. d, Comparison of pegRNAs with OPED editing score higher or lower than 10 on the HT-test (n = 1,935 (≥10) and n = 2,522 (<10)), Type-test (n = 294 (≥10) and n = 109 (<10)) and Position-test (n = 123 (≥10) and n = 77 (<10)) datasets. The bars indicate the minimum, the median and the maximum. ***P < 0.001, two-sided Wilcoxon rank-sum test (from left to right, <1.0 × 10−308, 4.4 × 10−21, 6.0 × 10−10). e, Performance comparison of OPED with DeepPE, Easy-Prime and PRIDICT on these datasets. f, Validation of OPED using 6 replicates of PE2 efficiencies at 33 endogenous sites. Only the Endo-BR3 (biological replicate 3 of these endogenous sites) dataset (n = 20) is shown here. g,h, Validation of OPED using datasets in HCT116 (g, abbreviated as HCT) and MDA-MB-231 (h, abbreviated as MDA) cells. Only BR2 (biological replicate 2) and TR1 (technical replicate 1) of each cell line (HCT, n = 75; MDA, n = 74) are shown here. i, Validation of OPED using a PE2 dataset (n = 199) encompassing endogenous editing sites in HEK293T cells provided by ref. 1. j,k, Line plots showing the trend between the OPED editing score (orange) and the measured editing efficiency (black) for two pegRNA sets targeting HEK4 (j) and RNF2 (k) sites, respectively. The details of the two pegRNA sets can be found in Supplementary Table 2. l,m, Line plots showing correlations between the nucleotides in the 3′ extension (PBS (l) and RTT (m)) and the measured editing efficiency. The red pentagon represents P for testing non-correlation <0.001, while the blue triangle represents P > 0.4.
Furthermore, we tested OPED on another dataset comprising 199 endogenous sites obtained from different laboratories1. Despite differences in experimental conditions, OPED yielded r = 0.469, R = 0.510, MAE = 6.30% and RMSE = 7.50% (Fig. 2i), and led to a 2.9-fold increase in the editing efficiency (Extended Data Fig. 3a). We visualized eight pegRNA sets targeting genomic loci HEK4, RNF2, HEK3 and EMX1 from the 199 endogenous sites (Supplementary Table 2). Specifically, for two pegRNA sets targeting HEK4 and RNF2, respectively, we observed high correlation between predicted editing scores and measured editing efficiencies (Fig. 2j,k and Extended Data Fig. 3b,c). Notably, the pegRNA with the highest editing score ranked first in terms of efficiency, while the second-highest pegRNA occupied the second rank overall. Similar trends were observed for the remaining six pegRNA sets targeting the four genomic sites (Extended Data Fig. 3d–o).
In summary, these results convincingly demonstrated the accuracy and generalizability of OPED in predicting pegRNA efficiencies across diverse scenarios, encompassing different edit types, edit positions, endogenous sites, cell lines and laboratory settings.
Intrinsic interpretability of OPED
To elucidate the nucleotide contribution of pegRNA in predicting editing efficiencies, we extracted the attention weights assigned to each nucleotide in the PBS and RTT from OPED’s attention network, and then correlated these attention weights with the observed editing efficiencies based on data from library 1 of ref. 4. Notably, the first seven nucleotides in the PBS, relative to the nick site, exhibited significant negative correlations. Conversely, the eighth to thirteenth nucleotides showed significant positive correlations, while the fourteenth to fifteenth nucleotides displayed no significant correlation (Fig. 2l). These findings support the recommendation of a 13 nt PBS, aligning with previous reports1,4. Similarly, in the RTT, the first 12 nucleotides from the nick site showed significant positive correlations, whereas nucleotides 13 to 20 showed significant negative correlations (Fig. 2m). This observation suggests that a 12 nt RTT would be a suitable choice, consistent with previous research1,4. The intrinsic interpretability of OPED has shed light on the nucleotide-level factors influencing pegRNA efficiency.
Validation on PRIDICT datasets
Recent work generated various PE datasets in the PRIDICT study20. To assess the generalizability of OPED on these datasets, we retrained OPED using the training subset from their library 1 and validated OPED on the test subset (Methods). The retrained OPED obtained r = 0.912, R = 0.905, MAE = 7.78% and RMSE = 11.50% for predicting editing efficiencies (Fig. 3a), r = 0.810, R = 0.826, MAE = 4.82% and RMSE = 7.76% for predicting unintended editing rates (Fig. 3b) and r = 0.925, R = 0.913, MAE = 7.87% and RMSE = 11.53% for predicting unedited rates (Extended Data Fig. 4a). Furthermore, pegRNAs with higher predicted editing scores resulted in a 4.3-fold increase in editing efficiencies (Extended Data Fig. 4b), while pegRNAs with lower unintended editing scores exhibited a 7.9-fold decrease in unintended editing rates (Extended Data Fig. 4c). We compared OPED with other models for predicting editing efficiencies (Fig. 3c) and unintended editing rates (Fig. 3d). OPED consistently outperformed these models, confirming its generalizability for accurate predictions of both editing efficiencies and unintended editing rates. We tested OPED on datasets20 encompassing scenarios with or without the tevopreQ1 modification8 and with or without MMR inhibition10 in MMR-deficient HEK293T and MMR-proficient K562 (human chronic myelogenous leukaemia) and U2OS (human osteosarcoma) cells. We correlated the editing efficiencies of different datasets with each other and with OPED predictions (Fig. 3e). Interestingly, OPED showed slightly higher performance on tevopreQ1 pegRNAs compared with unmodified pegRNAs. Moreover, the MMR pathway inhibition considerably improved OPED performance in K562 and U2OS cells. OPED consistently outperformed PRIDICT (Fig. 3f) and substantially increased editing efficiencies across all scenarios and cell lines (2.8–9.4-fold; Extended Data Fig. 4d–g). Furthermore, we validated OPED performance in K562 and U2OS cells with PEmax10,20. OPED performed comparably to PE2 on PEmax (Extended Data Fig. 4h) and substantially increased editing efficiencies across all scenarios in both cell lines (4.0–9.0-fold; Extended Data Fig. 4i–l). To further explore OPED performance in vivo in living animals, we tested OPED in vivo in hepatocytes of the mouse liver (Fig. 3g), and 9.1-fold (tevopreQ1 pegRNAs) and 19.2-fold (unmodified pegRNAs) increases in the editing efficiencies were observed based on OPED scores (Fig. 3h). Overall, these comprehensive validations demonstrated the robust generalizability and superior performance of OPED.

a,b, Validation of OPED for predicting editing efficiencies (a) and unintended editing rates (b) using the library 1 test dataset (n = 18,485) in HEK293T cells from ref. 20 in the PRIDICT study. c, Performance comparison of OPED with DeepPE, Easy-Prime and PRIDICT on the test dataset for predicting editing efficiencies. d, Performance comparison of OPED with PRIDICT on the test dataset for predicting unintended editing rates. e, Correlation between different experimental conditions (dominant-negative MLH1 (MLH1dn) and tevopreQ1) and cell lines (HEK293T, K562 and U2OS). f, Performance comparison of OPED with PRIDICT across different experimental conditions and cell lines. g, Performance of OPED across different experimental conditions in mouse liver. h, Comparison of pegRNAs with OPED editing score higher or lower than 50 in the mouse liver dataset (unmodified pegRNAs, n = 486 (≥50) and n = 383 (<50); tevopreQ1 pegRNAs, n = 483 (≥50) and n = 428 (<50)). The bars indicate the minimum, the median and the maximum. ***P < 0.001, two-sided Wilcoxon rank-sum test (from left to right, 1.6 × 10−27, 3.6 × 10−32).
Experimental validation on ClinVar pathogenic variants
The prioritization of pegRNAs for intended edits can be determined by ranking OPED editing scores. Considering the wide usage of PE3/PE3b and ePE editing systems, we integrated a rule-based hierarchical ranking of sgRNAs from PrimeDesign17 into OPED to optimize the secondary nicking sgRNA (Methods). To evaluate the practical effects of OPED’s optimized PE designs (pegRNAs for PE2, pegRNA and sgRNA combinations for PE3/PE3b and ePE), 30 different human pathogenic genetic mutations from the ClinVar22 database, including ten single nucleotide variants (SNVs), ten insertions and ten deletions, were installed into HEK293T cells with the first-ranked PE designs recommended by OPED previously trained on datasets from ref. 4 (Supplementary Table 3 and Methods). Overall, 28 of the 30 (93.33%) targeted loci showed varying editing efficiencies across different editing systems, variant types and positions, compared with untreated cells (Fig. 4a–c and Extended Data Fig. 5a). Specifically, these optimized PE designs enabled efficient introduction of desired mutations into the genome with editing frequencies of up to 29.30%, 82.84% and 90.05% for PE2, PE3/PE3b and ePE, respectively. On average, PE3/PE3b and ePE increased the editing efficiency 6.9-fold and 11.8-fold, respectively, compared with PE2. Among these 30 targeted loci, we observed the installation of the desired edit at average editing efficiencies of 10% or more for 8 (26.67%) loci and at average efficiencies of 1–10% for 14 (46.67%) loci for PE2 (Extended Data Fig. 5b), at average efficiencies of 20% or more for 10 (33.33%) loci and at average efficiencies of 1–20% for 18 (60.00%) loci for PE3/PE3b (Extended Data Fig. 5c), and at average efficiencies of 30% or more for 12 (40.00%) loci and at average efficiencies of 10–30% for 10 (33.33%) loci for ePE (Extended Data Fig. 5d). We used the GATK tool30 for variant calling and discovered desired variants with high quality (Extended Data Fig. 5e). We quantified mutations and produced intuitive visualizations of experimental outcomes for these editing systems (Fig. 4d–f) and observed that the first-ranked PE design introduced few by-products across the reference amplicons (Extended Data Fig. 6a–c and Extended Data Fig. 7a–i).

a–c, The measured editing efficiencies for the installation of 30 human pathogenic genetic variants, including ten SNVs (a), ten insertions (b) and ten deletions (c), from the ClinVar database in HEK293T cells, using the optimized PE designs recommended by OPED. Mean ± s.d. of n = 3 independent biological replicates are shown. d–f, Editing outcomes with optimized PE designs for the PE2 (d), PE3/PE3b (e) and ePE (f) editing systems in installing pathogenic SNVs (top), insertions (middle) and deletions (bottom) at example loci. Allele frequency tables and corresponding sequencing read counts after merging the three biological replicates are shown. Nucleotides are indicated by unique colours (A, green; C, orange; G, yellow; T, purple). Substitutions are shown in bold font. Red rectangles highlight inserted sequences. Horizontal dashed lines indicate deleted sequences. The vertical dashed line indicates the cleavage site of pegRNA.
For further comparisons and validations, we edited a subset of eight targeted loci using the first-ranked PE designs recommended by OPED, three machine leaning-based models and rule-based PrimeDesign. OPED substantially increased average editing efficiencies compared with DeepPE, Easy-Prime, PRIDICT and PrimeDesign (PE2, 6.8-fold, 4.5-fold, 2.9-fold and 2.1-fold, respectively; PE3/PE3b, 24.6-fold, 22.3-fold, 11.7-fold and 7.9-fold, respectively; ePE, 44.2-fold, 47.5-fold, 16.7-fold and 3.4-fold, respectively; Fig. 5a–c), while attaining higher purity (Extended Data Fig. 8a–c). Furthermore, we edited a subset of six targeted loci using the top-3-ranked PE designs recommended by OPED. These top-3-ranked PE designs show variations in pegRNAs, while most of the sgRNAs remain identical. We observed that the first-ranked designs consistently showed comparable purity (Extended Data Fig. 8d,e) and higher average editing efficiencies compared with the second-ranked and third-ranked designs (PE3/PE3b, 2.8-fold and 8.5-fold, respectively; ePE, 1.6-fold and 1.8-fold, respectively; Fig. 5d,e), demonstrating the power of OPED for optimizing both pegRNAs and sgRNAs. Similarly, we edited the same six targeted loci using combinations of the first-ranked pegRNA and top-3-ranked sgRNAs recommended by OPED. It was observed that first-ranked sgRNA led to comparable purity (Extended Data Fig. 8f,g) and notably increased average efficiency compared with the second-ranked and third-ranked sgRNAs (PE3/PE3b, 3.9-fold and 3.7-fold, respectively; ePE, 2.9-fold and 3.3-fold, respectively; Fig. 5f,g), demonstrating the power of OPED for optimizing sgRNAs.

a–j, The measured editing efficiency. Mean ± s.d. of n = 3 independent biological replicates are shown. a–c, Comparative assessments against four state-of-the-art tools (DeepPE, Easy-Prime, PRIDICT and PrimeDesign) using the optimized PE designs for the PE2 (a), PE3/PE3b (b) and ePE (c) editing systems in installing eight pathogenic variants. d,e, Comparative assessments using the top-3-ranked PE designs (top-3-ranked pegRNA and the corresponding first-ranked sgRNA) recommended by OPED for the PE3/PE3b (d) and ePE (e) editing systems in installing six pathogenic variants. f,g, Comparative assessments using combinations of the first-ranked pegRNA and top-3-ranked sgRNAs recommended by OPED for the PE3/PE3b (f) and ePE (g) editing systems. h, The measured editing efficiencies for the installation of six pathogenic variants using the optimized PE designs recommended by OPED for non-canonical NG PAM. i,j, Comparative assessments using mpknot epegRNAs recommended by OPED and ref. 8 for PE2 (i) and PE3/PE3b (j).
To experimentally validate the performance of OPED on targets with non-canonical NG (N denotes any nucleotide and G represents guanine) protospacer adjacent motif (PAM), we edited the same six targeted loci using the optimized PE design recommended by OPED for NG PAM. Despite the efficiency drop compared with NGG PAM, these optimized PE designs continued to demonstrate notable effectiveness compared with untreated controls (Fig. 5h and Extended Data Fig. 8h). In addition, we employed the OPED-optimized PE designs with mpknot modification to edit additional four targeted loci provided by ref. 8 (Methods). These engineered pegRNAs (epegRNAs) recommended by OPED showed comparable or slightly higher average efficiencies compared with those provided by ref. 8 (PE2, 1.0-fold; PE3/PE3b, 1.2-fold; Fig. 5i,j), while maintaining comparable purity (Extended Data Fig. 8i,j).
OPEDVar database and OPED web application
To illustrate the utility of OPED, we filtered 77,738 human pathogenic genetic variants from ClinVar and designed candidate pegRNAs and sgRNAs to correct or install these pathogenic mutations. For correction with NG PAM, a total of 778,791,772 candidate pegRNAs were identified with an average of 10,018 candidate pegRNAs per pathogenic variant (Fig. 6a), reflecting the complexity of pegRNA selection. Similar outcomes were observed when installing mutations with NG PAM or correcting/installing mutations with NGG PAM (Extended Data Fig. 9a–c and Supplementary Table 4). A comprehensive collection of 2 billion candidate pegRNAs was predicted and ranked by OPED, and the top-10 optimized PE designs of each event were selected to construct the OPEDVar database (http://bicdb.ncpsb.org.cn/OPED/OPEDVar/) (Fig. 6b). Analysis of all first-ranked pegRNAs from OPEDVar revealed significant distinctions between their characteristics and those of all the candidates (Fig. 6c–e). Specifically, when correcting 51,473 SNVs with NG PAM, the median lengths of PBS, RTT and edit-to-nick distance of the corresponding first-ranked pegRNAs were 9 nt, 12 nt and 5 nt, respectively (Fig. 6c), consistent with previous reports1,4. Similar results were obtained across other conditions, including correction/installation with NG/NGG PAM (Fig. 6d,e and Extended Data Fig. 9d–l). To enhance accessibility and user-friendliness of OPED, we developed an intuitive web application (http://bicdb.ncpsb.org.cn/OPED/) to facilitate the design of pegRNAs and sgRNAs for intended edits (Fig. 6f).

a, Distribution of the number of candidate pegRNAs per pathogenic variant with an NG PAM for correction. Horizontal black lines indicate the averages. b, Home page, query page and query result of the OPEDVar database. c–e, Distribution of PBS length, RTT length and edit-to-nick distance of all candidate pegRNAs (blue) and first-ranked pegRNAs predicted by OPED (green) for each variant to correct pathogenic SNVs (c), insertions (d) and deletions (e) with an NG PAM. Horizontal black lines indicate the medians. ***P < 0.001, two-sided Wilcoxon rank-sum test. c, Distribution of n = 527,326,019 candidate pegRNAs and n = 51,473 first-ranked pegRNAs (P < 1.0 × 10−308, P< 1.0 × 10−308, P < 1.0 × 10−308, respectively). d, Distribution of n = 18,269,053 candidate pegRNAs and n = 1,833 first-ranked pegRNAs (P = 1.2 × 10−221, P < 1.0 × 10−308, P < 1.0 × 10−308, respectively). e, Distribution of n = 233,196,700 candidate pegRNAs and n = 24,432 first-ranked pegRNAs (P < 1.0 × 10−308, P < 1.0 × 10−308, P < 1.0 × 10−308, respectively). f, Home page, parameter set-up, workflow and optimized PE designs of OPED web application. OPED takes a single sequence encoding both the original reference and desired edited sequences as input and outputs the recommended optimized PE designs to introduce the edit of interest.
Discussion
PE substantially expands the scope and capabilities of genome editing and can correct most human pathogenic variants. The comprehension of its editing specificity is paramount to potential therapeutic applications31,32. However, the optimization of pegRNAs for the desired edit presents challenges. Inspired by the natural language processing techniques, we developed a customized transformer-and-attention-based OPED model (Fig. 1) for the efficiency prediction and design optimization of pegRNAs. To improve its accuracy and generalizability, we introduced transfer learning to pre-train and fine-tune OPED. By working directly with raw nucleotide sequences, OPED automatically learns a comprehensive and interpretable representation of the DNA–pegRNA pair, thereby eliminating the need for predefined features. The tailored transformer adeptly handles variable-length sequences and effectively characterizes the spatial interdependencies between nucleotides and the complementary base pairing characteristics between the target DNA and pegRNA. The tailored attention network calculates an attention weight for each nucleotide and subsequently consolidates pertinent information based on these weights (Extended Data Fig. 1). The intrinsic interpretability of OPED provided nucleotide-level insights into the factors influencing pegRNA efficiency (Fig. 2l,m). Comprehensive validations on various published datasets demonstrated the accuracy and generalizability of OPED in predicting pegRNA efficiencies across diverse scenarios, encompassing different edit types, edit positions, endogenous sites, laboratory settings, tevopreQ1 conditions, MMR inhibition conditions, cell lines in vitro and mouse hepatocytes in vivo (Figs. 2 and 3). OPED consistently leads to a substantial efficiency increase based on the predicted editing scores and outperforms previous state-of-the-art models in these scenarios. After integrating a rule-based ranking strategy into OPED for the prioritization of sgRNAs, we tested OPED by installing a variety of ClinVar pathogenic variants into the human genome using three different editing systems. Experimental outcomes revealed that first-ranked PE designs suggested by OPED efficiently installed the desired variants, highlighting the versatility and efficacy of OPED across diverse edit types and editing systems (Fig. 4). Comparative analyses against four PE design tools, along with additional optimized designs produced by OPED, further underscored the superior performance and optimization capabilities of OPED for both pegRNAs and sgRNAs (Fig. 5a–g). In addition, we experimentally confirmed the effectiveness of OPED on targets with NG PAM and mpknot epegRNAs (Fig. 5h–j). To facilitate the utilization of OPED, we constructed the OPEDVar database of optimized PE designs for correcting or installing over 77,000 ClinVar pathogenic variants (Fig. 6b) and developed a web version of OPED to streamline the process of designing pegRNAs and sgRNAs for desired edits (Fig. 6f). It is important to note that OPED is trained solely on PE2 datasets with canonical NGG PAM. Therefore, although OPED has undergone experimental validations demonstrating its capacity to optimize both pegRNAs and sgRNAs across diverse scenarios including NG PAM, its ability to predict efficiencies for other PE versions is limited and its performance for NG PAM needs more experimental validation. In future studies, we propose the comprehensive and systematic optimization of pegRNA and additional sequences such as scaffold sequence, nicking sgRNA, 3′ structured RNA motif and linker sequence8 using artificial intelligence techniques, provided sufficient data become available. Moreover, the effector of PE containing a Cas9 nickase fused to an engineered reverse transcriptase can be improved by new artificial intelligence-based techniques33,34,35.
Methods
Design of the OPED network
For the pegRNA structure, we designed OPED, a nucleotide language model to predict the pegRNA editing efficiency and design the optimized pegRNA. Only the raw nucleotide sequences of the target DNA (including 20 nt protospacer), variable-length PBS and RTT were used as the input of OPED, and the reverse complements of PBS and RTT were extracted in the input module. Subsequently, K-mer (K = 1,2,3) sequences of these sequences could be easily extracted. Thus, for an M nt sequence, a K-mer sequence of length M − K + 1 was obtained. Then, we used learned word embeddings to convert each K-mer to a vector of dimension dmodel. Furthermore, to incorporate the positional order of the K-mer sequence, we added positional encoding24 to the embeddings with sine and cosine functions of different frequencies:
where pos is the position and i is the dimension. To learn general high-level features, one transformer encoder was employed to extract features of the target sequence, and two transformer decoders were specifically tailored to extract features of PBS and RTT by aligning them to the target sequence, respectively. Multihead attention in the transformer was calculated as follows:
where \({{{\mathrm{head}}}}_{i}={{\mathrm{Attention}}}\left(Q{W}_{i}^{\,Q},K{W}_{i}^{\,K},V{W}_{i}^{\,V}\right)={{\mathrm{SoftMax}}}\left(\frac{Q{W}_{i}^{\,Q}{(K{W}_{i}^{\,K})}^{T}}{\sqrt{{d}_{k}}}\right)V{W}_{i}^{\,V}\), parameter matrices \({W}_{i}^{\,Q}\in {R}^{{d}_{{{\mathrm{model}}}}\times {d}_{k}}\), \({W}_{i}^{\,K}\in {R}^{{d}_{{{\mathrm{model}}}}\times {d}_{k}}\), \({W}_{i}^{V}\in {R}^{{d}_{{{\mathrm{model}}}}\times {d}_{v}}\), \({W}^{\,O}\in {R}^{{{{hd}}_{v}\times d}_{{{\mathrm{model}}}}}\) and \({d}_{k}={d}_{v}={d}_{{{\mathrm{model}}}}/h\). Q, K and V matrices symbolize the repositories of queries, keys and values associated with the input, respectively. The matrix W represents the pertinent parameter matrix responsible for these projections. The superscript O indicates the output, and T denotes matrix transposition. The symbol R denotes the set of real numbers. The variables dk and dv refer to the dimensions of keys and values, respectively, while h signifies the number of parallel attention heads. Layer normalization36 in the transformer was applied as:
where γ and β are learnable parameters and ε is a small constant. The variable x represents the input of the layer normalization, while E[x] denotes the expected value or mean of the variable x. Position-wise feed-forward network (FFN) in the transformer is applied as the following:
where W1, W2, b1 and b2 are learnable parameters. After obtaining the hidden vector \({h}_{i}(i=1,2,\ldots ,{M}-K+1)\) of each K-mer in the sequence through the customized transformer, we customized an additional attention network containing a feed-forward layer to map the hidden vector of each K-mer into a scalar score, and then applied a SoftMax function to calculate the attention weight of each K-mer in the sequence based on the score. The attention weight was calculated as follows:
where MLP is a feed-forward multilayer perceptron. The attention weight ai explains the importance of ith K-mer in the sequence. By calculating the weighted summation of the hidden vector of each K-mer and the corresponding attention weight, a sequence description vector was obtained:
The sequence description vector is the feature extraction and generalization of the whole sub-sequence. All sequence description vectors corresponding to the target sequence, PBS and RTT were concatenated into a comprehensive description vector representing the target DNA and pegRNA pair. Finally, we used a multilayer fully connected feed-forward network to perform a nonlinear transformation on the comprehensive description vector to output the predicted editing score of the pegRNA. As experimental conditions strongly influence the absolute editing levels, the OPED editing score was set to a score between 0 and 100 rather than the percentual editing rate. The rectified linear unit function serves as the chosen nonlinear activation function and dropout37 was used to prevent overfitting.
Public datasets for training and validation
The OPED model was trained and evaluated using HEK293T cell datasets previously published by ref. 4. These datasets encompass two PE2 libraries denoted as library 1 and library 2. Library 1 encompasses 43,149 different pairs of target sequences and pegRNAs that induced a transversion mutation from G to C at position +5 from the nicking site. Ref. 4 randomly divided library 1 into two subsets: HT-training (38,692 pegRNAs) and HT-test (4,457 pegRNAs). Library 2 encompasses 5,752 different pairs of pegRNA-encoding sequences and corresponding target sequences for various edit types and positions and was randomly split into Type-training (3,375 pegRNAs), Type-test (403 pegRNAs), Position-training (1,774 pegRNAs) and Position-test (200 pegRNAs). Both library 1 and library 2 datasets comprise 47-nt-wide target sequences, 17–37 nt 3′ extension (RTT and PBS) sequences and 20 predefined pegRNA features computed from the nucleotide sequences, including melting temperature, GC counts and minimum self-folding free energy. While ref. 4 used both the nucleotide sequences and predefined features to predict PE2 efficiency, OPED solely utilized the nucleotide sequences (47 nt target sequences, RTT sequences and PBS sequences) as inputs to predict PE2 efficiency. To train the OPED model, we employed the HT-training, Type-training and Position-training datasets as training sets. To evaluate the performance of OPED, we employed independent test sets, namely, HT-test, Type-test and Position-test datasets. Furthermore, additional datasets from ref. 4 containing 6 replicates at 33 endogenous sites and 4 replicates in two additional cell types (HCT116 and MDA-MB-231) were also utilized to validate OPED.
For further validation, we used PE2 datasets encompassing 199 endogenous editing sites in HEK293T cells provided by ref. 1. These datasets consist of pegRNAs with a 20 nt spacer sequence, variable-length RTT sequence and PBS sequence. We employed BLAT38 to align the 20 nt spacer sequence to assembly hg38 and extracted the 350 nt target sequence (165 nt neighbouring sequence + 20 nt protospacer + 165 nt neighbouring sequence). Subsequently, the 350-nt target sequence, RTT sequence and PBS sequence were used as inputs of OPED to predict editing scores. The editing values of ref. 1 were analysed and formatted by ref. 19 and downloaded from their GitHub repository.
A recent work generated a comprehensive lentiviral library (library 1) in HEK293T cells in the PRIDICT study20. Library 1 encompasses a total of 92,423 pegRNA–target site pairs, comprising 57,920 single base replacements, 28,420 insertions and 6,083 deletions. Similar to the approach undertaken in ref. 20, library 1 was randomly split into 80% training (73,938 pegRNAs) and 20% test (18,485 pegRNAs) subsets. The library 2 consists of 1,938 pegRNAs with or without the tevopreQ1 modification and with or without MMR inhibition in HEK293T, K562 and U2OS cells, as well as in vivo in hepatocytes of the mouse liver. In addition, ref. 20 provided a library 2 screen in K562 and U2OS cells with PEmax. All datasets from ref. 20 contain the 99 nt target sequence, RTT sequence, PBS sequence, and dozens of predefined features such as melting temperature and minimum free energy. Only the nucleotide sequences (99 nt target sequences, RTT sequences and PBS sequences) of all these datasets were used by OPED to predict a probability distribution on three outcomes associated with edited, unedited and unintended edit proportions.
Training of the OPED model
Due to the absence of a sufficiently large prime-editing dataset encompassing diverse edit types and positions during the development of OPED, we employed a transfer learning approach to pre-train and subsequently fine-tune the OPED model, enhancing its accuracy, generalizability and interpretability in predicting pegRNA efficiency.
Given the abundance of data in the library 1 dataset from ref. 4, OPED was initially trained on the HT-training set of library 1. The selection of optimal hyperparameters was achieved by employing fivefold cross-validation, leading to the determination of the following hyperparameter values. The number of epochs, batch size and embedding size were set to 200, 512 and 64, respectively. In the transformer encoder and decoder, the dimension of the feed-forward network was set to 2,048, while a single sub-encoder layer was employed in the encoder and a single sub-decoder layer in the decoder. The multihead-attention models comprised 64 heads, and a dropout value of 0.1 was applied. For the regression module, a multilayer fully connected feed-forward network was employed, comprising three hidden layers with 512, 64 and 512 hidden units, respectively. The mean squared error (squared L2 norm) was employed as the loss function, and the Adam optimization algorithm39 was utilized with a learning rate of 0.001 and weight decay (L2 penalty) of 0.1. Using the aforementioned optimal hyperparameters, we trained the OPED model on the HT-training set with random initialization from scratch. Evaluation on the independent HT-test set yielded a Pearson correlation coefficient (r) of 0.793, Spearman correlation coefficient (R) of 0.820, MAE of 3.95% and RMSE of 5.59%. The MAE and RMSE were defined as follows:
where yi is the measured editing efficiency of the ith pegRNA and \({\bar{y}}_{i}\) is calculated by dividing the predicted editing score by 100.
However, the performance of the aforementioned intermediate model was considerably lacking when applied to the Type-test and Position-test sets from library 2, which consists of target sequences and pegRNAs encompassing various edit types and edit positions. To improve its generalization capability, we further fine-tuned the intermediate model using resampling and transfer learning technologies. Specifically, we randomly sampled 5,000 pegRNAs with replacement from HT-training, Type-training and Position-training sets, respectively, and then merged them into a comprehensive training set containing diverse edit types and positions. Instead of random initialization, we initialized the OPED model with the pre-trained parameters of the intermediate model. Moreover, we altered the downstream fully connected feed-forward regression network to 6 hidden layers, each containing 1,024, 2,048, 2,048, 1,024, 1,024 and 256 hidden units, respectively. Subsequently, we fine-tuned the OPED model on the merged general training set. The fine-tuned OPED model was then preserved for all subsequent validations and applications, excluding its utilization in the evaluation of PRIDICT datasets.
Furthermore, we retrained the OPED model using the recently published training dataset (73,938 pegRNAs) by ref. 20. In this retraining process, we used the Kullback–Leibler divergence as the loss function and modified the output layer of the regression module to accommodate three simultaneous outcomes: edited, unedited and unintended edit proportions. To determine optimal hyperparameters, we performed fivefold cross-validation and adjusted the fully connected feed-forward regression network to include 3 hidden layers comprising 1,024, 512 and 2,048 hidden units, respectively. The retrained OPED model was exclusively utilized to assess its performance across diverse PRIDICT datasets.
Comparison of OPED with alternative methods
To facilitate comprehensive comparisons, we developed other nine different machine learning approaches using the same features as DeepPE. These approaches include a customized deep learning model integrating bidirectional gated recurrent unit40 in conjunction with an attention mechanism24, support vector regression41, MLP42, K-nearest neighbours43, decision tree44, random forest45, AdaBoost46, gradient boosted decision trees47 and XGBoost48. We trained each of these nine methods on HT-training, Type-training and Position-training datasets, respectively, and then evaluated them on the corresponding HT-test, Type-test and Position-test dataset.
Extension of OPED to optimize the secondary nicking sgRNA
The lack of sufficiently large PE3/PE3b datasets has limited the training of state-of-the-art machine leaning-based models, including DeepPE and PRIDICT, to PE2 datasets. As a result, these models have been restricted to the pegRNA efficiency prediction and design optimization, without supporting PE3/PE3b and ePE systems. To address this limitation and enable OPED to optimize both pegRNA and sgRNA, we have integrated the rule-based hierarchical ranking of sgRNAs from PrimeDesign17 into the OPED framework, targeting the optimization of the secondary nicking sgRNA. Specifically, when a candidate pegRNA is designed by OPED for the intended edit, all candidate nicking sgRNAs are searched on the non-edited strand within a user-defined range determined by two hyperparameters: the minimum sgRNA-nick-to-pegRNA-nick distance (default 0 bp) and the maximum sgRNA-nick-to-pegRNA-nick distance (default 100 bp). Subsequently, the hierarchical ranking of sgRNAs from PrimeDesign is performed by first using the sgRNA annotations (PE3b seed -> PE3b non-seed -> PE3), and then using deviations from the sgRNA-nick-to-pegRNA-nick distance hyperparameter (default 75 bp). The prioritization of pegRNAs for the intended edit is determined by ranking the editing scores predicted by OPED. Similarly, the prioritization of sgRNAs for the corresponding pegRNA is determined by the integrated hierarchical ranking in OPED. Ultimately, OPED recommends the combination of the first-ranked pegRNA and its corresponding first-ranked sgRNA as the first-ranked PE design. This extension empowers OPED with the capability to design optimized sgRNAs in addition to optimized pegRNAs, expanding its utility in the field of genome-editing research.
Optimized PE designs by other tools
Comparative experiments were conducted by introducing eight ClinVar pathogenic variants using the recommended optimized PE designs provided by OPED, three machine learning-based models (DeepPE, Easy-Prime and PRIDICT) and the rule-based PrimeDesign in different editing systems (PE2, PE3/PE3b and ePE). In the case of DeepPE, it was employed to predict the efficiencies of candidate pegRNAs designed by OPED for the desired variant. Subsequently, all candidate pegRNAs were ranked based on DeepPE’s predicted efficiencies, and the first-ranked pegRNA was selected. However, considering that DeepPE solely focuses on pegRNA prediction, we employed the integrated hierarchical ranking in OPED to optimize the corresponding sgRNA for the DeepPE-selected pegRNA. For Easy-Prime, it supports the optimization of both pegRNAs and sgRNAs. Upon inputting the desired variant, the first-ranked pegRNAs and sgRNAs predicted by Easy-Prime were directly used. Regarding PRIDICT, although it is limited to pegRNA optimization, its program and website list optional sgRNAs for the PE3 system. The authors clarify that nicking sgRNAs are not part of the PRIDICT score or their paper but are listed for increased user experience. Therefore, we directly used the first-ranked pegRNAs and sgRNAs predicted by PRIDICT upon inputting the desired variant. As for PrimeDesign, the first-ranked pegRNAs and sgRNAs predicted by PrimeDesign were directly used when inputting the desired variant. After obtaining measured editing efficiencies of optimized PE designs recommended by various PE design tool for installing these ClinVar variants, we proceeded to calculate the editing-efficiency fold-change achieved by OPED compared with each benchmark PE tool (DeepPE, Easy-Prime, PRIDICT and PrimeDesign) for each individual variant. Subsequently, we conducted a mean aggregation of these fold-change values across these ClinVar variants, thus furnishing an averaged representation of the editing-efficiency fold-change.
Experimental validation of epegRNAs designed by OPED
A previous study developed epegRNAs to improve prime-editing efficiency8. The epegRNAs were constructed by incorporating one of two structured RNA motifs (tevopreQ1 or mpknot) to the 3′ terminus of pegRNAs to enhance pegRNA stabilization. The performance of OPED on epegRNAs with tevopreQ1 was validated in HEK293T, K562 and U2OS cells using the published datasets from ref. 20. To assess the performance of OPED on epegRNAs with mpknot, we edited four targeted loci (EMX1, FANCF, VEGFA and PRNP) provided by ref. 8 using the optimized PE designs recommended by OPED and ref. 8.
Plasmid construction
The pCMV-PE2 plasmid was purchased from Addgene (132775). The pGL3-U6-sgRNA-EGFP (Enhanced Green Fluorescent Protein) plasmid was purchased from Addgene (107721). The pCMV-PE2-P2A-hMLH1dn plasmid was purchased from Addgene (174827). The pCMV-NG-PE plasmid was constructed based on SpCas9-NG, which was purchased from Addgene (138566). The pegRNA plasmid was constructed according to a previous report15. To construct pegRNA expression plasmids, the plasmid backbone was amplified from pGL3-U6-sgRNA-EGFP using KOD OneTM PCR Master Mix -Blue- (TOYOBO) (Supplementary Table 5). The amplicon was then cut by BsaI-HFv2 (NEB) for overhangs. Scaffold sequences were synthesized as long oligos directly. For spacer oligos, the top strand oligo includes 5′ ACCG and 3′ GTTTT overhangs, while the bottom strand oligo comprises a 5′ CTCTGAAAA overhang (Supplementary Table 6). Canonical pegRNA 3′ extension, including PBS and RTT sequences, was synthesized on an oligo (the top strand oligo included 5′ GTGC overhang while the bottom strand oligo included 5′ AAAC overhang). The pegRNAs of ePE were produced according to our previous study3. The epegRNAs were produced according to a previous report8.
Cell culture, transfection and collection
HEK293T (ATCC CRL-3216) cells were cultured with Dulbecco’s modified Eagle medium (Hyclone) supplemented with 10% fetal calf serum (Bi) and incubated at 37 °C with 5% CO2. Cells were routinely passaged at a ratio of 1:3 at 90% confluency by digesting with 0.25% pancreatin (add EDTA). For plasmid transfection, cells were seeded in 24-well plates and transfected with 1.3 μg (900 ng pCMV-PE2 plasmid, 300 ng pegRNA plasmid and 100 ng nicking sgRNA plasmid) plasmids per well at approximate 70% confluency using EZ Trans (Shanghai Life iLab Biotech) according to the manufacturer’s protocols. After 72 h of transfection, cells were collected for EGFP+ cells by fluorescence-activated cell sorting.
Genomic DNA extraction and targeted deep sequencing
The genomic DNA of EGFP+ cells was extracted using QuickExtract DNA Extraction Solution (Lucigen) according to the manufacturer’s protocols. Then, the sequences around the target sites were amplified from the isolated DNA with Phanta Max Super-Fidelity DNA Polymerase (Vazyme) and primers (Supplementary Table 7). The amplicons with different barcodes were subjected to deep sequencing on an Illumina HiSeq X Ten platform (2 × 150 PE) by Annoroad Gene Technology. To evaluate the prime-editing efficiency, CRIPResso2 (V2.0.43)49 was used to analyse the sequenced amplicons. All values are presented as mean ± s.d.
OPEDVar database
The ClinVar22 variant summary was accessed on 29 September 2021. We chose Homo sapiens genome assembly GRCh38/hg38 and filtered all variants by allele ID to remove duplicates and by clinical significance to focus on pathogenic variants. The total number of filtered ClinVar variants was 77,738, and then we separated the variants into SNV, insertion and deletion by their variant type. For both the installation and correction of these pathogenic variants, all candidate pegRNAs and sgRNAs of each variant were enumerated with the following criteria: (1) maximum distance of 50 nt from the editing site to the pegRNA nicking site, (2) NGG PAM or NG PAM, (3) minimum homology of 5 nt downstream of the edit, (4) minimum PBS length of 8 nt and maximum PBS length of 18 nt, (5) minimum RTT length of 8 nt and maximum RTT length of 68 nt, (6) minimum sgRNA-nick-to-pegRNA-nick distance of 0 nt and maximum sgRNA-nick-to-pegRNA-nick distance of 100 nt. Then, OPED previously trained on datasets from ref. 4 was used to predict the editing scores of all candidate pegRNAs and rank all candidate pegRNAs for each variant. The top-10 optimized pegRNAs and sgRNAs (top-10-ranked pegRNAs and the corresponding first-ranked sgRNAs) for the installation or correction of each pathogenic variant were stored in a database named OPEDVar. OPEDVar is a comprehensive and searchable database for more than 77,000 pathogenic human genetic variants, making the optimized PE designs recommended by OPED for all ClinVar pathogenic variants more accessible. In OPEDVar, using the ClinVar allele ID, gene ID, gene symbol or HUGO Gene Nomenclature Committee ID, the top-10 optimized PE designs for the corresponding pathogenic mutations were readily available across a range of PBS (8–18 nt) and RTT (8–68 nt) lengths and distance from the edit site to the pegRNA nick site (1–50 nt) on both DNA strands.
OPED web application
To make OPED available and user-friendly, we developed a web application of OPED for any intended edit and provided the results of OPED for a given target sequence or a given position of a target site. After entering a target sequence or the position of a target site along with the intended edit, this web tool identified all candidate pegRNAs and sgRNAs for the intended edit on both DNA strands with the user-set hyperparameters including the maximum edit-to-nick distance, PAM type, maximum PBS length, minimum PBS length, maximum RTT length, minimum RTT length, number of optimized pegRNAs, minimum sgRNA-nick-to-pegRNA-nick distance and maximum sgRNA-nick-to-pegRNA-nick distance, and then provided a corresponding number of optimized PE designs by the predicted pegRNA editing score ranking of OPED that was previously trained on datasets from ref. 4.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The public datasets we used were from refs. 1,4,20, and the usages are fully illustrated in Methods. Editing values of ref. 1 were analysed and formatted by ref. 19 and downloaded from their GitHub repository (https://github.com/YichaoOU/easy_prime). The deep-sequencing data from this study have been submitted to the National Center for Biotechnology Information Sequence Read Archive database under accession number PRJNA882795.
Code availability
The web portal of OPED is accessible at http://bicdb.ncpsb.org.cn/OPED/. The source codes of OPED are freely available on GitHub (https://github.com/wenjiegroup/OPED), Zenodo (https://doi.org/10.5281/zenodo.8133309) and Code Ocean (https://doi.org/10.24433/CO.9224036.v1)50. Deep-sequencing data were analysed with CRISPResso2 with prime-editing mode (https://github.com/pinellolab/CRISPResso2) and GATK4 (https://github.com/broadinstitute/gatk/).
References
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Chen, P. J. & Liu, D. R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. https://doi.org/10.1038/s41576-022-00541-1 (2022).
Liu, Y. et al. Enhancing prime editing by Csy4-mediated processing of pegRNA. Cell Res. 31, 1134–1136 (2021).
Kim, H. K. et al. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 39, 198–206 (2021).
Li, X. et al. Highly efficient prime editing by introducing same-sense mutations in pegRNA or stabilizing its structure. Nat. Commun. 13, 1669 (2022).
Zhang, G. et al. Enhancement of prime editing via xrRNA motif-joined pegRNA. Nat. Commun. 13, 1856 (2022).
Liu, B. et al. A split prime editor with untethered reverse transcriptase and circular RNA template. Nat. Biotechnol. https://doi.org/10.1038/s41587-022-01255-9 (2022).
Nelson, J. W. et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 40, 402–410 (2022).
Ferreira da Silva, J. et al. Prime editing efficiency and fidelity are enhanced in the absence of mismatch repair. Nat. Commun. 13, 760 (2022).
Chen, P. J., Hussmann, J. A., Yan, J., Knipping, F. & Liu, D. R. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184, 5635–5652.e29 (2021).
Peterka, M. et al. Harnessing DSB repair to promote efficient homology-dependent and -independent prime editing. Nat. Commun. 13, 1240 (2022).
Anzalone, A. V. et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 40, 731–740 (2022).
Choi, J. et al. Precise genomic deletions using paired prime editing. Nat. Biotechnol. 40, 218–226 (2022).
Lin, Q. et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 38, 582–585 (2020).
Liu, Y. et al. Efficient generation of mouse models with the prime editing system. Cell Discov. 6, 27 (2020).
Chow, R. D., Chen, J. S., Shen, J. & Chen, S. A web tool for the design of prime-editing guide RNAs. Nat. Biomed. Eng. 5, 190–194 (2021).
Hsu, J. Y. et al. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun. 12, 1034 (2021).
Hwang, G.-H. et al. PE-Designer and PE-Analyzer: web-based design and analysis tools for CRISPR prime editing. Nucleic Acids Res. 49, W499–W504 (2021).
Li, Y., Chen, J., Tsai, S. Q. & Cheng, Y. Easy-Prime: a machine learning-based prime editor design tool. Genome Biol. 22, 235 (2021).
Mathis, N. et al. Predicting prime editing efficiency and product purity by deep learning. Nat. Biotechnol. https://doi.org/10.1038/s41587-022-01613-7 (2023).
Lundberg, S. M. et al. From local explanations to global understanding with explainable AI for trees. Nat. Mach. Intell. 2, 56–67 (2020).
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
Vaswani, A. et al. Attention is all you need. in Advances in Neural Information Processing Systems 30 (eds Guyon, I. et al.) (NIPS, 2017).
Brown, T. et al. Language models are few-shot learners. in Advances in Neural Information Processing Systems 33 (eds Larochelle, H., Ranzato, M., Hadsell, R., Balcan, M. F. & Lin, H.) 1877–1901 (NeurIPS, 2020).
He, H. Imbalanced Learning (Wiley-IEEE, 2013).
Davison, A. C. & Hinkley, D. V. Bootstrap Methods and Their Application (Cambridge Univ. Press, 1997).
Pan, S. J., Tsang, I. W., Kwok, J. T. & Yang, Q. Domain adaptation via transfer component analysis. IEEE Trans. Neural Netw. 22, 199–210 (2011).
Yosinski, J., Clune, J., Bengio, Y. & Lipson, H. How transferable are features in deep neural networks? Adv. Neural Inf. Process. Syst. 27, 3320–3328 (2014).
Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Preprint at bioRxiv https://doi.org/10.1101/201178 (2018).
Jin, S. et al. Genome-wide specificity of prime editors in plants. Nat. Biotechnol. 39, 1292–1299 (2021).
Gao, R. et al. Genomic and transcriptomic analyses of prime editing guide RNA-independent off-target effects by prime editors. CRISPR J. 5, 276–293 (2022).
Kim, N. et al. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 38, 1328–1336 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Ba, J., Kiros, J. R. & Hinton, G. E. Layer normalization. Preprint at https://arxiv.org/abs/1607.06450 (2016).
Hinton, G. E., Srivastava, N., Krizhevsky, A., Sutskever, I. & Salakhutdinov, R. R. Improving neural networks by preventing co-adaptation of feature detectors. Comput. Sci. 3, 212–223 (2012).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res 12, 656–664 (2002).
Kingma, D. & Ba, J. Adam: a method for stochastic optimization. Preprint at https://arxiv.org/abs/1412.6980 (2014).
Cho, K. et al. Learning phrase representations using RNN encoder–decoder for statistical machine translation. Preprint at https://arxiv.org/abs/1406.1078 (2014).
Chang, C. C. & Lin, C. J. LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2, 3 (2007).
Bengio, Y. & Glorot, X. Understanding the difficulty of training deep feed forward neural networks. in Proceedings of the Thirteenth International Conference on Artificial Intelligence and Statistics 249–256 (2010).
Hall, P., Park, B. U. & Samworth, R. J. Choice of neighbor order in nearest-neighbor classification. Ann. Stat. 36, 2135–2152 (2008).
Speybroeck, N. Classification and regression trees. Int. J. Public Health 57, 243–246 (2012).
Breiman, L. Random forests. Machine Learning 45, 5–32 (2001).
Drucker, H. Improving Regressors Using Boosting Techniques (Morgan Kaufmann, 1997).
Friedman, J. H. Greedy function approximation: a gradient boosting machine. Ann. Stat. 29, 1189–1232 (2001).
Chen, T. & Guestrin, C. XGBoost: a scalable tree boosting system. in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 785–794 (2016).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Liu, F. et al. Design of prime editing guide RNAs with deep transfer learning. Code Ocean https://doi.org/10.24433/CO.9224036.v1 (2023).
Acknowledgements
This work was supported in part by the National Key Research and Development Project of China (2021YFC2302400 to W.S.), the National Natural Science Foundation of China (31901064, 81830101 and 32272848 to F.L., S.W. and X.W., respectively), the General Project of Chongqing Natural Science Foundation of China (CSTB2022NSCQ-MSX1059 to F.L.), the Intelligent Medicine Research Project of Chongqing Medical University (ZHYXQNRC202103 to F.L.) and the Shaanxi Double-chain Fusion Key Project (2022GD-TSLD-46 to X.W.). The computational platform of OPED was supported by Y. Zhu in the Bioinformatics Platform of China National Center for Protein Sciences (Beijing), in conjunction with Supercomputing Center of Chongqing Medical University.
Author information
Authors and Affiliations
Contributions
F.L. and W.S. conceived the project. F.L. designed and developed OPED and conducted the method comparisons. X.W. and W.S. designed the experiments. S.H., Z.S., and Y.L. performed the experiments. F.L. analysed the data and constructed the database. F.L., J.H., S.H. and J.D. plotted the figures. J.H. developed the web interface. F.L. and Z.S. wrote the paper. X.W., W.S. and Y.L. revised the paper. X.C. and X.H. provided feedback and guidance. S.W., X.W. and W.S. provided conceptual advice and supervised the work. All of the authors reviewed and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Jacob Huth, in collaboration with the Nature Machine Intelligence team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Details of three major modules in OPED.
a, Input module. Only the target sequence (including 20-nt protospacer), PBS sequence and RTT sequence of variable length are used as input, and the input module performs preprocessing, embedding and positional encoding of these raw nucleotide sequences. b, Feature learning module employing customized transformer and attention networks to learn the comprehensive and interpretable features of the target sequence and pegRNA pair. c, Regression module outputting the predicted editing score through a multilayer fully-connected feedforward network.
Extended Data Fig. 2 Validation of OPED on datasets from Kim et al.
a-e, Validation of OPED using six replicates of PE2 efficiencies at 33 endogenous sites in HEK293T cells. Except Endo-BR3 dataset, the others are shown. The pegRNA number is n = 31, 30, 28, 28, and 28 for datasets Endo-BR1-TR1 (a), Endo-BR1-TR2 (b), Endo-BR2-TR1 (c), Endo-BR2-TR2 (d), and Endo-BR2-TR3 (e), respectively. f, Comparison of pegRNAs with OPED editing score higher or lower than 10 on Endo-BR1-TR1 (n = 14 ( ≥ 10) and n = 17 ( < 10)), Endo-BR1-TR2 (n = 14 ( ≥ 10) and n = 16 ( < 10)), Endo-BR2-TR1 (n = 13 ( ≥ 10) and n = 15 ( < 10)), Endo-BR2-TR2 (n = 14 ( ≥ 10) and n = 14 ( < 10)), Endo-BR2-TR3 (n = 14 ( ≥ 10) and n = 14 ( < 10)), and Endo-BR3 datasets (n = 8 ( ≥ 10) and n = 12 ( < 10)). Whiskers indicate the minimum, the median, and the maximum. ***, **, and * represent the P value of two-sided Wilcoxon rank-sum test <0.001, <0.01, and <0.05, respectively (\(4.8\times {10}^{-4},5.6\times {10}^{-4},1.9\times {10}^{-3},1.8\times {10}^{-3},1.8\times {10}^{-3},2.5\times {10}^{-2}\), from left to right). g-l, Validation of OPED using datasets in HCT116 (abbreviated as HCT, n = 72, 75, and 75 for HCT-BR1-TR1, HCT-BR1-TR2, and HCT-BR2-TR2, respectively) and MDA-MB-231 (abbreviated as MDA, n = 71, 73, and 75 for MDA-BR1-TR1, MDA-BR1-TR2, and MDA-BR2-TR2, respectively) cells. Except BR2 and TR1 of each cell line, the others are shown. m-n, Comparison of pegRNAs with OPED editing score higher or lower than 10 on HCT-BR1-TR1 (n = 33 ( ≥ 10) and n = 39 ( < 10)), HCT-BR1-TR2 (n = 34 ( ≥ 10) and n = 41 ( < 10)), HCT-BR2-TR1 (n = 34 ( ≥ 10) and n = 41 ( < 10)), HCT-BR2-TR2 (n = 34 ( ≥ 10) and n = 41 ( < 10)), MDA-BR1-TR1 (n = 33 ( ≥ 10) and n = 38 ( < 10)), MDA-BR1-TR2 (n = 34 ( ≥ 10) and n = 39 ( < 10)), MDA-BR2-TR1 (n = 34 ( ≥ 10) and n = 40 ( < 10)), and MDA-BR2-TR2 datasets (n = 34 ( ≥ 10) and n = 41 ( < 10)). Whiskers indicate the minimum, the median, and the maximum. *** represents the P value of two-sided Wilcoxon rank-sum test <0.001 (m, \(1.8\times {10}^{-7},6.2\times {10}^{-8},6.4\times {10}^{-9},3.5\times {10}^{-8}\); n, \(8.3\times {10}^{-8},1.6\times {10}^{-8},1.2\times {10}^{-8},1.2\times {10}^{-8}\) (from left to right)).
Extended Data Fig. 3 Validation of OPED on datasets from Anzalone et al.
a, Comparison of pegRNAs with OPED editing score higher or lower than 13 on a PE2 dataset encompassing 199 endogenous sites in HEK293T cells provided by Anzalone et al. (n = 127 ( ≥ 13) and n = 72 ( < 13)). Violin plots show the efficiency distributions of corresponding pegRNAs. Whiskers indicate the minimum, the median, and the maximum. *** represents the P value of two-sided Wilcoxon rank-sum test <0.001 (\(1.1\times {10}^{-13}\)). b-c, Validation of OPED using two pegRNA sets targeting HEK4 (b, referred as HEK4_2a, n = 9) and RNF2 (c, referred as RNF2_2b, n = 11) sites. d-i, Line plots showing the trend between the OPED editing score (orange) and the measured editing efficiency (black) for six pegRNA sets targeting HEK4, RNF2, HEK3, and EMX1. j-o, Validation of OPED using six pegRNA sets targeting HEK4 (j, referred as HEK4_2b, n = 14), RNF2 (k, referred as RNF2_2a, n = 9), HEK3 (l, referred as HEK3_2a, n = 10; m, referred as HEK3_2b, n = 11) and EMX1 (n, referred as EMX1_2a, n = 9; o, referred as EMX1_2b, n = 11) sites.
Extended Data Fig. 4 Validation of OPED on datasets from Mathis et al.
a, Validation of OPED for predicting unedited rates using the library 1 test dataset (n = 18,485) in HEK293T cells from Mathis et al. b-g and i-l, Violin plots showing pegRNAs with score higher or lower than 50. Whiskers indicate the minimum, the median, and the maximum. *** represents the P value of two-sided Wilcoxon rank-sum test <0.001. b, Comparison on the library 1 test dataset (n = 8,634 ( ≥ 50) and n = 9,851 ( < 50), \(P < 1.0\times {10}^{-308}\)). c, Comparison of unintended editing rates on the test dataset (n = 273 ( ≥ 50) and n = 18,212 ( < 50), \(P=7.7\times {10}^{-157}\)). d-g, Comparison on datasets in different experimental conditions and cell lines. d, unmodified pegRNAs without MMR inhibition (HEK293T, n = 523 ( ≥ 50) and n = 392 ( < 50), \(P=5.0\times {10}^{-119}\); K562, n = 503 ( ≥ 50) and n = 373 ( < 50), \(P=1.4\times {10}^{-41}\); U2OS, n = 499 ( ≥ 50) and n = 366 ( < 50), \(P=2.0\times {10}^{-46}\)). e, tevopreQ1 pegRNAs without MMR inhibition (HEK293T, n = 487 ( ≥ 50) and n = 421 ( < 50), \(P=1.5\times {10}^{-107}\); K562, n = 472 ( ≥ 50) and n = 400 ( < 50), \(P=6.0\times {10}^{-45}\); U2OS, n = 468 ( ≥ 50) and n = 384 ( < 50), \(P=1.6\times {10}^{-41}\)). f, unmodified pegRNAs with MMR inhibition (HEK293T, n = 521 ( ≥ 50) and n = 392 ( < 50), \(P=3.8\times {10}^{-120}\); K562, n = 515 ( ≥ 50) and n = 391 ( < 50), \(P=2.7\times {10}^{-102}\); U2OS, n = 499 ( ≥ 50) and n = 365 ( < 50), \(P=8.1\times {10}^{-75}\)). g, tevopreQ1 pegRNAs with MMR inhibition (HEK293T, n = 487 ( ≥ 50) and n = 422 ( < 50), \(P=2.5\times {10}^{-106}\); K562, n = 483 ( ≥ 50) and n = 427 ( < 50), \(P=2.3\times {10}^{-96}\); U2OS, n = 468 ( ≥ 50) and n = 384 ( < 50), \(P=1.3\times {10}^{-65}\)). h, Correlation between different experimental conditions and cell lines when using PEmax. i-l, Comparison on datasets in different experimental conditions and cell lines when using PEmax. i, unmodified pegRNAs without MMR inhibition (K562, n = 511 ( ≥ 50) and n = 393 ( < 50), \(P=6.6\times {10}^{-36}\); U2OS, n = 501 ( ≥ 50) and n = 368 ( < 50), \(P=7.8\times {10}^{-48}\)). j, tevopreQ1 pegRNAs without MMR inhibition (K562, n = 480 ( ≥ 50) and n = 426 ( < 50), \(P=8.8\times {10}^{-42}\); U2OS, n = 470 ( ≥ 50) and n = 386 ( < 50), \(P=6.9\times {10}^{-40}\)). k, unmodified pegRNAs with MMR inhibition (K562, n = 514 ( ≥ 50) and n = 392 ( < 50), \(P=9.1\times {10}^{-95}\); U2OS, n = 500 ( ≥ 50) and n = 367 ( < 50), \(P=5.1\times {10}^{-72}\)). l, tevopreQ1 pegRNAs with MMR inhibition (K562, n = 482 ( ≥ 50) and n = 427 ( < 50), \(P=8.5\times {10}^{-86}\); U2OS, n = 470 ( ≥ 50) and n = 382 ( < 50), \(P=6.8\times {10}^{-59}\)).
Extended Data Fig. 5 Statistics and quality of 30 pathogenic variants after installation.
a, Distribution of editing efficiencies (mean of three biological replicates) with optimized PE designs of OPED for PE2, PE3/PE3b and ePE editing systems to install 30 human pathogenic genetic variants (n = 30) from the ClinVar database in HEK293T cells. Boxplots represent the 25th, 50th and 75th percentiles. The upper whisker extends from the hinge to the largest value no further than 1.5 × IQR from the hinge (where IQR is the inter-quartile range, or distance between the first and third quartiles). The lower whisker extends from the hinge to the smallest value at most 1.5 × IQR of the hinge. b-d, Bar plots showing the detail of mean editing efficiencies with optimized PE designs of OPED for PE2 (b), PE3/PE3b (c) and ePE (d) editing systems at the 30 target sites. e, Heatmap showing the Phred-scaled quality score of each pathogenic variant called by the GATK tool. The quality is calculated as -10×log10 Probability(the called variant is wrong), and high-quality scores indicate high-confidence calls. Each column indicates a pathogenic variant, and each row indicates an editing system (PE2, PE3/PE3b and ePE) with a biological replicate.
Extended Data Fig. 6 Frequency of mutations across the entire amplicon through the installation of pathogenic variants.
a-c, Frequency of substitutions (green), insertions (red), and deletions (purple) across the entire amplicon after editing with optimized PE designs for PE2 (a), PE3/PE3b (b) and ePE (c) editing systems in installing pathogenic SNVs (left), insertions (middle) and deletions (right). The vertical dashed line indicates the cleavage site of pegRNA. The orange box indicates the spacer of pegRNA. The grey box indicates the quantification window, and only modifications that overlap with the quantification window are considered.
Extended Data Fig. 7 Frequency distribution of sequence modifications through the installation of 30 pathogenic variants.
a-i, Frequency distribution of alleles after editing with optimized PE designs for PE2 (a-c), PE3/PE3b (d-f) and ePE (g-i) editing systems to install pathogenic SNVs (a, d, g), insertions (b, e, h) and deletions (c, f, i). Left panel, the frequency distribution of sequence modifications that increase read length with respect to the reference amplicon, classified as insertions (positive indel size). Middle panel, the frequency distribution of sequence modifications that reduce read length with respect to the reference amplicon, classified as deletions (negative indel size). Right panel, the frequency distribution of sequence modifications that do not alter read length with respect to the reference amplicon, which are classified as substitutions (number of substituted positions shown).
Extended Data Fig. 8 Purity of optimized PE designs by OPED.
a-j, Bar plots showing the percentage of total edited reads with unintended indels (number of unintended indel reads / number of total edited reads). Mean ± s.d. of n = 3 independent biological replicates are shown. a-c, Comparative assessments against four state-of-the-art tools (DeepPE, Easy-Prime, PRIDICT, and PrimeDesign) using the optimized PE designs for PE2 (a), PE3/PE3b (b) and ePE (c) editing systems in installing eight pathogenic variants. d-e, Comparative assessments using the top-3-ranked PE designs (top-3-ranked pegRNA and the corresponding first-ranked sgRNA) recommended by OPED for PE3/PE3b (d) and ePE (e) editing systems in installing six pathogenic variants. f-g, Comparative assessments using combinations of the first-ranked pegRNA and top-3-ranked sgRNAs recommended by OPED for PE3/PE3b (f) and ePE (g) editing systems. h, Installation of six pathogenic variants using the optimized PE designs recommended by OPED for noncanonical NG PAM. i-j, Comparative assessments using mpknot epegRNAs recommended by OPED and Nelson et al. for PE2 (i) and PE3/PE3b (j).
Extended Data Fig. 9 Analysis of optimized pegRNAs for correcting/installing pathogenic variants with an NG/NGG PAM.
a-c, Distribution of the number of candidate pegRNAs per pathogenic variant with an NG PAM for installation (a), with an NGG PAM for correction (b) and installation (c). Horizontal black lines indicate the averages. d-l, Distribution of PBS length, RTT length and edit-to-nick distance of all candidate pegRNAs (blue) and first-ranked pegRNAs predicted by OPED (green) for each variant. Horizontal black lines indicate the medians. *** indicates the P value of the two-sided Wilcoxon rank-sum test <0.001. d-f, Installing pathogenic SNVs (d), insertions (e) and deletions (f) with an NG PAM. d, Distribution of n = 542,781,954 candidate pegRNAs and n = 51,473 first-ranked pegRNAs (\(P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308}\), respectively). e, Distribution of n = 16,815,216 candidate pegRNAs and n = 1,833 first-ranked pegRNAs (\(P=1.1\times {10}^{-178},P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308}\), respectively). f, Distribution of n = 253,831,281 candidate pegRNAs and n = 24,432 first-ranked pegRNAs (\(P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308}\), respectively). g-i, Correcting pathogenic SNVs (g), insertions (h) and deletions (i) with an NGG PAM. g, Distribution of n = 144,531,827 candidate pegRNAs and n = 50,761 first-ranked pegRNAs (\(P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308}\), respectively). h, Distribution of n = 4,660,997 candidate pegRNAs and n = 1,773 first-ranked pegRNAs (\(P=7.2\times {10}^{-51},P < 1.0\times {10}^{-308},P=1.6\times {10}^{-59}\), respectively). i, Distribution of n = 64,266,906 candidate pegRNAs and n = 23,936 first-ranked pegRNAs (\(P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308}\), respectively). j-l, Installing pathogenic SNVs (j), insertions (k) and deletions (l) with an NGG PAM. j, Distribution of n = 152,685,709 candidate pegRNAs and n = 50,924 first-ranked pegRNAs (\(P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308}\), respectively). k, Distribution of n = 4,399,142 candidate pegRNAs and n = 1,780 first-ranked pegRNAs (\(P=7.4\times {10}^{-39},P < 1.0\times {10}^{-308},P=9.1\times {10}^{-47}\), respectively). l, Distribution of n = 71,950,692 candidate pegRNAs and n = 23,946 first-ranked pegRNAs (\(P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308},P < 1.0\times {10}^{-308}\), respectively).
Supplementary information
Supplementary Information
Supplementary Tables 1–7.
Supplementary Table 1
Performance comparison of OPED with nine alternative machine learning models on the HT-test, Type-test and Position-test datasets.
Supplementary Table 2
Eight published pegRNA sets at genomic sites HEK4, EMX1, RNF2 and HEK3 for validation of OPED.
Supplementary Table 3
The optimized PE designs of OPED to install 30 human pathogenic genetic variants from the ClinVar database with the PE2, PE3/PE3b and ePE editing systems.
Supplementary Table 4
Analysis of candidate pegRNAs for correcting or installing ClinVar pathogenic variants with an NG or NGG PAM.
Supplementary Table 5
Primer sequences used for constructing the pegRNA–GFP plasmid.
Supplementary Table 6
Primer sequences of the scaffold.
Supplementary Table 7
Primer sequences used for cell genomic DNA amplification and deep sequencing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, F., Huang, S., Hu, J. et al. Design of prime-editing guide RNAs with deep transfer learning. Nat Mach Intell 5, 1261–1274 (2023). https://doi.org/10.1038/s42256-023-00739-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-023-00739-w
This article is cited by
-
Interpretable model of CRISPR–Cas9 enzymatic reactions
Nature Computational Science (2023)
