
Abstract
Mitochondrial DNA (mtDNA) encodes protein subunits and translational machinery required for oxidative phosphorylation (OXPHOS). Using repurposed whole-exome sequencing data, in the present study we demonstrate that pathogenic mtDNA mutations arise in tumours at a rate comparable to those in the most common cancer driver genes. We identify OXPHOS complexes as critical determinants shaping somatic mtDNA mutation patterns across tumour lineages. Loss-of-function mutations accumulate at an elevated rate specifically in complex I and often arise at specific homopolymeric hotspots. In contrast, complex V is depleted of all non-synonymous mutations, suggesting that impairment of ATP synthesis and mitochondrial membrane potential dissipation are under negative selection. Common truncating mutations and rarer missense alleles are both associated with a pan-lineage transcriptional programme, even in cancer types where mtDNA mutations are comparatively rare. Pathogenic mutations of mtDNA are associated with substantial increases in overall survival of colorectal cancer patients, demonstrating a clear functional relationship between genotype and phenotype. The mitochondrial genome is therefore frequently and functionally disrupted across many cancers, with major implications for patient stratification, prognosis and therapeutic development.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

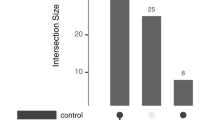
Data availability
All data not available for download on public repositories as described in Methods are available on GitHub (https://github.com/reznik-lab/mtdna-mutations).
Code availability
R code to regenerate all figures is available on GitHub (https://github.com/reznik-lab/mtdna-mutations) with the relevant data and instructions to execute the code.
References
Hornshøj, H. et al. Pan-cancer screen for mutations in non-coding elements with conservation and cancer specificity reveals correlations with expression and survival. NPJ Genom. Med. 3, 1 (2018).
Ju, Y. S. et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 3, e02935 (2014).
Yuan, Y. et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 52, 342–352 (2020).
Stewart, J. B. et al. Simultaneous DNA and RNA mapping of somatic mitochondrial mutations across diverse human cancers. PLoS Genet. 11, e1005333 (2015).
Grandhi, S. et al. Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue-specific signals of relaxed and positive selection. Hum. Mol. Genet. 26, 2912–2922 (2017).
Hopkins, J. F. et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 8, 656 (2017).
To, T.-L. et al. A compendium of genetic modifiers of mitochondrial dysfunction reveals Intra-organelle buffering. Cell 179, 1222–1238.e17 (2019).
Birsoy, K. et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015).
Samuels, D. C. et al. Finding the lost treasures in exome sequencing data. Trends Genet. 29, 593–599 (2013).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Li, H. et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Collura, R. V., Auerbach, M. R. & Stewart, C. B. A quick, direct method that can differentiate expressed mitochondrial genes from their nuclear pseudogenes. Curr. Biol. 6, 1337–1339 (1996).
Cheng, D. T. et al. Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 17, 251–264 (2015).
Bolze, A. et al. Selective constraints and pathogenicity of mitochondrial DNA variants inferred from a novel database of 196,554 unrelated individuals. Preprint at bioRxiv https://doi.org/10.1101/798264 (2019).
Chang, M. T. et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163 (2016).
Chang, M. T. et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 8, 174–183 (2018).
Triska, P. et al. Landscape of germline and somatic mitochondrial DNA mutations in pediatric malignancies. Cancer Res. 79, 1318–1330 (2019).
Gopal, R. K. et al. Early loss of mitochondrial complex I and rewiring of glutathione metabolism in renal oncocytoma. Proc. Natl Acad. Sci. USA 115, E6283–E6290 (2018).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Alston, C. L. et al. A novel mitochondrial MTND5 frameshift mutation causing isolated complex I deficiency, renal failure and myopathy. Neuromuscul. Disord. 20, 131–135 (2010).
Castellana, S. et al. High-confidence assessment of functional impact of human mitochondrial non-synonymous genome variations by APOGEE. PLoS Comput. Biol. 13, e1005628 (2017).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Martínez-Reyes, I. et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292 (2020).
El-Hattab, A. W., Adesina, A. M., Jones, J. & Scaglia, F. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 116, 4–12 (2015).
Gorman, G. S. et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 77, 753–759 (2015).
Gopal, R. K. et al. Widespread chromosomal losses and mitochondrial DNA alterations as genetic drivers in Hürthle cell carcinoma. Cancer Cell 34, 242–255.e5 (2018).
Terzioglu, M. et al. MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 17, 618–626 (2013).
Spagnolo, M. et al. A new mutation in the mitochondrial tRNA(Ala) gene in a patient with ophthalmoplegia and dysphagia. Neuromuscul. Disord. 11, 481–484 (2001).
Horváth, R., Reilmann, R., Holinski-Feder, E., Ringelstein, E. B. & Klopstock, T. The role of complex I genes in MELAS: a novel heteroplasmic mutation 3380G>A in ND1 of mtDNA. Neuromuscul. Disord. 18, 553–556 (2008).
Agip, A.-N. A. et al. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 25, 548–556 (2018).
Joshi, S. et al. The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep. 13, 1895–1908 (2015).
Ganly, I. et al. Integrated genomic analysis of Hürthle cell cancer reveals oncogenic drivers, recurrent mitochondrial mutations, and unique chromosomal landscapes. Cancer Cell 34, 256–270.e5 (2018).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Yaeger, R. et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 33, 125–136.e3 (2018).
Smith, A. L. et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 1, 976–989 (2020).
Yan, H. et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009).
Baysal, B. E. et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851 (2000).
Tomlinson, I. P. M. et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30, 406–410 (2002).
Fendt, S.-M., Frezza, C. & Erez, A. Targeting metabolic plasticity and flexibility dynamics for cancer therapy. Cancer Discov. 10, 1797–1807 (2020).
Gammage, P. A. & Frezza, C. Mitochondrial DNA: the overlooked oncogenome? BMC Biol. 17, 53 (2019).
Priolo, C. et al. Impairment of gamma-glutamyl transferase 1 activity in the metabolic pathogenesis of chromophobe renal cell carcinoma. Proc. Natl Acad. Sci. USA 115, E6274–E6282 (2018).
Gammage, P. A., Moraes, C. T. & Minczuk, M. Mitochondrial genome engineering: the revolution may not be CRISPR-ized. Trends Genet. 34, 101–110 (2018).
Trifunovic, A. et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004).
Mok, B. Y. et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637 (2020).
Cancer Genome Atlas Research Network et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
Hyman, D. M. et al. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov. Today 20, 1422–1428 (2015).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Chakravarty, D. et al. Oncokb: a precision oncology knowledge base. JCO Precis. Oncol. https://doi.org/10.1200/PO.17.00011 (2017).
Sonney, S. et al. Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput. Biol. 13, e1005867 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Zhu, A., Ibrahim, J. G. & Love, M. I. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019).
Benjamini, Y. et al. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Sergushichev, A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. Preprint at bioRxiv https://doi.org/10.1101/060012 (2016).
Liu, Y. et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 33, 721–735.e8 (2018).
Ellrott, K. et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e7 (2018).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Jurcik, A. et al. CAVER Analyst 2.0: analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 34, 3586–3588 (2018).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA 98, 10037–10041 (2001).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 (2004).
Acknowledgements
We thank the members of the Reznik, Gammage and Taylor laboratories for discussion and support. We also thank L. Finley, K. Birsoy, J. Blaza, C. Winchester and N. Rusk for their feedback. A.N.G., M.K., W.K.C., A.A.H., M.F.B, B.S.T. and E.R. were supported by the National Cancer Institute (NCI) Cancer Center Support (grant no. P30 CA008748). W.K.C. was supported by a National Institutes of Health (NIH) award (no. T32 GM132083). K.C.L. was supported by an F31 Predoctoral Fellowship from the NCI (award no. 7F31CA247528-02). B.S.T. was supported by the NIH (award nos. U54 OD020355, R01 CA207244, R01 CA204749 and R01 CA245069), as well as the American Cancer Society, Anna Fuller Fund and the Josie Robertson Foundation. E.R. was supported by the Geoffrey Beene Cancer Research Center Grant Award, Department of Defense Kidney Cancer Research Program (no. W81XWH-18-1-0318), and a Kidney Cancer Association Young Investigator Award. P.A.G. was supported by core funding from CRUK BI (nos. A17196 and A31287).
Author information
Authors and Affiliations
Contributions
A.N.G., P.A.G. and E.R. conceived the study. M.K., W.K.C., K.L., A.A.H., M.F.B. and B.S.T. assisted with genomic data analysis. A.N.G. analysed protein structures. A.N.G., P.A.G. and E.R. wrote the manuscript, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
B.S.T. reports receiving honoria and research funding from Genentech and Illumina, and advisory board activities for Boehringer Ingelheim and Loxo Oncology, a wholly owned subsidiary of Eli Lilly, Inc. All stated activities were outside the work described in the present study. He is currently an employee of Loxo Oncology. P.A.G. is a shareholder of Pretzel Therapeutics Inc. The remaining authors declare no competing interests.
Additional information
Peer review information Nature Metabolism thanks Navdeep Chandel, Young Seok Ju and Carlos Moraes for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: George Caputa.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Baseline demographics of cohort and aspects of sequencing coverage.
a, The demographic distributions of patient age, race, gender, mtDNA somatic mutation status, history of neoadjuvant treatment, mtDNA coverage, and tumor sample type for each of the cancer types included in our analysis. Somatic mutation status is annotated among the subset of samples with ≥90% paired tumor-normal mtDNA sequencing coverage (see Methods: classifying sample mtDNA variant status); mtDNA status distributions are shown for cancer types >10 such samples. Cancer types are ordered by increasing proportions of samples with VUS or truncating mtDNA mutations. b, Cancer type mtDNA coverage variation based on sequencing center. Center, the average percentage of mtDNA (among regions considered in our study) with sufficient coverage for calling mutations, compared between different cancer types in our cohort. Dot color indicates the sequencing center from which the exome sequencing data originate. Top, density histograms of the average % mtDNA coverage for each sequencing center. Samples sequenced at the Broad Institute are uniquely depleted for mtDNA off-target coverage. c, mtDNA coverage from off-target reads at each position. The number of samples for which the given mtDNA position was sequenced to at least 5 reads (top, the depth threshold used in our analyses) and 20 reads (bottom, for comparison). Red, the number of samples using unpaired tumor-only data, applicable only for protein-truncating variants which were always assumed to be of somatic origin; blue, the number using only matched-normal samples; green, the number of samples with coverage in both tumor and matched-normal samples at the given position (applicable for all non-truncating variants which required evidence that the variant was absent in the matched normal to be classified as somatic). Purple, the number of whole-genome sequenced samples available from ICGC/PCAWG for comparison. d, Proportion of samples with detectable mutations is not biased by cancer type sequencing coverage. There is no correlation between the fraction of well-covered samples in a cancer type and the proportion of well-covered samples with a detectable somatic mtDNA mutation. Cancer types with ≥30 well-covered samples shown, P-value and 95% confidence intervals from linear regression.
Extended Data Fig. 2 Strand-specific mutational signatures in our dataset.
The frequency of somatic SNVs on the light or heavy mtDNA strand with each of the 96 possible mutational signatures with trinucleotide contexts (among n = 3,872 SNVs). Blue bars indicate the prevalence of mutational signatures for heavy-strand encoded SNVs (substitutions at C or T central nucleotides); red bars indicate those for light-strand encoded SNVs (substitutions at G or T nucleotides, which were standardized to their C or T complementary nucleotide). The most prevalent mutational signatures are labeled. The underlined central position is mutated with the single nucleotide substitution labeled in the tile below.
Extended Data Fig. 3 Analysis of mutation burden in normal tissues and of tumor mtDNA mutation burden with nuclear mutagenic processes.
a, Comparison of heteroplasmies between truncating variants detected in tumor tissue, adjacent normal tissue, and blood. P-values from two-sided Wilcoxon-rank sum test. Boxes are centered at the median and extended to from 25th-percentile to 75th-percentile; whiskers extend from 25/75th-percentiles to the largest value within 1.5 × IQR (interquartile range, 75th-percentile - 25th-percentile. b, Rate of truncating variants in TCGA tumors compared to matched non-malignant tissue, matched blood, and unmatched saliva samples from HelixMTdb. Truncating variants arise at 10-80-fold higher rate in tumors relative to normal tissues. Error bars are exact binomial 95% confidence intervals. P-values are from two-sided two-sample z-tests. c, The percentage of rescued truncating variants in TCGA that are recapitulated in orthogonal RNA sequencing from the same tumor sample. d, Correlation between heteroplasmies of rescued truncating variants in DNA and orthogonal RNA sequencing. Pearson correlation coefficient as shown. e, Mitochondrial and nuclear tumor mutation burdens (TMB, mutations/Mb) are shown for each well-covered tumor, among cancer types with n ≥ 100 samples. Nuclear TMBs are calculated based on mutations to 468 cancer-associated genes and their total exonic-sequence length. Pearson correlation coefficients r indicate no linear correlation between mitochondrial and nuclear TMBs were observed for any cancer type tested. f, TMBs for somatic mtDNA mutations and mutations to cancer-associated genes are compared between microsatellite stable (MSS) and microsatellite unstable (MSI-High) tumors, for both (n colorectal cancer: MSI=65, MSS=318; n stomach adenocarcinomas: MSI=75, MSS=256). Although MSI-High tumors have elevated TMB for nuclear cancer genes, there is no effect on mtDNA TMB. Moreover, mtDNA TMB is similar to (or exceeds) that of nuclear cancer associated genes in both cancer types. Error bars are 95% exact Poisson confidence intervals.
Extended Data Fig. 4 Age- and tumor stage-associations of somatic mtDNA mutations across cancer types.
Heatmap shows tumor mutation burden (total mutations/total covered Mbps) for samples of each tumor type (a) combined across varying patient age at time of diagnosis and (b) tumor pathologic stage. Gray tiles indicate cancer type/age combinations with fewer than 3 patients; cancer types shown had at least 2 non-gray tiles. Right column: Spearman correlation coefficient r indicating correlation between age or pathologic stage and tumor mutation burden. Asterisks denote statistically significant correlations based on FDR-corrected P-values from a Student’s t-distribution.
Extended Data Fig. 5 Molecular features of truncating variants at homopolymeric loci.
a,b, Enrichment for truncating variants in CI and non-truncating in CIII when restricted to mutations with 20+ reads supporting the alternate allele. Error bars are 95% Poisson exact confidence intervals; P-values from two-sided Poisson tests. c, Comparison of frameshift indel homopolymer hotspots detected among indels supported by a minimum of 20 alt-reads (Y-axis) to those with a minimum support of 5 alt-reads (X-axis). d, Percentage of cases per cancer type with truncating frameshift indels at any of 6 indel hotspot loci. Plotted cancer types had ≥ 20 well-covered samples (n=4,432 paired tumor and matched-normal samples total). Bar height indicates the fraction of samples with any indels at homopolymer hotspot out of the total number of well-covered samples for the given cancer type; numbers above bars indicate the total number of cases. e, Validation of homopolymeric indel hotspot loci. The proportion of samples in TCGA (X-axis) or PCAWG (excluding samples also in TCGA, Y-axis) with frameshift indels at 73 homopolymeric regions. The 6 indel hotspot loci are colored red and labeled. y=x is shown as a dashed line. Pearson correlation coefficient r as indicated. f, Breakdown of homopolymer loci and their hotspot incidence rates by mitochondrial complex. Heatmap tile shading indicates overall mutation rate (total number of mutants across homopolymer loci divided by the total number of samples with sufficient sequencing coverage). Fractions in tile labels are the number of homopolymer hotspots divided by the total number of homopolymer loci. Right, histogram of the total number of loci with each homopolymer length. g, The percentage of all truncating variants which arose at 6 homopolymer hotspot loci in TCGA tumor samples and in saliva-derived normal samples from HelixMTdb. Error bars are 95% binomial confidence intervals.
Extended Data Fig. 6 Homopolymer hotspots for frameshift indels in TCGA and MSK-IMPACT cohorts.
a, Analysis of 73 homopolymer loci for enrichment of protein-truncating indels in TCGA samples in shown on X-axis, and in MSK-IMPACT samples on Y-axis. 5 out of 6 originally reported hotspot loci are enriched in both TCGA and MSK-IMPACT (green), while 1 was only enriched in TCGA samples (orange). Two additional candidate hotspot loci are unique to the MSK-IMPACT dataset (recurrently observed in TCGA, but not reaching statistical significance). b, Heatmap indicates the proportion of truncating indels at each of the 8 homopolymer hotspots to affect samples of different cancer types (that is rows are proportion of samples with indels at a given homopolymer summing to 1; clustered by hierarchical clustering). Right, histogram of the total number of affected samples in TCGA and MSK-IMPACT data. Note that the CIV hotspot discovered in MSK-IMPACT data preferentially arises in lung and prostate cancers.
Extended Data Fig. 7 Validation of VUS pathogenicity and tRNA mutation recurrence.
a, VUSs only observed in tumors are more likely to be pathogenic. Bars compare the clinical significance (annotated by ClinVar) of SNVs observed somatically in tumors but never in patients’ matched-normal samples against SNVs never observed in either tumor or matched-normal samples. P-value from a two-sided Cochran-Armitage trend test. b, Validation of tRNA structural hotspots in PCAWG. The number of samples with SNVs in tRNAs at the indicated cloverleaf structural position, bottom; top, the statistical enriched of the given position for mutations. Position 31 Q-value=0.014, n=196 tRNA mutations among 1,951 PCAWG samples.
Extended Data Fig. 8 mtDNA mutations produce transcriptional phenotypes.
a,b, Transcriptional dysregulation attributed to truncating (a) and VUS (b) mtDNA variants. Heatmaps shows directional significance of dysregulation of a given geneset in tumors with truncating or VUS mtDNA variants among the given cancer type; −log10(Q-value) > 2 indicates significant up-regulation in mutated compared to wild-type samples, < −2 indicates significant down-regulation. Histograms on the right show the number of wild-type samples and mutated samples used in calculating differentially expressed genes and dysregulated genesets. c, Difference in mtDNA mutation status between colorectal cancer consensus molecular subtypes. Left, the proportion of samples with wild-type mtDNA (that is no somatic mutations), VUS (any non-truncating) or truncating variants among colorectal tumors with each consensus molecular subtype (CMS) is shown. Right, histogram of the number of well-covered colorectal tumors. There was a statistically significant difference in mtDNA mutation status between different CMS classifications (P=0.03, Chi-squared test, n=415 samples total, error bars are 95% exact binomial confidence intervals).
Extended Data Fig. 9 mtDNA mutations are protective in colorectal cancer patients in the MSK-IMPACT cohort.
a, Multivariate survival analysis based on Cox proportional hazards regression demonstrating the effect of VUS or truncating mtDNA mutations (relative to wild-type) on colorectal cancer patient overall survival in the MSK-IMPACT cohort. b, Same as in (a) but treating VUS and truncating mtDNA mutations as a single class compared to wild-type. Error bars are 95% confidence intervals from Cox proportional-hazards regression, n=172 MSK-IMPACT patients.
Extended Data Fig. 10 Repurposing whole-exome and clinical sequencing data optimizes sample size at the expense of sensitivity for low-heteroplasmy variants.
a, The number of different classes of mtDNA variants detected from either repurposed TCGA samples using our approach, or using only whole-genome sequenced tumors from PCAWG, stratified by heteroplasmy < 30% or ≥ 30%. Labels above bars indicate the exact number. b, Comparison of the difference in gene expression between (1) high-heteroplasmy truncating mutations and wild-type tumors (X-axis) and (2) low-heteroplasmy truncating mutations and wild-type tumors (Y-axis). c, Strengths and use-cases of three common tumor DNA sequencing modalities for mtDNA mutation analysis. WGS-based approaches are optimal for studying low-heteroplasmy variants and identifying structural variants and mtDNA copy number, while whole-exome and targeted gene-panel-based approaches optimize sample size, detection of recurrent variants, and clinical associations.
Supplementary information
Supplementary Tables
Supplementary Tables 1–6
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gorelick, A.N., Kim, M., Chatila, W.K. et al. Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA. Nat Metab 3, 558–570 (2021). https://doi.org/10.1038/s42255-021-00378-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-021-00378-8
This article is cited by
-
Mitochondrial DNA mutation enhances sensitivity to immunotherapy in melanoma
Nature Cancer (2024)
-
Mitochondrial DNA mutations drive aerobic glycolysis to enhance checkpoint blockade response in melanoma
Nature Cancer (2024)
-
Role of mitochondrial alterations in human cancer progression and cancer immunity
Journal of Biomedical Science (2023)
-
Pan-tissue mitochondrial phenotyping reveals lower OXPHOS expression and function across cancer types
Scientific Reports (2023)
-
Time to hit pause on mitochondria-targeting cancer therapies
Nature Medicine (2023)
