
Abstract
Sepsis-induced cardiomyopathy (SICM) is common in septic patients with a high mortality and is characterized by an abnormal immune response. Owing to cellular heterogeneity, understanding the roles of immune cell subsets in SICM has been challenging. Here we identify a unique subpopulation of cardiac-resident macrophages termed CD163+RETNLA+ (Mac1), which undergoes self-renewal during sepsis and can be targeted to prevent SICM. By combining single-cell RNA sequencing with fate mapping in a mouse model of sepsis, we demonstrate that the Mac1 subpopulation has distinct transcriptomic signatures enriched in endocytosis and displays high expression of TREM2 (TREM2hi). TREM2hi Mac1 cells actively scavenge cardiomyocyte-ejected dysfunctional mitochondria. Trem2 deficiency in macrophages impairs the self-renewal capability of the Mac1 subpopulation and consequently results in defective elimination of damaged mitochondria, excessive inflammatory response in cardiac tissue, exacerbated cardiac dysfunction and decreased survival. Notably, intrapericardial administration of TREM2hi Mac1 cells prevents SICM. Our findings suggest that the modulation of TREM2hi Mac1 cells could serve as a therapeutic strategy for SICM.
Similar content being viewed by others


Main
Sepsis affects approximately 49 million individuals and causes over 11 million fatalities worldwide annually1,2. The majority of patients with sepsis display aberrant cardiac function, usually termed SICM3. As an important contributor to organ dysfunction, SICM is characterized by a reduced ejection fraction (EF) and is associated with high mortality in patients. Of note, transiently reversible myocardial dysfunction is also observed in SICM, suggesting that cardiac homeostasis can be restored after the septic stress4,5,6; however, the mechanisms facilitating heart rehabilitation during sepsis have not been fully elucidated.
Immune cells play vital roles in regulating tissue homeostasis following cardiac injury7. In heart tissue, macrophages are dominant immune cells, which are highly heterogeneous and diversified in lineage8,9,10. During the past decade, bone-marrow-derived macrophages (circulating) have been regarded as the only primary phagocytes, which are involved in, for example, cardiac tissue healing and homeostasis11; however, emerging evidence indicates that cardiac-resident macrophages (CRMs) play fundamental roles in the clearance of necrotic cells and debris, promotion of angiogenesis, restriction of inflammation and tissue remodeling during the cardiac injury process, which are independent of circulating macrophages10,12,13. Very recently, a study showed that CRMs removed the decaying mitochondria ejected by cardiomyocytes to maintain heart health14.
A septic heart has a high energy requirement under stress, including elevated body temperature, hypoxia, tachycardia and systemic cytokine storm. In line with this pathological change, cardiac mitochondrial dysfunction has been demonstrated in many animal studies15,16. Clinically, mitochondrial dysfunction correlates directly with poor sepsis outcomes17,18. Therefore, the maintenance of cardiac mitochondrial homeostasis is essential for the recovery of SICM; however, how immune cells serve as a manipulator of mitochondrial quality control in the septic heart remains completely unknown.
In the present study, by combining single-cell RNA sequencing (scRNA-seq) with fate-mapping techniques, we profiled dynamic changes of cardiac immune cells, especially cardiac macrophages, in the murine septic heart. We identified a CD163+RETNLA+ Mac1 subpopulation with self-renewal ability that was critical for supporting the cardiac function under septic stress. Deep-phenotypic and functional analysis revealed that the Mac1 subpopulation was characterized by endocytic transcriptomic signatures and by high TREM2 expression. In addition, we observed that the TREM2hi macrophage subpopulation actively scavenged cardiomyocyte-ejected mitochondria and therefore promoted cardiac homeostasis in sepsis. Trem2 ablation in macrophages impaired the ability of Mac1 cell self-renewal, which resulted in the accumulation of dysfunctional mitochondria in the extracellular space of myocardium and exacerbated cardiac function following sepsis. Finally, intrapericardial administration of TREM2hi Mac1 cells improved cardiac function and prevented SICM. These findings provide a perspective for the development of TREM2-targeted immunotherapies of SICM.
Results
Single-cell characterization of immune cells in septic heart
To explore the mechanisms of reversible myocardial dysfunction during septic stress, we established the septic murine model (cecal ligation and puncture (CLP) model) and profiled the septic heart by echocardiography and cardiac injury biomarkers. As expected, cardiac function deteriorated steadily upon sepsis progression and became significantly worse 3 d after CLP. Subsequently, cardiac function recovered gradually (Extended Data Fig. 1a–d). Congruently, cardiac injury parameters (lactate dehydrogenase (LDH), troponin I, Anp and Bnp) also showed comparable dynamic changes (Extended Data Fig. 1e–h). Owing to the crucial role of inflammation in the pathology of SICM, cardiac inflammatory markers were measured by a multiplex protein array. The levels of granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, interleukin (IL)-4, IL-1α, IL-9, interferon (IFN)-γ, IL-1β, IL-21 and IL-10 in the heart reached a peak 3 d after CLP and then decreased slowly as time progressed (Extended Data Fig. 1i). These results indicate that the dynamic changes of cardiac function during sepsis progression are consistent with the inflammatory environment of cardiac tissue.
Next, we characterized the types and states of immune cells involved in SICM using scRNA-seq. Metabolically active nucleated CD45+ cells were separated by fluorescence-activated cell sorting (FACS) from 14 mice at different time points. A total of 29,537 quality-control-positive cells were selected for further analysis, which generated on average about 9,362 mapped reads and 2,718 genes per cell (Fig. 1a and Extended Data Fig. 2a). As expected, unbiased clustering analysis revealed multiple clusters of cardiac immune cells (Fig. 1b), which included macrophages (Adgre1 and Fcgrt), monocytes (Plac8 and Chil3), neutrophils (S100a8/a9), natural killer (NK)/T cells (Cd3e, Klrk1 and Il7r), B cells (Igkc and Cd79a) and cycling cells (Mki67 and Stmn1) (Fig. 1c,d and Extended Data Fig. 2b,c). Among them, macrophages were the most abundant cells with a rich set of subclusters in the heart. By comparing changes of the immune cells at each time point, we found that the percentages of macrophages slightly decreased, whereas the heterogeneity of macrophage subclusters was more substantial after CLP. Meanwhile, large numbers of neutrophils and monocytes were recruited into the myocardium after sepsis (Fig. 1e,f). In addition to the findings from scRNA-seq, flow cytometry analysis confirmed the parallel changes in cardiac immune cells during sepsis progression (Extended Data Fig. 2d,e).

a, Schematic diagram showing scRNA-seq pipeline of murine cardiac immune cells during sepsis progression. SS, steady state. b, Uniform Manifold Approximation and Projection (UMAP) plots of 29,537 cardiac immune cells allocated into 15 clusters from the hearts of 14 WT mice at SS, 3, 7 and 21 d after CLP. Cardiac cells from 3–4 mice were mixed as one sample. c, UMAP plots of cardiac immune cells colored by cell type. d, Heat map showing selected cluster marker genes (top, color-coded by cluster and condition) with exemplar genes and cell-type annotation labeled. e, UMAP plots of cardiac immune cells from WT mice in different time points of sepsis, annotated by cell type as in c. Each time point is contributed by either three (SS or 21 d) or four mice (3 or 7 d). f, Bar plots showing the distribution of immune cell types from WT mice at different time points of sepsis (Extended Data Figs. 1 and 2).
Mac1 is associated with restoration of cardiac function
Given that the monocyte-macrophage compartment represents the largest and most significantly altered population of cardiac immune cells during sepsis, we performed an unsupervised cluster analysis to investigate the heterogeneity and role of cardiac monocyte-macrophages. Five macrophage subclusters (Mac1–5), two monocyte subclusters (Mon1 and Mon2) and a dendritic cell (DC) subcluster were identified (Fig. 2a). Mac1 was characterized by the expression of Retnla, Lyve1, Cd163 and Folr2, which resembled the signatures of cardiac tissue-resident macrophages19. Mac2 expressed relatively high levels of antigen-presentation genes, the characteristics of which were consistent with the previously reported major histocompatibility complex (MHC)-II cluster19,20. Mac3 was enriched in Cxcl13, Ccl8, Saa3 and a few IFN-stimulated genes (Ifit3 and Irf7). This cluster might be pro-inflammatory and contained IFN-inducible cells21. Mac4 was identified by the expression of Cd72 and Acp5, which were confirmed as markers of pathologically pro-inflammatory cells22. Mac5 corresponded to CCR2+ macrophages12,19,23, which also expressed high levels of antigen-presentation genes (H2-Aa and H2-Eb1). Mon1 was the classical monocyte cluster with high expression of Ly6c2, whereas Mon2 was the nonclassical monocyte cluster with low expression of Ly6c2 (refs. 24, 25). The DC cluster was characterized by high levels of Xcr1, Naaa and Irf8, which were reported as markers of classical DCs26 (Fig. 2b and Extended Data Fig. 3a). We next compared the dynamic differences in relative proportions of the eight subclusters among different stages of SICM. Notably, the Mac1 and Mac2 subclusters were predominantly presented in hearts at steady state, whereas the levels of the Mac1 subset were dramatically reduced 3 d after CLP. The levels of Mac3 and Mac4, meanwhile, remarkably increased 3 d after CLP; however, as cardiac function gradually recovered 7 and 21 d after CLP, the Mac1 subcluster was restored (Fig. 2c,d). We also used flow cytometry and immunofluorescence to confirm scRNA-seq results by selecting CD163 and RETNLA as markers for Mac1. Consistently, the relative frequency and absolute numbers of Mac1 decreased 3 d after CLP and were restored 7 and 21 d after CLP (Fig. 2e,f and Extended Data Fig. 3b–d). Subsequently, correlation analysis found that the absolute numbers of Mac1 were positively correlated with EF of the heart and negatively correlated with serum cardiac troponin I (cTnI) levels during sepsis progression (Fig. 2g,h). Taken together, these data reveal a distinct CD45+CD11b+F4/80+CD163+RETNLA+ Mac1 subcluster that is associated with the restoration of cardiac dysfunction after septic stress.

a, UMAP plots of 24,058 monocyte-macrophages allocated to the eight clusters from Fig. 1c. b, Heat map showing top 20 cluster marker genes (top, color-coded by cluster and condition) with exemplar genes and cluster annotations labeled. c, UMAP plots of the monocyte-macrophage compartment from WT mice at SS, 3, 7 and 21 d after CLP, annotated by cluster as in a. d, Bar plots showing cluster distribution within the monocyte-macrophage compartment from WT mice after CLP. e, Representative contour plots showing flow cytometric analysis of cardiac CD45+CD11b+F4/80+CD163+RETNLA+ macrophages from WT mice after CLP. f, Quantification of CD163+RETNLA+ macrophages per mg of cardiac tissue. Bars show mean ± s.e.m. Two-sided P values were determined by one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. g,h, Correlation of cardiac CD163+RETNLA+ macrophage numbers with EF and cTnI levels. Data are presented as two-tailed Spearman’s rank correlation. Dashed lines represent 95% confidence intervals. Each symbol represents one animal (SS, n = 8 mice; CLP 3 d, n = 8 mice; CLP 7 d, n = 9 mice; CLP 21 d, n = 10 mice) (f,g). Results represent four independent experiments in this figure (Extended Data Fig. 3).
Developmental trajectory and lineage tracing of Mac1 subset
To determine the potential relationship between monocytes and the Mac1 subcluster, we examined the dynamic immune states and cell transitions of the monocyte-macrophage compartment by the Monocle algorithm27. In pseudotime analysis, we downsampled monocyte-macrophages to 1,500 cells to provide a clear cell developmental trajectory. We observed that Mac1 cells localized at the beginning of the pseudotime trajectory, whereas Mon1, Mon2 and DC cells were at the end of the trajectory. In addition, the majority of Mac2 cells were at the terminus of the pseudotime trajectory, whereas Mac3 cells, similarly to the Mac1 cells, were localized at the initiating branch (Fig. 3a,b and Extended Data Fig. 4a). Tracking the alterations of gene expression across monocyte-macrophage compartments revealed developmental patterns defining the phenotype and function of macrophage subsets. In short, the Mac1 cluster was characterized by upregulated expression levels of Cd163, Lyve1, Retnla, Mrc1, Gas6 and Folr2 and by downregulated expression levels of monocyte genes (Plac8 and Chil3). The Mon1, Mon2, Mac5 and DC clusters were characterized by upregulated expression of Ccr2, Il1b, Chil3 and Plac8, whereas the Mac2 cluster was characterized by high expression of antigen-presenting genes (H2-Aa and H2-Ab1). These cell clusters displayed the reduced expression of the Mac1 gene profile (Fig. 3c and Extended Data Fig. 4b).

a, The Monocle prediction of the monocyte-macrophage developmental trajectory with Seurat’s cluster information in Fig. 2a mapped alongside. b, The Monocle prediction of monocyte-macrophage developmental trajectory with each Seurat-based cluster shown separately. c, Heat map of top 50 DEGs along with the pseudotime. The relative position of individual subsets across pseudotime is illustrated below. d, Representative contour plots showing the expression of tdTomato (CX3CR1) on gated CD45+CD11b+F4/80+CD163+RETNLA+ macrophages, CD45+CD11b+F4/80+ (non-CD163+RETNLA+) macrophages and CD45+CD11b+F4/80−LY6C+ monocytes. Graph showing the percentage of tdTomato+ cells isolated from hearts of WT mice at each time point along sepsis. NS, not significant. e, Representative contour plots showing the expression of tdTomato (CX3CR1) and CD163 on gated CD45+CD11b+F4/80+RETNLA+ macrophages and graph showing absolute numbers of CD45+CD11b+F4/80+CD163+RETNLA+tdTomato+ macrophages isolated from hearts of WT mice at each time point following sepsis. f, Representative histograms showing the expression of Ki67 in CD45+CD11b+F4/80+CD163+RETNLA+ macrophages and graph showing quantification of mean fluorescence intensity (MFI) of Ki67 at SS, 3 and 7 d after CLP. Dots represent individual subjects (d–f); SS, n = 5 mice; CLP 3 d, n = 6 mice; CLP 7 d, n = 7 mice. Data are shown as mean ± s.e.m.; two-sided P values were determined by one-way ANOVA, followed by Games-Howell’s (d) or Sidak’s (d–f) multiple comparison test; results represent three independent experiments (Extended Data Fig. 4).
To confirm the above pseudotime analysis results, we further used a tamoxifen-inducible Cx3cr1CreERT2–IRES–YFP mouse line crossed with Rosa26-stop-tdTomato reporter mice (termed Cx3cr1CreERT2:Rosa26tdTom; Extended Data Fig. 4c) to track Mac1 cells in SICM. tdTomato fluorescence signals of these reporter mice reliably distinguished CRMs from recruited monocyte-derived macrophages19. Around 95% of circulating monocytes (CD115+CD11b+) and ~89% cardiac macrophages (F4/80+CD11b+) were tdTomato+ 1 week after the first administration of tamoxifen. Three weeks after the administration of tamoxifen, circulating monocytes were gradually replaced by tdTomato− monocytes, whereas their contribution to cardiac macrophages was negligible. At the time point of 5 weeks, almost 85% macrophages and ~98% CD163+RETNLA+ Mac1 cells in the heart retained tdTomato+, which was then maintained at similar levels (Extended Data Fig. 4d). Six weeks after administration of tamoxifen, Cx3cr1CreERT2:Rosa26tdTom mice had CLP surgery. We observed that more than 95% of the CD163+RETNLA+ Mac1 cells remained as tdTomato+ 3 and 7 d after CLP (Fig. 3d). Accordingly, the proportion and the absolute numbers of tdTomato+ Mac1 cells in the septic heart were remarkably reduced 3 d after CLP and partly recovered 7 d after CLP (Fig. 3e). To determine the proliferation of Mac1 cells in vivo, Ki67 expression was assessed. tdTomato+ Mac1 cells exhibited high expression of Ki67 3 d after CLP compared to those at steady state, indicating that Mac1 cells presented an actively proliferative state under septic stress (Fig. 3f and Extended Data Fig. 4e). These data suggest that CD163+RETNLA+ Mac1 cells are self-renewing CRMs, independent of replenishment from circulating monocytes and display proliferative bursts during sepsis.
TREM2 is essential for Mac1 cells remodeling during SICM
We next sought to explore the functional characteristics of the Mac1 subset in SICM. Gene Ontology (GO) enrichment analysis revealed that 156 differentially expressed genes (DEGs) upregulated in Mac1 were related to phagocytosis and endocytosis in biological terms (Fig. 4a). Of note, phagocytosis-related gene Trem2 was upregulated in Mac1 cells relative to other macrophages. (Extended Data Fig. 5a). We then examined the expression of Trem2 in all cell clusters and observed that Trem2 was particularly abundant in the Mac1 subset (Fig. 4b and Extended Data Fig. 5b). Immunofluorescence staining showed the expression of TREM2 on Mac1 cells as well as colocalization with CD163 (Fig. 4c). Flow cytometry analysis showed that CD163+RETNLA+ Mac1 cells expressed higher levels of TREM2 compared to other macrophages (Fig. 4d and Extended Data Fig. 5c). In addition, to determine the role of TREM2 for the remodeling of the macrophage subset in SICM, we performed CLP in Trem2-deficient (Trem2−/−) mice and wild-type (WT) littermate controls and analyzed their cardiac monocyte-macrophage compartments by scRNA-seq. Unsupervised cluster analysis of monocyte-macrophage compartments in Trem2−/− and WT mice showed similar types and proportions of subclusters at steady state and 3 d after CLP, respectively (Extended Data Fig. 5d). Notably, we observed that the proportion of Mac1 cells both significantly decreased in WT and Trem2−/− mice 3 d after CLP (Extended Data Fig. 5e). At day 7 after CLP, Mac1 cells recovered to normal counts in WT mice, but the proportion of Mac1 cells in Trem2−/− mice could not be restored (Fig. 4e,f). Flow cytometry analysis confirmed that the percentage and absolute number of CD163+RETNLA+ Mac1 cells in Trem2−/− mice did not recover at day 7 after CLP compared to WT controls (Fig. 4g). In contrast, the proportions of other cardiac immune cell compartments were not affected by Trem2 deficiency, with the exception of reduction in the proportion of macrophages in Trem2−/− mice 3 and 7 d after CLP and a modest increase in the proportion of neutrophils 7 d after CLP (Extended Data Fig. 5f). Moreover, we revealed that the proliferative capability of CD163+RETNLA+ CRMs was significantly decreased in Trem2−/− mice compared to WT controls 3 d after CLP indicated by Ki67 expression (Fig. 4h). These results collectively suggest that TREM2 is a marker for the Mac1 subset and essential for the remodeling of Mac1 cells in septic hearts.

a, Dot plots showing the top 20 biological processes for the upregulated DEGs of Mac1 analyzed by GO. Red arrows indicate endocytosis-related pathways. b, UMAP plots and violin plots showing the expression of Trem2 in the monocyte-macrophage compartment. c, Representative immunofluorescence images of CD163 (green), TREM2 (red) and nuclei (blue) in cardiac tissue from WT mice at SS (n = 5) and 7 d after CLP (n = 5). The dotted lines indicate CD163+TREM2+ macrophages. Scale bars, 20 μm. d, Representative histograms of flow cytometric analysis of TREM2 expression in cardiac CD45+CD11b+F4/80+CD163+RETNLA+ and CD45+CD11b+F4/80+(non-CD163+RETNLA+) macrophages from WT mice at SS and 7 d after CLP. e, UMAP plots showing cell clustering results corresponding to Fig. 2a for a total of 13,405 monocytes-macrophages in WT and Trem2 knockout (KO) hearts at 7 d after CLP. f, Cluster distribution within the monocyte-macrophage subsets in WT and Trem2-KO hearts at 7 d after CLP. g, Percentages and absolute numbers of CD163+RETNLA+ macrophages analyzed by flow cytometry from hearts of WT (n = 6) and Trem2-KO (n = 6) mice at 7 d after CLP. h, Representative contour plots showing the expression of Ki67 on gated CD45+CD11b+F4/80+CD163+RETNLA+ macrophages and graph showing percentages of Ki67-expressing cells in WT (n = 5–6) and Trem2-KO (n = 6–7) hearts at SS and 3 d after CLP. Every symbol represents a mouse (g,h). Data are presented as mean ± s.e.m.; two-sided P values were determined by unpaired t-test (g) and one-way ANOVA with Sidak’s multiple comparisons test (h). Results represent three independent experiments (c,d,g,h). See also Extended Data Fig. 5.
Sepsis induces extracellular exopher accumulation in heart
Cardiomyocytes ejected dysfunctional mitochondria in dedicated vesicles referred to as ‘exophers’, which are characterized as 3.5 ± 0.1 μm in mean diameter and 31.0 ± 2.5 μm3 in mean volume14, similar to structures described in Caenorhabditis elegans neurons28. Cardiac macrophages are indispensable in maintaining heart homeostasis by cleaning these subcellular particles and defective mitochondria14. Our data suggested that homeostasis of macrophages in septic hearts was disrupted. We speculated that disrupted homeostasis of macrophages could impair the removal of cardiomyocyte-derived damaged mitochondria during sepsis. First, αMHCCre mice were crossed with Rosa26TdTom mice and only the cardiomyocytes of generated mice (referred as CardRED mice) expressed red fluorescence. We observed that the accumulation of some subcellular particles, defined as exophers, colocalized with mitochondrial protein Tom20 in the extracellular space of septic CardRED mice hearts (Fig. 5a,b and Supplementary Video 1). Transmission electron microscopy (TEM) analysis revealed a significant accumulation of free mitochondria in the periphery of cardiomyocytes 3 d after CLP (Extended Data Fig. 6a,b). In addition, these exophers were purified with flow cytometry14. We observed that cardiac exophers were positive for mitochondrial probes MitoTracker Green and MitoNIR (Extended Data Fig. 6c,d), also displayed the reduced membrane potential and lost responsiveness to hyperpolarizing agents (Extended Data Fig. 6e). These results suggest that sepsis causes the accumulation of dysfunctional mitochondria ejected by cardiomyocytes in the extracellular space of the heart.

a, Immunofluorescence images and 3D reconstruction showing presence of Tom20+ material in cardiomyocyte-derived exophers from hearts of septic CardRED mice. b, Presence of Tom20+ material in cardiomyocyte-derived exophers from hearts of CardRED mice at SS and 3 d after CLP. Graph showing exopher numbers per field of view (FOV); n = 6 per group. c, TEM image of a mononuclear cell taking up mitochondria by extended pseudopods in septic hearts (CLP 7 d). d, Images showing cardiomyocyte-derived exophers (red) containing mitochondria (Tom20, white) present in TREM2+ macrophages (green) from hearts of CardRED mice (n = 4). e, Cardiomyocyte-derived mitochondria (mt-Dendra2, green) present in TREM2+ macrophages (red) from hearts of MitoCard mice (n = 4). f, Cardiomyocyte-derived mitochondria (mt-Dendra2, green) localized in lysosomes (LAMP1, white) of TREM2+ macrophages (red) in hearts of MitoCard mice (n = 3). g, TREM2+ macrophages (green) took up cardiomyocyte-derived mitochondria (mt-Keima-458, cyan) and some mitochondria in an acidic environment (mt-Keima-561, red) from hearts of AAV9-Tnnt2-mt-Keima infected mice. h, CD163+ macrophages (green) engulfed cardiomyocyte-derived mitochondria in hearts of WT and Trem2−/− mice infected with AAV9-Tnnt2-mt-Keima, respectively. Graph showing percentages of mt-Keima+ mitochondria in CD163+ macrophages (n = 5 per group). For each animal, we randomly selected five visualization areas and five Mac1 cells were analyzed. The ratio of the area of mt-Keima mitochondria in a single Mac1 cell to the area of the same Mac1 cell was calculated. The ratio in the KO group was normalized to the ratio of the control group. i, The incorporation of cardiomyocyte-derived tdTomato protein in CD45+CD11b+F4/80+CD163+RETNLA+ macrophages and CD45+CD11b+F4/80−LY6C+ monocytes from WT → CardRED or KO → CardRED chimeras 7 d after CLP. Graph showing quantification of MFI of tdTomato (n = 6–7 per group). j, Presence of Tom20+ material (cyan) in cardiomyocyte-derived exophers (red) from hearts of WT → CardRED and KO → CardRED chimeras 7 d after CLP. Graph showing the exopher numbers per FOV (n = 6 per group). Scale bars are indicated in the images. Bars show as mean ± s.e.m. (b,h,j) and median with interquartile range (i). Two-sided P values were determined by unpaired t-test (b,h,j) and Mann–Whitney U-test (i). Results represent four (b), three (d–f) and two (g–j) independent experiments (Extended Data Figs. 6 and 7).
TREM2hi Mac1 cells eliminate cardiac exophers during sepsis
Given that free mitochondria and mitochondrial DNA (mtDNA) can elicit cardiac damage, elimination of exophers containing cardiomyocyte-derived mitochondria is important to maintain cardiac homeostasis and restore optimal cardiac function29,30. We next explored the mechanism for the clearance of dysfunctional mitochondria in the septic heart. TEM images showed that some cells adjacent to cardiomyocytes displayed a similar size and morphology to macrophages. These macrophages stretched out pseudopods, which surrounded the mitochondria-containing vesicles in the periphery of septic cardiomyocytes (Fig. 5c and Extended Data Fig. 6f). Consistently, confocal and three-dimensional (3D) images showed that TREM2+ cell-engulfed exophers (red) contained mitochondria in septic hearts of CardRED mouse (Fig. 5d and Supplementary Video 2).
To quantify the phagocytic activity of TREM2hi Mac1 (CD45+CD11b+F4/80+CD163+) cells, flow cytometry analysis with the hearts of CardRED mice revealed the higher incorporation of cardiomyocyte-derived tdTomato protein in Mac1 cells compared to non-Mac1 cells (Extended Data Fig. 6g). Further, cardiomyocyte-specific mitochondria reporter mice (MitoCard) were generated by crossing mtD2Flox/Flox with αMHCCre/+ mice31 and confocal images showed that cardiomyocyte-derived mitochondria localized in TREM2+ macrophages (Fig. 5e and Supplementary Video 3). Flow cytometry analysis also showed that TREM2hi Mac1 cells incorporated more cardiomyocyte-derived mitochondria during SICM (Extended Data Fig. 6h). In addition, we performed TUNEL staining with hearts of septic WT mice and found that the percentage of TUNEL+cTnI+ cells in total cells (TUNEL+cTnI+/4,6-diamidino-2-phenylindole (DAPI)) was very low (around 1%) on day 3 after sepsis (Extended Data Fig. 6i). These results excluded the possibility that TREM2hi cells incorporate apoptotic material derived from the septic heart.
To elucidate the fate of cardiomyocyte-derived mitochondria in Mac1 cells, we performed immunostaining of TREM2 with lysosome marker Lamp1 with the heart sections of septic MitoCard mice and observed that mt-Dendra2 signals in TREM2+ cells partially localized in LAMP1+ lysosomes (Fig. 5f and Supplementary Video 4). Next, we packaged adeno-associated virus serotype 9 (AAV9)-Tnnt2-mt-Keima virus and intramyocardial injection of AAV9 virus was performed to ensure the specific expression of Keima protein in the mitochondria (mt-Keima) of cardiomyocytes (Extended Data Fig. 7a)32. Immunofluorescence staining revealed that abundant mt-Keima+ particles concentrated in TREM2+ macrophages (Fig. 5g). Keima is pH-sensitive and its peak of the excitation spectrum can shift from the shorter (green, in the neutral environment) to longer (red, in the acidic environment) wavelength33. Video and 3D images showed that mt-Keima in TREM2+ macrophages exhibited both green and red fluorescence signals in the hearts of septic WT mice injected with AAV9-Tnnt2-mt-Keima virus (Fig. 5g and Supplementary Video 5), suggesting that mitochondria taken up by Mac1 cells may be delivered to the acidic lysosomal environment. Together, these data indicate that TREM2hi Mac1 cells have robust activity in eliminating of cardiomyocyte-derived dysfunctional mitochondria.
Trem2 deficiency impairs defective mitochondria removal
scRNA-seq data analysis revealed that the majority of the endocytic gene signatures such as Wwp1, Ccr5, Cd36, Eps15, Cltc, Mrc1, Ccl24, Dab2 and Folr2 in the whole macrophage population decreased in Trem2−/− mice compared to WT cells (Extended Data Fig. 7b,c). We then examined the effect of TREM2 on the uptake of cardiomyocyte-derived mitochondria by macrophages. The intramyocardial injection of AAV9-Tnnt2-mt-Keima virus was carried out with WT and Trem2−/− mice, respectively. Trem2−/− septic mice displayed the reduced uptake of cardiomyocytic mitochondria and Keima by CD163+ macrophages (Fig. 5h and Supplementary Video 5). Next, we transplanted bone marrow cells from WT or Trem2−/− mice into irradiated CardRED recipient mice and 8 weeks later, CLP surgery was performed (Extended Data Fig. 7d). We found that WT → CardRED CD163+RETNLA+ Mac1 cells scavenged more cardiomyocyte-derived material than Trem2−/−→CardRED CD163+RETNLA+ Mac1 cells 7 d after CLP (Fig. 5i). Consistently, immunofluorescence showed that more cardiomyocyte-derived mitochondria accumulated in the heart of Trem2−/−→CardRED chimeras (Fig. 5j). In addition, Trem2-deficient mice exhibited severe accumulation of free mitochondria in the extracellular space of cardiomyocytes 7 d after CLP (Extended Data Fig. 7e), but this was not found in Trem2-deficient mice at steady state (Extended Data Fig. 7f). These data suggest that TREM2 facilitates the scavenging of damaged mitochondria by Mac1 cells in septic hearts.
Trem2 deficiency exacerbates cardiac dysfunction in sepsis
To determine the physiological importance of TREM2hi Mac1 cells in SICM, we compared survival rate, cardiac systolic function, cardiac injury markers and inflammation between WT and Trem2−/− mice after CLP. As shown in Fig. 6a, the mortality of Trem2−/− septic mice was notably increased. Moreover, echocardiography assay revealed that Trem2−/− mice had worse cardiac function indicated by lower EF, fractional shortening (FS) and cardiac output (CO), especially 7 and 21 d after CLP, compared to WT littermate controls (Fig. 6b and Extended Data Fig. 8a,b). The concentrations of cTnI and LDH in the serum as well as messenger RNA levels of Anp and Bnp in heart tissues were all increased in Trem2−/− septic mice (Fig. 6c and Extended Data Fig. 8c–e). Also, Trem2 deficiency significantly elevated mRNA levels of pro-inflammatory mediators after CLP (Extended Data Fig. 8f–i).

a, Survival analysis of WT (n = 18) and Trem2−/− (n = 30) mice after CLP performance was monitored for 7 d. b,c, WT and Trem2−/− mice were subjected to CLP and cardiac function was examined at SS and 3, 7 and 21 d after CLP. Graph showing EF % (b) measured by echocardiography (n = 7–8 mice for each group) and the levels of cTnI (c) in the serum (n = 5–8 mice for each group). d, Representative M-mode echocardiography images and EF % of WT → WT chimeras (Trem2+/+Ch) and Trem2−/−→WT chimeras (Trem2−/−Ch) 7 d after CLP. e, Representative continuous-wave Doppler echocardiography images and E/A ratio of Trem2+/+Ch and Trem2−/−Ch chimeras 7 d after CLP. f, Graph showing CO measured by echocardiography. g,h, Graphs showing the levels of cTnI (g) and LDH (h) in the serum of Trem2+/+Ch and Trem2−/−Ch chimeras 7 d after CLP (d–h, n = 11 mice for each group). i, Graphs showing protein levels of ANP and BNP in the serum of Trem2+/+Ch and Trem2−/−Ch chimeras 7 d after CLP. j, Graphs showing protein levels of tumor necrosis factor (TNF)-α, IL-1β, IL-6 and CCL2 in the heart tissues of Trem2+/+Ch and Trem2−/−Ch chimeras 7 d after CLP (i,j, n = 10 mice for each group). k, Representative TEM images (left) and mitochondria injury score (right) of Trem2+/+Ch and Trem2−/−Ch chimeras 7 d after CLP. Scale bars, 5 μm. Bar graph showing mitochondria injury score per FOV. Each symbol represents a FOV of TEM images. Each group has four mice and five FOVs were randomly selected for assay from each animal. l,m, Graphs showing the levels of ATP (l) in heart tissue lysates and the levels of serum lactate (m) of Trem2+/+Ch and Trem2−/−Ch chimeras 7 da after CLP (l,m, n = 11 mice for each group). Each symbol represents one animal (b–j,l,m). Bars show as mean ± s.e.m. (b–f,h–l) and median with interquartile range (g,m). Two-sided P values were determined by Kaplan–Meier log-rank test (a), two-way ANOVA with Sidak’s multiple comparisons test (b,c), unpaired t-test (d–f,h–k) and Mann–Whitney U-test (g,m). Results represent at least four (b,c) and three (d–m) independent experiments (Extended Data Fig. 8).
Bone-marrow-derived macrophages contribute to the resident microglia pool in chimeric mice34,35. Irradiated recipients (CD45.1 background) were transplanted with bone marrow cells from WT or Trem2−/− mice (CD45.2 background), respectively (Extended Data Fig. 8j). Flow cytometry assay showed that the efficiency of Mac1 cells reconstitution was over 90% in irradiated chimeras 8 weeks after bone marrow transplantation (BMT) (Extended Data Fig. 8k, l). Flow cytometry and real-time–PCR results revealed that Trem2−/−→WT chimeras (Trem2−/−Ch) displayed lower TREM2 protein level in cardiac macrophages and Trem2 mRNA level in heart tissues than WT → WT chimeras (Trem2+/+Ch), respectively (Extended Data Fig. 8m). Eight weeks after BMT, chimeric mice were subjected to CLP. We found that Trem2-specific deficiency in macrophages exacerbated cardiac dysfunction, myocardial injury and cardiac inflammation following sepsis (Fig. 6d–j and Extended Data Fig. 8n,o). In addition, Trem2−/−Ch chimeras exhibited aggravated mitochondrial damage (Fig. 6k), reduced ATP levels (Fig. 6l) and increased lactate levels (Fig. 6m). These results suggest that TREM2 is essential for Mac1 to protect the cardiac function after sepsis.
Transplantation of TREM2hi Mac1 cells protects SICM
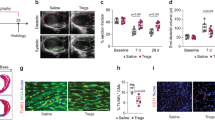
Finally, we sought to examine whether treatment with Mac1 cells could protect cardiac function after sepsis. The sorted TREM2hi Mac1 and non-Mac1 cells from the hearts of CD45.1 mice were respectively transplanted into the pericardial space of CD45.2 mice immediately after CLP. The control animals were intrapericardially injected with the same volume of Matrigel (MG) (Fig. 7a). Flow cytometry analysis showed that the transplanted Mac1 and non-Mac1 cells can survive and persist in recipient hearts for at least 7 d after CLP (Extended Data Fig. 9a). The proportion of Mac1 cells in total CD45+ cells was significantly increased in the hearts of Mac1 cell-injected mice at the examined time points compared to MG-injected mice (Extended Data Fig. 9b,c). Similarly, the proportion of non-Mac1 cells in total CD45+ cells was also increased in the hearts of non-Mac1-injected mice at the examined time points compared to MG-injected mice (Extended Data Fig. 9d,e). Furthermore, Mac1 and non-Mac1 cells were labeled with CellTracker Orange (CMTMR) and then transferred into the pericardial cavity immediately after CLP. After 3 d, histological assay indicated that the transplanted cells were widely distributed in the myocardium (Extended Data Fig. 9f). In addition, we intrapericardially transferred the CMTMR-labeled Mac1 cells into MitoCard mice. Three days later, confocal and 3D images showed that injected cells engulfed cardiomyocyte-derived mitochondria (Extended Data Fig. 9g and Supplementary Video 6). These data collectively show that the transplanted Mac1 cells can enter the myocardium, survive in the heart of recipient mice for at least 7 d and engulf cardiomyocyte-derived mitochondria.

a, Schematic illustration of TREM2hi Mac1 cells transplantation in WT mice. TREM2hi Mac1 (CD45+CD11b+F4/80+CD163+RETNLA+) and non-Mac1 (CD45+CD11b+ F4/80+ and non-CD163+RETNLA+) cells isolated from WT mice were mixed with MG and transplanted into the pericardial cavity of WT mice (2 × 105 cells per animal) immediately after CLP. Control mice were injected with MG only. Mice were killed and analyzed 3 d after the transplantation. b–e, Representative M-mode echocardiography images (b) and graphs showing EF % (c), FS % (d) and CO (e) measured by echocardiography (b–e, +MG, n = 5 mice; +non-Mac1, n = 9 mice; +Mac1, n = 10 mice). f,g, Graphs showing levels of cTnI and LDH in the serum. h, Graphs showing mRNA levels of Anp and Bnp in the heart tissues. i, Graphs showing protein levels of ANP and BNP in the serum. j, Graphs showing mRNA levels of Tnfα, Il1β, Ccl2 and Il6 in heart tissues. k, Graphs showing protein levels of TNF-α, IL-1β, IL-6 and CCL2 in heart tissues. l, Representative TEM images (left) and mitochondria injury score (right). Scale bars, 5 μm. Each symbol represents a FOV. Each group has four mice and five FOV were randomly selected for assay from each animal. m, Graph showing the levels of ATP in heart tissue lysates. n, Graph showing the levels of serum lactate. (f–k,m,n, +MG, n = 5 mice; +non-Mac1, n = 9 mice; +Mac1, n = 9 mice). Each symbol represents one animal in c–k, n, m. Bars show as mean ± s.e.m. Two-sided P values were determined by one-way ANOVA followed by Games-Howell’s (c–h,j,n,m) or Tukey’s (i,k) multiple comparisons test. Results represent four independent experiments (b–n). See also Extended Data Figs. 9 and 10.
Compared to intrapericardial transplanted non-Mac1 cells or MG groups, hearts of septic mice that were transplanted with TREM2hi Mac1 cells showed significantly enhanced cardiac function (Fig. 7b–e). Consistently, the transplantation with TREM2hi Mac1 cells improved cardiac injury (Fig. 7f–i), inflammation (Fig. 7j,k), mitochondrial damage (Fig. 7l), increased ATP levels (Fig. 7m) and reduced lactate levels (Fig. 7n).
Next, WT Mac1 cells and Trem2−/− Mac1 cells were isolated from the hearts of WT or Trem2−/− mice and intrapericardially transplanted (2 × 105 cells per mouse) into the hearts of Trem2−/− mice immediately after CLP (Extended Data Fig. 10a). Three and 7 d later, echocardiography revealed that transplantation with WT Mac1 cells significantly enhanced cardiac function of septic mice compared to Trem2−/− mice injected with Trem2−/− Mac1 cells or MG groups (Extended Data Fig. 10b–d). Consistently, the transplantation with WT Mac1 cells improved cardiac injury, inflammation (Extended Data Fig. 10e–j), enhanced ATP levels (Extended Data Fig. 10k) and reduced lactate levels (Extended Data Fig. 10l). Taken together, these results suggest that administration of TREM2hi Mac1 cells may be a potential therapeutic approach for prevention and rescue of cardiac dysfunction in sepsis.
Discussion
Patients with SICM have a high mortality rate4,36,37 and their septic hearts are associated with a high energetic metabolism and catastrophic mitochondrial damage5,38. Macrophages are involved in cardiac inflammation, efferocytosis, tissue remodeling and homeostasis11,13,39; however, the roles of distinct macrophage subsets in the septic heart have not been fully elucidated. In the present study, we investigated a cardiac macrophage subset in septic and steady-state hearts, which was featured by high expression of TREM2 and the ability of self-renewal, defined as TREM2hi Mac1 cells. The high expression of TREM2 is essential for the function of Mac1 cells in scavenging defective mitochondria and protecting the septic heart (Extended Data Fig. 10m). In addition, intrapericardial injection of CD163+RETNLA+TREM2hi Mac1 cells could prevent the cardiac dysfunction of septic mice.
Accumulating studies highlighted the heterogenous composition and tissue-specific changes of macrophages at steady state and different pathological settings such as myocardial infarction8,11,19,22. Our scRNA-seq data revealed the presence of five macrophage populations, Mac1–5, in the steady-state heart of mice, which were indicated by a previous study19. Mac1 and Mac2 subsets were the dominant cardiac macrophage subsets, which were featured by the expression of phagocytosis (Trem2) and antigen-presenting (H2-Aa)-related genes, respectively. The Mac3–5 subsets had low proportions and highly expressed genes related to IFN stimulation (Irf7), pathological inflammation (CD72) and chemokines (Ccr2), respectively. We found that the Mac1 subset exhibited a dynamic change, decreased at 3 d and recovered at 7 d after CLP, which was well correlated with the deterioration and recovery of cardiac function in sepsis. Both Mac3 and Mac4 subsets were significantly increased along with exacerbated systemic inflammatory responses and bone marrow mobilization. A recent study showed that in a transverse aortic constriction model, CD72 was a key marker of a pro-inflammatory macrophage subset, in which many genes overlapped with Mac4 identified in our study22, suggesting that the CD72-expressed Mac4 subset is closely associated with inflammation and cardiac injury in SICM.
Regarding the origin, cardiac macrophages can be subdivided into embryo-derived resident CCR2− subsets (resident) and hematopoietic-derived recruited CCR2+ subsets (circulating)11,12,13. CCR2− subsets are replenished through local proliferation, mediate metabolic stability and promote cardiac recovery after injury14,19. In contrast, recruited CCR2+ subsets enhance inflammation and oxidative stress40. Dick et al. found that a TIMD4+LYVE1+MHC-IIloCCR2− resident cardiac macrophage subset, which was maintained by self-renewal, inhibits adverse remodeling and promotes the recovery of cardiac function after myocardial infarction14. In line with this study, we identified a subset of CD163+RETNLA+ CRMs (Mac1 cells), which was remarkably decreased upon septic stress. Upon SICM progression, resident Mac1 cells displayed a self-renewal capability and presented a protective role in septic heart. A recent manuscript also showed that the number of cardiac macrophages was decreased 1 d after CLP and then increased to supra-normal levels by 28 d41. The recovery of cardiac macrophage numbers was dominated by local proliferation, whereas peripheral recruitment accounted for only 6.2%41. These findings suggest an essential role of self-renewing resident Mac1 cells in protecting cardiac function in sepsis.
As a transmembrane receptor of the immunoglobulin superfamily, the activation of TREM2 leads to DAP12 phosphorylation and consequently promotes cell proliferation and survival, modulates phagocytosis and counteracts inflammation42,43. We observed that the Mac1 subset features TREM2hi and its proliferation was enhanced during sepsis. Furthermore, Trem2 deficiency did not affect the number of Mac1 cells in the healthy heart but significantly impaired the restoration of these macrophages in the septic heart. Our previous study showed that TREM2 was barely detectable in the liver, lung and spleen of steady-state mice, whereas TREM2 expression was significantly increased in these tissues after CLP and blockage of TREM2 exacerbated polymicrobial sepsis44, suggesting its protective effect against sepsis. The specific function of TREM2+ macrophages in other organs requires further investigation. Previous studies demonstrated that Trem2 deficiency resulted in a decrease of macrophage proliferation in pathological conditions45,46 and induced cell cycle arrest47. Similarly, we noticed that Trem2 deficiency remarkably decreased the proliferation of Mac1 cells in SICM. Our findings suggest that TREM2 play essential roles in the recovery of SICM through the reconstitution of CD163+RETNLA+ Mac1 subpopulation.
Extracellular mitochondria and mtDNA can trigger inflammation and cause cardiac damage29,30. A recent study showed that CRMs cleared extruded mitochondrial debris to preserve the homeostasis of the long-lived cardiomyocytes and played protective function14. Our present findings indicated that dysfunctional mitochondria were remarkably increased in the extracellular space of myocardium, paralleled by cardiac dysfunction under septic stress. Consistent with previous results41, sepsis showed a very slight effect on the survival of cardiac cells. Mac1 cells exhibited a potent ability to remove cardiomyocyte-derived abnormal mitochondria in SICM. Moreover, Trem2 ablation downregulated the endocytic genes in CRMs and exacerbated the accumulation of defective mitochondria in the heart and cardiac dysfunction. In addition, intrapericardial administration of CD163+RETNLA+TREM2hi macrophages could survive and engulf cardiomyocyte-derived defective mitochondria, which consequently protect against septic myocardial dysfunction. Our findings suggest that TREM2hi Mac1 cells had a powerful endocytosis function and preserved cardiac homeostasis by eliminating defective mitochondria during sepsis.
In summary, our study has revealed a CD163+RETNLA+ macrophage subset (Mac1), which features high expression of TREM2. TREM2 is essential for self-renewal and remodeling of Mac1 cells in the septic heart. TREM2hi Mac1 cells scavenge defective mitochondria and the transplantation of Mac1 cells contribute to the recovery of cardiac function during SICM. Our study highlights that maintaining the function of this subset by harnessing TREM2 could be a potential therapeutic strategy for cardiac diseases in clinic.
Methods
Mice
C57BL/6 WT mice were obtained from Shanghai SLAC Laboratory Animal Center. M. Colonna kindly provided Trem2−/− mice from Washington University in St. Louis. M. Shang from Zhejiang University kindly provided αMHCCre mice. The following mice were purchased from Jackson Laboratory: Rosa26-stop-tdTomato, Cx3cr1CreERT2–IRES–YFP, mtD2flox/+ (B6;129S-Gt(ROSA)26Sortm1(CAG−COX8A/Dendra2)Dcc/J) and B6;CD45.1 mice (B6.SJL-Ptprca Pepcb/BoyJ). αMHCCre mice were crossed with Rosa26-stop-tdTomato or mtD2flox/flox, to generate cardiomyocyte fluorescent reporter mice (referred to as CardRED mice) or cardiomyocyte-specific mitochondria fluorescent reporter mice (referred to as MitoCard mice). Mice were housed in a specific-pathogen-free environment, with a 12 h light–dark cycle and the ambient temperature was maintained at 22 °C. Mice were fed irradiated chow (SZS9126, XIETONG BIO-ENGINEERING) and sterile water. Cages were autoclaved and changed once a week. Only male mice were used in the experiments. The animal experiments were approved by the Animal Care and Use Committee of Zhejiang University School of Medicine and performed according to institutional guidelines (reference nos. 2017442 and 2021858).
Cecal ligation and puncture model
Anesthesia was administered to 8-week-old male mice with ketamine (50 mg kg−1) and xylazine (5 mg kg−1) intraperitoneally injected before abdominal depilation. Then mice were exposed to a 1.5-cm longitudinal cut in the lower quadrant of the abdomen and then the cecum was isolated. The distal three-quarters of the cecum was ligated with 4-0 silk sutures and two punctures were made in the ligated cecum with a 22-gauge needle, inducing sublethal CLP-induced sepsis. Then the cecum was placed back into the abdominal cavity after feces was extruded and the wound was sutured with a 4-0 silk suture. Each mouse was given 1 ml sterile saline after operation48. The survival rate was evaluated every 2 h for 7 d.
Fate mapping
To fate-label cardiac macrophage, Cx3cr1CreERT2–IRES–YFP mice were crossed to Rosa26-stop-tdTomato mice. At the age of 6 weeks, Cx3cr1CreERT2:Rosa26tdTom mice were administrated with 4 mg tamoxifen (Meilunbio) per mouse in corn oil and again 48 h later. After 5 weeks, mice were killed for assays.
Bone marrow chimeric mouse model
Bone marrow cells were obtained from femurs and tibias of 8-week-old donor male mice. First, bone marrow was flushed out with precooled PBS. After red blood cell lysis, a single-cell suspension was obtained through a 70-µm filter. A total of 1 × 107 bone marrow cells were transferred into lethally irradiated (8 Gy) recipient male mice intravenously. The chimeric mice were given antibiotics through drinking water for 1 week. After 8 weeks, the model construction was completed.
Generation and delivery of adeno-associated virus
Recombinant adeno-associated virus serotype 2/9 (AAV 2/9) vectors carrying mt-Keima with the cardiac-specific promoter cTnT were designed as previously described14 and manufactured by OBiO Technology. Each mouse was administered with a titer of 3 × 1011 AAV9-Tnnt2-mt-Keima virus via intra-cardiac injection. After 6 weeks, mice were used for CLP experiments.
RNA extraction and RT–qPCR
Total RNA was obtained from mouse tissues and cells using TRIzol (Ambion). Complementary DNA samples were synthesized with PrimeScript RT reagent kit (Takara) and gene expression was analyzed with qPCR using SYBR premix Ex Taq (Takara) on Lightcycler 480 system (Roche). Primer sequences used are listed in Supplementary Table 1.
Echocardiography
Echocardiography was administrated in mice under isoflurane anesthesia using a Vevo 2100 system (VisualSonics). Normal body temperature was maintained by a heating platform. Parasternal standard two-dimensional (2D) and M-mode short-axis view was used to measure the left ventricular (LV) internal dimensions at diastole (LVIDd) and systole (LVIDs), the LV internal volume at diastole (LVEDV) and systole (LVESV) and the heart rate (HR). The LV EF was acquired as ((LVEDV − LVESV) / LVEDV) × 100, the LV FS was acquired as ((LVIDd−LVIDs) / LVIDd) × 100 and CO was acquired as (LVEDV − LVESV) × HR. For diastolic dysfunction analysis, a 2D apical view was used by pulsed-wave Doppler. Early and late diastolic velocity peak waves (E and A, respectively) were detected and the E/A ratio was acquired. Image analysis was performed offline using Vevo LAB v.3.1.0 (VisualSonics).
Immunofluorescence
Fresh hearts were embedded with optimal cutting temperature compound and cut into 10-μm sections. After drying in air for 1 h, slides were fixed in 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 15 min and blocked with blocking buffer (3% BSA + 5% FBS + 0.1% Tween 20) for 30 min at room temperature. Then tissue sections were incubated at 4 °C overnight with primary antibodies against rat anti-CD68 (1:250 dilution; Abcam), mouse anti-CD163 (1:100 dilution; Santa Cruz), rat anti-TREM2 (1:100 dilution; R&D), rabbit anti-Tom20 (1:200 dilution; Abcam), rat anti-LAMP1 (1:50 dilution, DSHB) and cTnI (1:250 dilution; Abcam). After being washed three times with PBST for 5 min each, slides were incubated with conjugated secondary antibodies at room temperature for 2 h, followed by washing three times with DPBS and sealing with DAPI-containing blocking agent. To analyze cardiomyocyte apoptosis, TUNEL staining was performed with a TUNEL kit according to the manufacturer’s instructions. Images were acquired with Olympus OSR confocal microscope (OLYMPUS IX83-FV 3000-OSR) or Zeiss LSM 880 (with fast AiryScan) and processed with by ImageJ Pro Plus v.6.0 software (Media Cybernetics).
To analyze the cardiomyocyte-derived vesicles containing mitochondria, samples from CardRED mice were stained with rabbit anti-Tom20 primary antibody (1:250 dilution, Abcam) and Alexa fluor 488-conjugated donkey anti-rabbit secondary antibody (1:500 dilution, Invitrogen). Images were acquired using Zeiss LSM 880 (with AiryScan) at an interval of 0.38 μm per z-step (z = 7–8 μm) for 3D reconstructions. A 3D reconstruction analysis of above experiments was processed using Imaris software v.9.7 (Bitplane). After reconstructing cardiomyocytes, vesicles and mitochondria using a surface tool based on 0.2-mm detail and absolute intensity, we analyzed the distance between Tom20+tdTomato+ vesicles and cardiomyocytes to show that vesicles were outside of cardiomyocytes. Then we quantified Tom20+tdTomato+ vesicles on five random FOVs per sample.
Mt-Keima fluorescence was imaged in two channels by two sequential excitations (488 nm, green; 561 nm, red) using a Zeiss LSM 880 (Zeiss LSM 880 with fast AiryScan). Identical imaging settings were maintained to compare under different experimental conditions. All images were analyzed with ImageJ Pro Plus v.6.0 software (Media Cybernetics) to calculate the ratio of red fluorescent protein to green fluorescent protein area.
All antibodies applied are listed in Supplementary Table 2.
Transmission electron microscopy
Collected heart samples were obtained from the anterior ventricular wall and cut into 2-mm3 cubes (2 × 1 × 1 mm). These samples were fixed in 2.5% glutaraldehyde (pH 7.2) for 2 d at room temperature. Then the cubes were embedded with epoxypropane resin following standard methods. The slices were observed and scanned using a Tecnai G2 Spirit 120 kV TEM (Thermo FEI) with a charge-coupled device camera.
Semi-quantitative analysis of electron microscopic specimens
Myocardial mitochondria were observed using TEM and analyzed semi-quantitatively as described previously49. Five FOVs were randomly selected for photography at the same magnification and approximately 20 mitochondria in each field were randomly chosen for analysis. Each sample was analyzed for 100 mitochondria. Each mitochondrion was graded on a scale of 0–4 according to the degree of injury (higher scores indicated more severe injury).
Detection of molecular markers in serum or heart
According to the manufacturer’s instructions, serum lactate levels were detected using a Lactic Acid Assay kit (Nanjing Jiancheng), serum LDH levels were tested using a Lactate Dehydrogenase Assay kit (Nanjing Jiancheng) and serum cTnI levels were determined using a mouse cTnI ELISA kit (Cloud-Clone). ATP content in the heart was measured using an ATP Assay kit (Beyotime) and serum ANP and NT-proBNP levels were determined using mouse ELISA kits (USCN Life Science). IL-1β, IL-6, TNF-α and CCL2 protein levels in heart tissue lysates were determined using mouse ELISA kits (NOVUS (IL-1β and IL-6) and MULTI SCIENCES (TNF-α and CCL2)).
Isolation of cells from mouse heart tissue
Mice were anesthetized by isoflurane inhalation. Afterward, the hearts were perfused with 20 ml perfusion buffer (1× DPBS with 0.8 mM CaCl2) to remove peripheral blood from the chambers. Subsequently, atria and valves of the isolated heart were removed and ventricles were minced to ~1-mm cubes. The hearts were digested with 0.25 mg ml−1 of Liberase TL (Sigma-Aldrich), 20 μg ml−1 DNase I (Sinopharm Chemical Reagent) and 10 mM HEPES (Sigma-Aldrich) in serum-free DMEM (Gibco) at 37 °C for 15 min. Then, the tissue suspension was triturated using 1,000-μl micropipettes. The resulting cell suspension was obtained through a 70-μm filter to remove undigested tissue mass, washed with PBS containing 2% FBS and diluted in 1 ml HBSS. Then the cell suspension was spread on a 15% and 60% Percoll (Yeason) top layer and subjected to density gradient centrifugation at 400g for 20 min. The cell layer between the liquid surfaces was collected and washed with 10 ml DPBS to obtain single-cell suspensions for further experiments.
Flow cytometry and fluorescence-activated cell sorting
For flow cytometric analysis, the single-cell suspensions were stained with relative antibodies at 4 °C for 30 min in a final volume of 200 μl. Cells were washed with DPBS, resuspended in a final volume of 400 μl and filtered through a 40-μm filter. Samples were collected on a BD Fortessa (BD Biosciences) by BD FACSDiva software v.8.0.1 and analyzed with FlowJo v.10.6.2 software (TreeStar). Details of the immunostaining and gating strategy of immune cells and cardiac macrophage subpopulations in the septic heart are shown in Extended Data Figs. 2d and 3b.
For measurement of mitochondrial function, L-929 fibroblasts were used as a control to evaluate treatment responsiveness compared to isolated vesicles. Oligomycin (Cell Signaling Technology, 5 μM) was employed to promote mitochondrial membrane hyperpolarization. Carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (Sigma-Aldrich, 2 μM) was applied to uncouple oxidative phosphorylation in mitochondria and depolarize mitochondrial membranes. Both isolated vesicles and L-929 cells were administrated at 37 °C for 1.5 h. Then, cells or vesicles were stained in culture medium supplemented with 1× MitoNIR (Abcam), which is a fluorescent probe measuring mitochondrial membrane potential, for 20 min at 37 °C. Cells or vesicles were acquired by a BD Fortessa (BD Biosciences). MFI was analyzed using FlowJo software (TreeStar).
We assessed Ki67 expression by fixing and permeabilizing cells with a Foxp3 Transcription Factor Staining Buffer Set (Thermo Fisher Scientific) according to the manufacturer’s instructions and then staining cells with anti-Ki67 antibody as mentioned above.
For FACS sorting, the single-cell suspensions were stained with relative antibodies for 30 min at 4 °C and then were washed with DPBS. Pellets were resuspended with 200 μl DPBS. For scRNA-seq experiments, Calcein Blue AM (Thermo Fisher Scientific, 4 μg ml−1) and Vybrant DyeCycle Ruby (Thermo Fisher Scientific, 10 μM) were added in 200 μl solutions and were incubated for 10 min at 4 °C. After incubation, live nucleated CD45+ cells were obtained on a Beckman MoFlo Astrios EQ with a 100-μm nozzle (Beckman). For Mac1 transplantation experiments, TREM2hi Mac1 (CD45+ CD11b+F4/80+CD163+RETNLA+) and non-Mac1 (CD45+CD11b+F4/80+ and non-CD163+RETNLA+) cardiac macrophages were sorted and collected separately for further experiments.
Single-cell RNA-seq
Single cells were collected into RPMI 1640 containing 5% FBS. Trypan blue exclusion confirmed the cell viability. Sorted cells were counted and concentration adjusted to 700–1,200 cells μl−1. Then single-cell suspensions were loaded to a 10x Chromium to capture no more than 10,000 single cells by Chromium Single Cell 3′ Reagent kits v.3 (10x Genomics). The cells were partitioned into Gel Beads in the Chromium instrument, where cell lysis and barcoded reverse transcription of RNA occurred. DNA amplification and library construction were conducted. Libraries were sequenced on a Novaseq 6000 (Illumina) by LC-Bio Technology. The data were aligned to the GRCm38 reference genome using CellRanger v.4.0 (10x Genomics).
Dimension reduction, clustering and differentially expressed gene analysis
The CellRanger output was loaded into Seurat (v.3.1.1) for unsupervised clustering. All genes expressed in fewer than one cell were removed. Cells expressing fewer than 200 and more than 6,000 genes, unique molecular identifier counts more than 40,000 and the percent of mitochondrial DNA (mtDNA) gene expression more than 20% were excluded. Mitochondrial genes were excluded from the expression matrix.
To visualize the data, we used the Seurat package for further analysis. First, we used the LogNormalize method of the Normalization function of the Seurat package to assess the expression value of genes. Second, we performed principal-component analysis on the normalized expression matrix with highly variable genes identified by FindVariableGenes function. Based on the top ten principal components, we obtained the unsupervised cell cluster result by weighted shared nearest neighbor graph-based clustering method. To detect cluster-specific genes, we identified the marker genes by the bimod (Likelihood-ratio test) of the FindAllMarkers function in Seurat. Compared to other clusters, the genes whose expression was more than 25% of the cells and average log (fold change) >0.26 in the target cluster, were defined as marker genes. Cell types were defined based on known markers. Cells expressing non-immune cell markers were excluded.
Cell re-clustering, marker gene visualization, DEG analysis, GO enrichment analysis and other bioinformatics analyses were performed with the OmicStudio tools (https://www.omicstudio.cn/tool). DEGs were identified by the bimod with default parameters through the FindAllMarkers function under the following criteria: (1) log (fold change) >0.26; (2) P value <0.01; and (3) min.pct > 0.1.
Monocytes and macrophages from the four WT samples are shown in Fig. 2a–d. Fig. 4e,f shows Trem2−/− samples and WT littermate control samples 7 d after CLP, aggregated and analyzed as described above to compare the impact of Trem2−/− on the monocyte-macrophage compartment.
Single-cell trajectory analysis
For single-cell trajectory analysis, we used Monocle v.2.4.0 to investigate developmental trajectories between macrophage/monocyte subsets19,27. scRNA-seq data were scaled, normalized and clustered by the Seurat tool, then downsampled to 1,500 cells and loaded into a Monocle object. The DifferentialGeneTest function was used to infer DEGs from each cluster and we used the top 800 genes with the lowest q value to sequence the cells in pseudotime analysis. The developmental trajectory was plotted by plot_cell_trajectory command. The ‘root_state’ (the starting point) of the trajectory was defined as the terminal with lower expression of monocyte genes. After the cell trajectories were constructed, DEGs along the pseudotime were detected by the DifferentialGeneTest function. Changes in the top 50 genes through pseudotime with the lowest q values were visualized by a heat map using the plot_pseudotime_heatmap function. Plot_genes_in_pseudotime function was used to show the dynamic trend of the selected significant gene expression levels.
Preparation of cardiac vesicles for flow cytometry and sorting
CardRED mice hearts were cut into small pieces and digested in DPBS with 0.25 mg ml−1 of Liberase TL (Sigma-Aldrich), 20 μg ml−1 DNase I (Sinopharm Chemical Reagent) and 10 mM HEPES (Sigma-Aldrich) for 15 min at 37 °C. Then, tissue suspensions were acquired by gentle pipetting. We serially centrifuged these suspensions at 50g and 300g and discarded the pellet. The supernatant was then centrifuged at 1,000g and washed with DPBS. Pellets were resuspended with 200 μl DPBS. We used the endogenous expression of tdTomato, CD31 and Vybrant DyeCycle Ruby (Thermo Fisher Scientific, 10 μM) to define cardiac vesicles. The gating strategy for cardiac vesicles is shown in Extended Data Fig. 6c. The material was sorted in a Beckman MoFlo Astrios EQ (Beckman) with a 100-μm nozzle.
Intrapericardial injection of sorted cardiac macrophages
FACS-sorted cells were washed with DPBS and quantified. Cardiac macrophages (2 × 105 cells) were resuspended in 25 μl MG (Corning). After anesthetizing and intubating the recipient mice, the mouse chest was opened and MG containing cardiac macrophages was injected into the pericardial cavity using a 30-gauge needle. The control animals were only intrapericardially injected with 25 μl MG.
For cell survival and distribution experiments, the sorted Mac1 and non-Mac1 cells were stained with CMTMR (50 nM) at 37 °C for 20 min and then washed with DPBS. Labeled macrophages (2 × 105 cells) were resuspended in 25 μl MG and then transplanted into the pericardial cavity immediately after CLP. Three days later the mice were killed. Heart samples were collected and cryosections were made. Images were acquired using a Zeiss LSM 880 (with fast AiryScan) according to standard procedures and analyzed with ImageJ Pro Plus v.6.0 software.
Protein array assay
Total protein was isolated from the mouse heart using RIPA (Applygen) and the supernatant was analyzed by a glass-based and sandwich-based antibody microarray to measure 20 cytokines quantitatively (RayBiotech). Each cytokine was analyzed in quadruplicate per array. Then, 100 µl supernatant were loaded to each well, incubated overnight at 4 °C and then extensively washed. A biotin-labeled detection antibody was incubated for 2 h followed by the application of AlexaFluor 555-conjugated streptavidin at room temperature for 1 h. The slides were analyzed by an InnoScan 300 Scanner (Innopsys) with 532 nm excitation and 635 nm emission. We obtained raw data (images) from the scanner and integrated spot intensities (tab-delimited .txt file) using Mapix v.7.3.1 software. Data visualization was obtained by Q-Analyzer Software (RayBiotech). All raw data were log2-converted for statistical analysis.
Statistical analysis
All statistical analyses were performed using SPSS v.21.0 (IBM) or Prism v.8.0 (GraphPad Software). For comparison between two groups, normally distributed data were evaluated by the unpaired two-tailed Student’s t-test and data without a normal distribution were evaluated by the Mann–Whitney U-test. For more than two groups, normally distributed data were evaluated by one-way or two-way ANOVA, followed by Games-Howell’s, Sidak’s or Tukey’s multiple comparisons test and data without a normal distribution were evaluated by the Kruskal–Wallis with Dunn’s multiple comparison test. Correlations were analyzed by Pearson’s correlation coefficient. The survival rate was analyzed by Kaplan–Meier analysis and log-rank test. Data are presented as mean ± s.e.m. or median with interquartile range
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
scRNA-seq data for this study have been deposited at the Gene Expression Omnibus under accession code GSE190856. All data and materials are available in the paper and the supplementary information. Source data are provided with this paper.
References
Rudd, K. E. et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395, 200–211 (2020).
Singer, M. et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315, 801–810 (2016).
Ammann, P. et al. Elevation of troponin I in sepsis and septic shock. Intensive Care Med. 27, 965–969 (2001).
Bréchot, N. et al. Venoarterial extracorporeal membrane oxygenation to rescue sepsis-induced cardiogenic shock: a retrospective, multicentre, international cohort study. Lancet 396, 545–552 (2020).
Hollenberg, S. M. & Singer, M. Pathophysiology of sepsis-induced cardiomyopathy. Nat. Rev. Cardiol. 18, 424–434 (2021).
Lelubre, C. & Vincent, J.-L. Mechanisms and treatment of organ failure in sepsis. Nat. Rev. Nephrol. 14, 417–427 (2018).
Swirski, F. K. & Nahrendorf, M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 18, 733–744 (2018).
Heidt, T. et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 115, 284–295 (2014).
Pinto, A. R. et al. Revisiting cardiac cellular composition. Circ. Res. 118, 400–409 (2016).
Williams, J. W. et al. Macrophage biology, classification, and phenotype in cardiovascular disease: JACC macrophage in CVD series (part 1). J. Am. Coll. Cardiol. 72, 2166–2180 (2018).
Sansonetti, M. et al. Resident cardiac macrophages: crucial modulators of cardiac (patho)physiology. Basic Res. Cardiol. 115, 77 (2020).
Bajpai, G. et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24, 1234–1245 (2018).
Alvarez-Argote, S. and C.C. O’Meara, The evolving roles of cardiac macrophages in homeostasis, regeneration, and repair. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22157923 (2021).
Nicolás-Ávila, J. A. et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183, 94–109 (2020).
Piquereau, J. et al. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy 9, 1837–1851 (2013).
Brealey, D. et al. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am. J. Physiol. Regulatory, Integr. Comp. Physiol. 286, R491–R497. (2004).
Brealey, D. et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360, 219–223 (2002).
Pinto, B. B. et al. Improved survival in a long-term rat model of sepsis is associated with reduced mitochondrial calcium uptake despite increased energetic demand. Crit. Care Med. 45, e840–e848 (2017).
Dick, S. A. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39 (2019).
Farbehi, N. et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife https://doi.org/10.7554/elife.43882 (2019).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481–1487 (2017).
Ni, S.-H. et al. Single-cell transcriptomic analyses of cardiac immune cells reveal that Rel-driven CD72-positive macrophages induce cardiomyocyte injury. Cardiovasc. Res. 118, 1303–1320 (2022).
Martini, E. et al. Single-cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation 140, 2089–2107 (2019).
Epelman, S., Lavine, K. J. & Randolph, G. J. Origin and functions of tissue macrophages. Immunity 41, 21–35 (2014).
Narasimhan, P. B. et al. Nonclassical monocytes in health and disease. Annu. Rev. Immunol. 37, 439–456 (2019).
Forte, E. et al. Cross-priming dendritic cells exacerbate immunopathology after ischemic tissue damage in the heart. Circulation 143, 821–836 (2021).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Melentijevic, I. et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 542, 367–371 (2017).
Oka, T. et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 (2012).
Zhou, R. et al. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Pham, A. H., McCaffery, J. M. & Chan, D. C. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genes 50, 833–843 (2012).
Sun, N. et al. Measuring in vivo mitophagy. Mol. Cell 60, 685–696 (2015).
Sun, N. et al. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 12, 1576–1587 (2017).
Bruttger, J. et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity https://doi.org/10.1016/j.immuni.2015.06.012 (2015).
Mildner, A. et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 10, 1544–1553 (2007).
Jeong, H. S. et al. Risk factors and outcomes of sepsis-induced myocardial dysfunction and stress-induced cardiomyopathy in sepsis or septic shock: a comparative retrospective study. Medicine 97, e0263 (2018).
Poelaert, J. et al. Left ventricular systolic and diastolic function in septic shock. Intensive Care Med. 23, 553–560 (1997).
Martin, L. et al. The septic heart: current understanding of molecular mechanisms and clinical implications. Chest 155, 427–437 (2019).
Frodermann, V. & Nahrendorf, M. Macrophages and cardiovascular health. Physiol. Rev. 98, 2523–2569 (2018).
Bajpai, G. et al. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 124, 263–278 (2019).
Hoyer, F. F. et al. Tissue-specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity https://doi.org/10.1016/j.immuni.2019.10.010 (2019).
Deczkowska, A., Weiner, A. & Amit, I. The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell 181, 1207–1217 (2020).
Ulland, T. K. & Colonna, M. TREM2 - a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 14, 667–675 (2018).
Chen, Q. et al. Triggering receptor expressed on myeloid cells-2 protects against polymicrobial sepsis by enhancing bacterial clearance. Am. J. Resp.Crit. Care Med. 188, 201–212 (2013).
Seno, H. et al. Efficient colonic mucosal wound repair requires Trem2 signaling. PNAS 106, 256–261 (2009).
Wang, Y. et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675 (2016).
Zheng, H. et al. TREM2 promotes microglial survival by activating Wnt/β-catenin pathway. J. Neurosci. 37, 1772–1784 (2017).
Rittirsch, D. et al. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 4, 31–36 (2009).
Flameng, W. et al. Ultrastructural and cytochemical correlates of myocardial protection by cardiac hypothermia in man. J. Thorac. Cardiovasc. Surg. 79, 413–424 (1980).
Acknowledgements
We dedicate this paper to Prof. Andreas Hoeft (Department of Anesthesiology and Intensive Care Medicine, University Hospital of Bonn), who unfortunately passed away before the paper was submitted for publication. Prof. Hoeft played an important role in this study and we give special thanks for his contribution to its conceptualization. We also thank D. Neculai (Department of Cell Biology and Department of Pathology Sir Run Run Shaw Hospital, Zhejiang University School of Medicine) and M. Guan (Institute of Genetics, Zhejiang University) for helpful discussion and linguistic advice. We thank C. Yang (Center of Cryoelectron Microscopy, Zhejiang University); L. Xie and J. Li (Center of Electron Microscopy, Zhejiang University); S. Liu, G.Xiao, J. Xuan, X. Song and Y. Li (Core Facilities, School of Medicine, Zhejiang University) for technical assistance; L. Wang and H. Lou (Laboratory Animal Center, Zhejiang University) for support on animal studies; J. Zhao and J. Lin (The Second Affiliated Hospital, Zhejiang University School of Medicine) for help in echocardiography, X. Wang and H. Zhu (The First Affiliated Hospital, Zhejiang University School of Medicine) for help with cardiomyocyte isolation and statistical analysis. We also thank staff at LC-Bio Technology for sequencing services. This study was partially supported by the Natural Science Foundation of China (82230074, 81720108025 to X.F., 82072221 to K.Z., 81971809 to Y.J. and 92049108 to X.L.), the Natural Science Foundation of Zhejiang Province (LZ22H150002 to K.Z.) and the National Key Research and Development Program of China (2018YFC2001904 to X.F. and 2017YFE0196600 to X.L.).
Author information
Authors and Affiliations
Contributions
K.Z., Y.W., S.C., J.M. and X.F. conceptualized and designed the project. K.Z. and Y.W. conducted flow cytometry studies and performed scRNA-seq analysis. K.Z. S.C., J.M., H.Y., C.G. and Y.Z. performed in vivo studies. S.C., J.M., X.L., Y.J., X.C. and X.H. performed in vitro experiments. Y.J. and S.C. conducted statistical analysis of the data. A.H. provided critical insights and advice. X.F., X.L., K.Z., S.C., Q.C. and Y.W. obtained the data and wrote the manuscript. X.F., X.L. and Q.C. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Metabolism thanks Andres Hidalgo, José Ángel Nicolás and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ashley Castellanos-Jankiewicz, in collaboration with the Nature Metabolism team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 The dynamic changes of cardiac function, injury biomarkers and inflammatory cytokines in SICM, related to Fig. 1.
a-d, Representative echocardiography images (a) and EF% (b), FS% (c), and CO (d) measured by echocardiography in Wild-type (WT) mice at steady state (SS), 1, 3, 7, and 21 days after CLP (SS, n = 6 mice; CLP 3 d, n = 6 mice; CLP 7 d, n = 5 mice; CLP 21 d, n = 6 mice). e,f, Graphs showing the levels of cTnI (e) and LDH (f) in the serum. g,h, Graphs showing mRNA levels of Anp (g), Bnp (h) in the heart tissues; n = 5-6 (e,f,g,h) mice for each treatment. i, Heat map showing the scaled expression of 14 selected cytokines (row) across 22 heart tissue lysate samples (column), colored by different conditions. In b-h, each symbol represents one animal and data are presented as mean ± SEM. Two-sided P values were determined by one-way ANOVA followed by Games-Howell’s (b-d,f) or Tukey’s (g) multiple comparisons test, Kruskal–Wallis with Dunn’s multiple comparison test (e,h). Exact P values are shown. Results represent four independent experiments (b-h).
Extended Data Fig. 2 Single-cell characterization and flow cytometric analysis of the cardiac immune cells during sepsis progression, related to Fig. 1.
a, Violin plots of average unique molecular identifier (UMI) (left) and gene (right) numbers of scRNA-seq at different stages of sepsis. Each dot represents a cell. b, UMAP plots showing the expression of selected marker genes for the defined cell types in Fig. 1c. c, Violin plots showing the expression of selected marker genes in all defined cell types corresponding to Fig. 1c. d, Gating strategy of 10-color flow cytometry for identifying various types of immune cells in murine hearts. e, Graph showing percentages of cardiac immune cells during sepsis progression (n = 4 - 5 mice for each treatment). Each symbol represents one animal in e. Data are presented as mean ± SEM. Two-sided P values were determined by one-way ANOVA followed by Tukey’s multiple comparisons test, n.s., not significant. In e, experiments were performed four times.
Extended Data Fig. 3 Identification of Mac1 subset in the septic heart, related to Fig. 2.
a, UMAP plots showing the expression of selected marker genes for the monocyte-macrophage subpopulations. b, Gating strategy to identified Mac1 cells (CD45+CD11b+F4/80+CD163+RETNLA+). c, Graph showing percentages of CD163+RETNLA+ cardiac macrophages during sepsis progression (SS, n = 8 mice; CLP 3 d, n = 8 mice; CLP 7 d, n = 9 mice; CLP 21 d, n = 10 mice). Each symbol represents one animal. Bars show mean ± SEM. Two-sided P values were determined by one-way ANOVA with Games-Howell’s multiple comparisons test. d, Representative immunofluorescence images showing the presence of CD163 (green), CD68 (red), and nuclei (blue) in cardiac tissues during sepsis progression. n = 6 mice per group. Scale bars, 20 μm. Exact P values are shown. Results represent four independent experiments (c,d). SS, steady state.
Extended Data Fig. 4 Developmental trajectory and lineage tracing track the fate of Mac1 subset in SICM, related to Fig. 3.
a, Monocle prediction of monocyte-macrophage compartment developmental trajectory with pseudotime mapped along. b, Cluster-defining gene expression plotted along the pseudotime, with Seurat’s cluster mapped along. c, Schematic illustration of lineage tracking Mac1 cells in Cx3cr1CreERT2:Rosa26tdTom mice. d, Representative contour plots showing the expression of tdTomato (CX3CR1) on gated CD115+CD11b+ blood monocytes, CD45+CD11b+F4/80+ cardiac macrophages, CD45+CD11b+F4/80+CD163+RETNLA+ macrophages and graph showing percentages of tdTomato-labeled cells in those cells (0 week, n = 4 mice; 1 week, n = 5 mice; 3 weeks, n = 5 mice; 5 weeks, n = 2 mice; 6 weeks, n = 3 mice). Bars are mean ± SEM. e, Representative contour plots showing the expression of Ki67 on gated CD45+CD11b+F4/80+CD163+RETNLA+ macrophages and graph showing percentages of Ki67 expressing in CD45+CD11b+F4/80+CD163+RETNLA+ macrophages at steady state (SS), 3 days and 7 days after CLP. Each symbol represents one animal. Data are presented as mean ± SEM. Two-sided P values were determined by one-way ANOVA with Games-Howell’s multiple comparisons test. Exact P values are shown. Results represent five (d) and two (e) independent experiments.
Extended Data Fig. 5 TREM2 is highly expressed on Mac1 subset and essential for Mac1 remodeling during SICM, related to Fig. 4.
a, Volcano plots showing the gene expression fold change (x-axis, log2 scale) and p-value significance (y-axis, -log10 scale) in Mac1 subset of steady state (SS) versus Mac2 subset (left), Mac3 subset (middle) or Mac4 subset (right) of 3 days after CLP. Selected significant genes were labeled. Two-sided P values were determined by the Mann-Whitney U test. b, Violin plots showing the expression of Trem2 in all defined immune cell types corresponding to Fig. 1c. c, Graph showing the quantification of mean fluorescence intensity (MFI) of TREM2 expression in CD45+CD11b+F4/80+CD163+RETNLA+ and CD45+CD11b+F4/80+(non-CD163+RETNLA+) cardiac macrophages at steady state and 7 days post-CLP (n = 8 mice for each group). Each symbol represents one animal. Data are presented as mean ± SEM. Two-sided P values were determined by Kruskal–Wallis with Dunn’s multiple comparison test. d, UMAP plots showing cell clustering results of the monocyte-macrophage compartment from hearts of WT littermate controls and Trem2-KO mice at steady state and 3 days after CLP. Monocyte-macrophages under each condition are downsampled to 1000 cells. The subpopulations obtained by Seurat were identical to those previously identified in Fig. 2a according to their marker genes. e, Bar plots showing cluster distribution of the monocyte-macrophage subsets in WT littermate controls and Trem2-KO mice at steady state and 3 days after CLP. f, Graph showing percentages of cardiac immune cells in WT controls and Trem2-KO mice at 0, 3 and 7 days post-CLP (n = 4-5 mice for each treatment). Every symbol represents a mouse. Data are presented as mean ± SEM, Two-sided P values were determined by Mann-Whitney U test, n.s., not significant. Results represent three independent experiments (c,f).
Extended Data Fig. 6 Sepsis induces extracellular damaged mitochondria accumulation and TREM2hi Mac1 cells eliminate cardiomyocyte-derived mitochondria during sepsis, related to Fig. 5.
a, TEM images showing vesicles in hearts of WT mice 3 days post-CLP. b, TEM images showing cardiac interstitial mitochondria vesicles (red arrowheads) of WT mice at steady state (SS) or 3 days post-CLP. Bar graph showing free mitochondria numbers per field of view (FOV). Each symbol represents a FOV. Five fields of view are randomly selected for assay from each animal (n = 3). c, Schematic illustration of the gating strategy for collecting vesicles from hearts of mice. d, Flow cytometric analysis of purified vesicles stained with the mitochondria-selective probes MitoTracker Green and MitoNIR (n = 3 per group). e, Mitochondrial membrane potential in cardiac vesicles and cultured fibroblasts as assessed by MFI levels of MitoNIR in basal conditions and in the presence of depolarizing (FCCP) or hyperpolarizing (Oligomycin) agents (n = 3-4 per group). f, Pseudo-colored representative TEM images of two mononuclear cells (grey) taking up vesicles containing mitochondria (brown) from cardiomyocytes (green) of WT mice 7 days post-CLP. g, The incorporation of cardiomyocyte-derived tdTomato protein in CD45+CD11b+F4/80+CD163+RETNLA+ macrophages, CD45+CD11b+F4/80+ (non-CD163+RETNLA+) macrophages and CD45+CD11b+F4/80-LY6C+ monocytes from αMHCCre/+ (control) and αMHCCre/+:Rosa26TdTom (CardRED) mice at SS and 7 days post-CLP (control mice, n = 4; CardRED SS, n = 5; CardRED CLP 7d, n = 8). h, The incorporation of cardiomyocyte-derived mt-Dendra2 protein in CD45+CD11b+F4/80+CD163+RETNLA+ macrophages, CD45+CD11b+F4/80+(non-CD163+RETNLA+) macrophages and CD45+CD11b+F4/80-LY6C+ monocytes from αMHCCre/+ (control) and αMHCCre/+:mtD2Flox/Flox (MitoCard) mice at SS and 7 days post-CLP (control mice, n = 4; MitoCard SS, n = 6; MitoCard CLP 7d, n = 6). i, Proportions of TUNEL+ and TUNEL+cTnI+ cells in hearts of WT mice at SS and 3 days post-CLP (n = 6 mice per group). All scale bars are indicated in the images. Data are presented as mean ± SEM (b,e,g,h,i). Two-sided P values were determined by Mann-Whitney U test (b), or by Kruskal–Wallis with Dunn’s multiple comparison test (e,g,h,i). Results represent three (a,b,d,g-i) and two (e,f) independent experiments.
Extended Data Fig. 7 Trem2 deficiency impairs the uptake of cardiomyocyte-derived mitochondria by Mac1 subset in septic heart, related to Fig. 5.
a, Schematic illustration of the cardiomyocyte mitochondria labeled with AAV9-Tnnt2-mt-Keima virus. Right panel showing the mt-Keima fluorescence signals in hearts of WT mice at weeks 0, 3 and 6 after virus infection respectively. Keima-tagged mitochondria were indicated by different fluorescence in neutral (Keima 458 nm; green) and acidic (Keima 561 nm; red) environments. Scale bars, 20 μm. b, Scatter plots showing the average expression levels (log10(FPKM+1) scale) of total macrophage populations from WT littermate controls (y-axis) and Trem2-/- mice (x-axis). c, Violin plots showing the selected endocytic gene expression in total macrophage from WT littermate controls and Trem2-/- mice. d, Study protocol of bone marrow transplantation from WT or Trem2-/- donors into CardRED recipient mice. e,f, Representative TEM images showing cardiac interstitial mitochondria vesicles from WT and Trem2-/- mice at steady state (f) and 7 days post-CLP (e), Scale bars, 5 μm. Bar graph showing the quantification of free mitochondria numbers per field of view (FOV). Each symbol represents a FOV (n = 3 mice for each treatment). Five fields of view are randomly selected for assay from each animal. Data are presented as mean ± SEM (e,f). Two-sided P values were determined by Mann-Whitney U test (e,f). Results represent two independent experiments (e,f).
Extended Data Fig. 8 Trem2 deficiency exacerbates cardiac dysfunction in sepsis, related to Fig. 6.
a-i, WT and Trem2-/- mice were subjected to CLP and cardiac function was examined at steady state (SS) and 3, 7 and 21 days post-CLP. Graph showing FS% (a); CO (b) measured by echocardiography; Graph showing the levels of LDH in the serum (c); Graphs showing mRNA levels of Anp (d), Bnp (e), Tnfα (f), Il-1β (g), Ccl2 (h) and Il-6 (i) in the heart tissue lysates (a-b, n = 7–8 mice; c, n = 6-8 mice; d-i, n = 6-7 mice for each treatment). j, Study protocol of bone marrow transplantation (BMT) from WT and Trem2-/- donors (CD45.2 background) into WT recipients (CD45.1 background). k, Representative contour plots of CD45.1 and CD45.2 expressions on gated CD45+CD11b+F4/80+CD163+RETNLA+ Mac1 subset in recipient mice 1, 3, 5 and 8 weeks after BMT. l, Graph showing percentages of CD45.2+ cells in CD11b+F4/80+CD163+RETNLA+ Mac1 subset after BMT (n = 4-7 mice for each group).m, Representative contour plots of TREM2 expression on gated CD45+CD11b+F4/80+CD163+ macrophages in hearts of both WT→WT chimeras (Trem2+/+Ch) and Trem2-/-→WT chimeras (Trem2-/-Ch) 8 weeks after BMT. Graph showing Trem2 mRNA levels in heart tissue lysates (n = 6 mice for each group). n, Graphs showing mRNA levels of Anp and Bnp in the heart tissue lysates 7 days post-CLP (n = 11 mice for each group). o, Graphs showing mRNA levels of Tnfα, Il1β, Ccl2 and Il6 in the heart tissue lysates 7 days post-CLP (n = 11 mice for each group). Each symbol represents one animal. All data are presented as mean ± SEM. Two-sided P values were determined by Mann-Whitney U test (a-i,m-o) or by Kruskal-Wallis with Dunn’s multiple comparison test (l). Results represent at least four (a-i,l) and three (m-o) independent experiments.
Extended Data Fig. 9 Transplanted cardiac macrophages are detected by flow cytometry and immunofluorescence, related to Fig. 7.
a, TREM2hi Mac1 cells (CD45+CD11b+F4/80+CD163+RETNLA+) and non-Mac1 (CD45+CD11b+F4/80+ and non-CD163+RETNLA+) cells were isolated from CD45.1 mice. Mac1 or non-Mac1 cells (2 × 105 cells per mouse) were mixed with Matrigel (MG), and transplanted into the hearts of CD45.2 mice, respectively. Seven days after the transplantation, macrophages from hearts of recipients (CD45.2) were analyzed with flow cytometry assay. b,c, Graphs showing percentages of total Mac1 (b) and CD45.1+ Mac1 (c) cells within the CD45+ populations from hearts of recipients (CD45.2) at 1, 3, and 7 day(s) post the cell transplantation, respectively (n = 6 mice for each treatment). d,e, Graphs showing percentages of total non-Mac1 (d) and CD45.1+ non-Mac1 (e) cells within the CD45+ populations from hearts of recipients (CD45.2) at 1, 3, and 7 day(s) post the cell transplantation, respectively (n = 5-6 mice for each treatment). f,g, TREM2hi Mac1 cells (CD45+CD11b+F4/80+CD163+RETNLA+) and non-Mac1 (CD45+CD11b+F4/80+ and non-CD163+RETNLA+) cells were obtained from the hearts of WT mice by FACS and stained by CMTMR. Then, both types of cells (2 × 105 cells/mouse mixed with Matrigel) were respectively injected into the pericardial cavity of recipient mice immediately after CLP. f, Representative immunofluorescence images showing the transplanted cells in the epicardium (Epi), intermyocardium (Inter), and endocardium (Endo) of recipient mice at 3 days post the cell transplantation. CMTMR (red) signals indicating Mac1 or non-Mac1 cells; epicardial layer (E.L.); myocardium (Myo); inner layer (I.L.). Scale bar, 20 μm. g, Representative immunofluorescence images showing the transplanted CMTMR labeled Mac1 cells in MitoCard mice at 3 days post CLP. 3D reconstruction images of the dashed box area, illustrating that cardiomyocyte-derived mitochondria (mtDendra2+, green) presented in the transplanted cells (CMTMR+, red). Nuclei were shown in blue (DAPI). Scale bar, 10 μm. n = 6 mice per group (f,g). Bars show mean ± SEM, Two-sided P values were determined by one-way ANOVA with multiple comparisons (b-e). Results represent four (b-e) and two (f,g) independent experiments.
Extended Data Fig. 10 Intrapericardial administration of WT Mac1 cells protects SICM in Trem2−/− mice, related to Fig. 7.
a, Schematic illustration of Mac1 cells transplantation in Trem2-/- mice. Mac1 (CD45+CD11b+F4/80+CD163+RETNLA+) cells isolated from WT and Trem2-/- mice were mixed with Matrigel (MG), respectively, and then transplanted into the pericardial cavity of Trem2-/- mice (2×105 cells/animal) immediately after CLP. Control mice were injected MG only. Mice were analyzed 3 or 7 days after the transplantation. b-d, Graphs showing EF% (b), FS% (c) and CO (d) measured by echocardiography. e,f, Graphs showing the levels of cTnI (e) and LDH (f) in the serum. g,h, Graphs showing mRNA levels of Anp (g) and Bnp (h) in the heart tissue lysates. i,j, Graphs showing mRNA levels of Tnfα, Il1β, Ccl2 and Il6 in the heart tissue lysates at 3 (i) and 7 (j) days after CLP. k, Graph showing ATP content in heart tissue lysates. l, Graphs showing the levels of serum lactate. b-l, each symbol represents one animal (n = 6 mice for each treatment). All data are presented as mean ± SEM. Two-sided P values by Kruskal-Wallis with Dunn’s multiple comparison test, n.s., not significant. m, Schematic diagram showing TREM2hi Mac1 phagocytoses cardiomyocyte-derived mitochondria protect the septic heart. scRNA-seq uncovers a cardiac macrophage subset that was highly expressing TREM2 and CD163; Sepsis stimulates the release of massively cardiomyocyte-derived mitochondria; TREM2hi Mac1 cells scavenge defective mitochondria and protect the septic heart. Results represent two (b-l) independent experiments.
Supplementary information
Supplementary Information
Supplementary Tables 1–3 and legends for Supplementary Videos 1–6.
Supplementary Video 1
Sepsis induces the release of cardiomyocyte-derived exophers. 3D reconstruction of heart slices from CardRED mice show the presence of mitochondria (Tom20, cyan) in cardiomyocyte-derived exophers (red) and the accumulation of Tomato+ exophers colocalized with Tom20 in septic hearts.
Supplementary Video 2
Mac1 cells take up cardiac exophers containing mitochondria in SICM. 3D reconstruction of the heart slices from CardRED mice. Mac1 cells (TREM2, green) phagocytosed cardiomyocyte-derived exophers (red). Exophers in Mac1 cells included mitochondria (Tom20, white).
Supplementary Video 3
Cardiomyocyte-derived mitochondria transfer to Mac1 cells. 3D reconstruction of the heart slices from MitoCard mice. Mac1 cells (TREM2, red) took up cardiomyocyte-derived mitochondria (mtDendra2, green).
Supplementary Video 4
Cardiomyocyte-derived mitochondria processed with LAMP1+ phagolysosomes in Mac1 cells. 3D reconstruction of the heart slices from MitoCard mice. Cardiomyocyte-derived mitochondria (mtDendra2, green) phagocytosed by Mac1 cells (TREM2, red) partially localized in lysosomes (LAMP1, white).
Supplementary Video 5
Trem2 deficiency impairs the uptake of cardiomyocyte-derived mitochondria by Mac1 subset in septic heart. Part 1: Schematic illustration of the cardiomyocyte mitochondria labeled with AAV9-Tnnt2-mt-Keima virus. Keima-tagged mitochondria were indicated by different fluorescence in neutral (Keima 458nm, green) and acidic (Keima 561nm, red) environments. Part 2: 3D reconstruction showed that TREM2+ macrophages (green) took up cardiomyocyte-derived mitochondria (mtKeima-458, cyan) and some mitochondria in an acidic environment (mtKeima-561, red) in the hearts of AAV9-Tnnt2-mt-Keima-infected mice. Part 3: CD163+ macrophages (green) took up cardiomyocyte-derived mitochondria (mtKeima-458, cyan; mtKeima-561, red) in hearts of WT and Trem2−/− mice infected with AAV9-Tnnt2-mt-Keima.
Supplementary Video 6
Transplanted Mac1 cells engulf cardiomyocyte-derived mitochondria in SICM. 3D reconstruction of the heart slices from MitoCard mice. Injected Mac1 cells (CMTMR, red) phagocytosed cardiomyocyte-derived mitochondria (mtDendra2, green).
Source data
Source Data Fig. 1
Single-cell RNA-seq data and statistical source data.
Source Data Fig. 2
Single-cell RNA-seq data and statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Single-cell RNA-seq data and statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Single-cell RNA-seq data and statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, K., Wang, Y., Chen, S. et al. TREM2hi resident macrophages protect the septic heart by maintaining cardiomyocyte homeostasis. Nat Metab 5, 129–146 (2023). https://doi.org/10.1038/s42255-022-00715-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-022-00715-5
This article is cited by
-
Single-cell transcriptomics reveals writers of RNA modification-mediated immune microenvironment and cardiac resident Macro-MYL2 macrophages in heart failure
BMC Cardiovascular Disorders (2024)
-
Mitochondria from osteolineage cells regulate myeloid cell-mediated bone resorption
Nature Communications (2024)
-
Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets
Signal Transduction and Targeted Therapy (2024)
-
The crosstalk between mitochondrial quality control and metal-dependent cell death
Cell Death & Disease (2024)
-
Macrophage profiling in atherosclerosis: understanding the unstable plaque
Basic Research in Cardiology (2024)
