
Abstract
Colchicine has served as a traditional medicine for millennia and remains widely used to treat inflammatory and other disorders. Colchicine binds tubulin and depolymerizes microtubules, but it remains unclear how this mechanism blocks myeloid cell recruitment to inflamed tissues. Here we show that colchicine inhibits myeloid cell activation via an indirect mechanism involving the release of hepatokines. We find that a safe dose of colchicine depolymerizes microtubules selectively in hepatocytes but not in circulating myeloid cells. Mechanistically, colchicine triggers Nrf2 activation in hepatocytes, leading to secretion of anti-inflammatory hepatokines, including growth differentiation factor 15 (GDF15). Nrf2 and GDF15 are required for the anti-inflammatory action of colchicine in vivo. Plasma from colchicine-treated mice inhibits inflammatory signalling in myeloid cells in a GDF15-dependent manner, by positive regulation of SHP-1 (PTPN6) phosphatase, although the precise molecular identities of colchicine-induced GDF15 and its receptor require further characterization. Our work shows that the efficacy and safety of colchicine depend on its selective action on hepatocytes, and reveals a new axis of liver–myeloid cell communication. Plasma GDF15 levels and myeloid cell SHP-1 activity may be useful pharmacodynamic biomarkers of colchicine action.
Similar content being viewed by others


Main
Inflammation involves local activation and extravasation of circulating myeloid cells into damaged or infected tissues. Excessive inflammation is implicated as a causal factor in numerous diseases1, and drugs that treat inflammation are among the most used of all medicines. Nevertheless, many inflammatory diseases remain difficult to treat and current drugs have unwanted side effects2.
Colchicine is one of the oldest medicines still in common use. Historically, colchicine-containing extracts from Colchicum autumnale and Gloriosa superba were important traditional medicines3. Pure colchicine is still widely used to treat diverse inflammatory diseases, including gout, familial Mediterranean fever (FMF), pericarditis, Behçet’s disease and auto-inflammatory dermatitis. It recently demonstrated major potential preventive benefit in cardiovascular disease and COVID-19 mortality, with multiple clinical trials ongoing4,5,6. However, wider application of colchicine is limited by its narrow therapeutic index and lethal toxicity at high doses7. Determination of its therapeutic mechanism would provide biomarkers to guide optimal dosing in repurposing trials and targets for safer replacement drugs.
Colchicine binds to tubulin, inhibits microtubule assembly3,8 and decreases recruitment of myeloid cells into inflamed tissues9. It has long been assumed to act directly on circulating myeloid cells3. However, the concentrations of colchicine required to block chemotaxis of cultured neutrophils10 or inhibit inflammasome activation11 are much higher than those measured in patient plasma following safe and efficacious doses12. Thus, direct targeting of circulating myeloid cells may not be the therapeutic mechanism. A potential explanation for the safety and efficacy of colchicine, and its unique therapeutic profile compared to anti-microtubule drugs used in cancer13, is selective tissue distribution. Colchicine is known to distribute selectively to liver and kidney14. These organs were previously ignored in considerations of its therapeutic action. However, the liver secretes many plasma proteins that affect immune responses, including signalling proteins termed hepatokines15. Here we show that selective action in the liver, and hepatokine secretion, play central roles in colchicine therapeutic action.
Results
Colchicine inhibits inflammation through induced factors in plasma
To model the anti-inflammatory action of colchicine in mice, we first determined a dose that is bio-equivalent to human clinical doses by allometric scaling and measurement of diarrhoea, the dose-limiting toxicity in humans7 (Extended Data Fig. 1). The dose used throughout to model as safe was 0.4 mg kg–1 intraperitoneally (i.p.). Subsequent experiments showed that it was efficacious in inflammation models. Modelling of a toxic dose was performed at 2.4 mg kg–1 i.p., which is close to the median lethal dose16.
To measure the kinetics of anti-inflammatory action, mice were dosed with colchicine then challenged 1–12 h later by i.p. injection of zymosan, a pro-inflammatory yeast cell wall glycan, to trigger peritonitis. Neutrophil infiltration was scored 3 h after the challenge (Fig. 1a). Colchicine pretreatment 6 h before the challenge decreased peritoneal infiltration of Ly-6G+ neutrophils approximately fourfold, while 1- and 12-h pretreatment had no effect (Fig. 1a). Because colchicine is rapidly cleared from plasma14, circulating neutrophils would be most exposed to the drug in the 1-h pretreatment group. The delayed anti-inflammatory effect we observed in mice, which is consistent with the kinetics of gout pain reduction in humans17, suggests that colchicine might act indirectly on myeloid cells via changes in blood chemistry.

a, Kinetics of in vivo anti-inflammatory effects of colchicine (colc.). At x h (x = 1, 6 or 12) following colchicine treatment, mice were challenged with i.p. zymosan. Three hours later, peritoneal-infiltrated Ly-6G+ neutrophils were detected by FACS analysis. Only 6-h pretreatment of colchicine decreased neutrophil infiltration. Each dot represents one mouse, seven mice per condition. b–e, Colchicine indirectly inhibited neutrophil activation. b, Ex vivo neutrophil activation assays comparing the effects of colchicine itself, or of plasma from colchicine-treated mice, on inflammation markers in wild-type primary neutrophils following PMA challenge. Direct colchicine treatments were performed with vehicle plasma, so plasma concentrations were the same in all assays. Colchicine plasma (plasma from colchicine-treated mice) was collected at the indicated times after in vivo treatment. c, Neutrophil adhesion to plastic tubes. The y axis indicates cells remaining in suspension. FC, fold change. At least three samples per condition over three biological replicates. Veh. plasma, plasma from vehicle-treated mice. d, ROS generation detected with the ROS-sensitive dye, dihydrorhodamine 123, and measured by FACS analysis. gMFI, geometric mean fluorescence intensity. At least three samples per condition over three biological replicates. e, Pro-IL-1β gene expression by immunoblotting. Colchicine itself lacked activity in all assays, while plasma collected 6 h after colchicine treatment had strong anti-inflammatory activity in all assays. f, In vivo biomarker analysis. Colchicine-activated JNK selectively in the liver. The amounts of phosphorylated and total JNK in nine different tissues from four mice were measured by immunoblotting. Data are represented as mean ± s.d. Two-sided t-tests were used for statistical analysis. g–i, Colchicine selectively depolymerized microtubules in hepatocytes. g, Microtubules in hepatocytes (identified by HNF4+), Kupffer cells (identified by F4/80+) and other cell types (HNF4−, F4/80− and DAPI+). Images show frozen sections of livers fixed 6 h after vehicle or colchicine treatment, stained and imaged by confocal microscopy. The boxed areas are magnified and shown at the right. Scale bars, 10 μm. h, Microtubules in circulating leukocytes. Scale bar, 5 μm. i, Intensity measurement of microtubules. Approximately 700 hepatocytes, 200 Kupffer cells and 150 other liver cells from four livers following vehicle or colchicine treatment were quantified. AU, arbitrary units.
To investigate whether colchicine acts directly or indirectly on neutrophils, we developed ex vivo activation assays (Fig. 1b). Primary neutrophils from the bone marrow of wild-type, untreated mice were pre-incubated with plasma from vehicle- or colchicine-treated mice, with or without colchicine added ex vivo. We then challenged neutrophils with the standard pro-inflammatory trigger, phorbol 12-myristate 13-acetate (PMA), which activates multiple inflammatory signalling pathways. Neutrophil activation was assayed in three ways: cell adhesion, reactive oxygen species (ROS) generation and expression of the pro-inflammatory cytokine pro-IL-1β. Colchicine mixed with control plasma from vehicle-treated mice had little effect (Fig. 1c–e), showing no direct action of colchicine in these assays. Plasma collected 6 h after colchicine dosing, but not 1 or 18 h, robustly lowered all three activation markers (Fig. 1c–e). These data support the indirect action of colchicine via unknown substances in plasma. These substances are unlikely to be colchicine metabolites since it is excreted mostly unmetabolized14. The known major metabolite, 3-desmethylcolchicine, did not block pro-IL-1β expression in our ex vivo assay (Supplementary Fig. 1a). The colchicine-induced, anti-inflammatory factors in plasma were heat labile and had a molecular weight >3 kDa (Supplementary Fig. 1b,c), suggesting they were proteins.
Colchicine selectively targets hepatocytes
We next implemented a biomarker strategy to identify tissues in which colchicine has a direct action, using activation of c-Jun N-terminal kinase (JNK) as a reporter of cell stress induced by microtubule depolymerization18. Among nine tissues from treated mice, only the liver showed an increase in the ratio of phosphorylated (pJNK) to total JNK 6 h after a safe and efficacious dose (Fig. 1f and Supplementary Fig. 2). We then tested whether microtubules are depolymerized in the liver, using immunofluorescence. The liver contains multiple cell types. Tubulin immunofluorescence revealed that hepatocytes (identified by HNF4+ nuclei19) exhibited tubulin depolymerization (Fig. 1g,i and Extended Data Fig. 2). Microtubules in F4/80+ Kupffer cells and other liver cells remained intact. Furthermore, we imaged microtubules in circulating leukocytes from colchicine-treated mice and found no depolymerization at the safe dose (Fig. 1h,i and Extended Data Fig. 3). Quantitative intensity measurements confirmed selective depolymerization in hepatocytes (Fig. 1i). At a toxic dose, colchicine lost this cell-type selectivity and caused depolymerization of microtubules in all liver cell types and circulating blood cells (Extended Data Figs. 3 and 4). These data show that colchicine, at a safe and efficacious dose, acts selectively in hepatocytes and support the premise that colchicine does not directly target myeloid cells in vivo. Hepatocytes express drug transporters that concentrate drugs and xenobiotics20 and colchicine is known to concentrate in the liver14, so selective targeting probably arises from selective drug distribution.
Pharmacokinetic data support liver-selective action
To test whether hepatocyte or myeloid cell targeting is more likely in humans, we performed pharmacokinetics-pharmacodynamics (PK-PD) modelling using measured plasma colchicine concentrations in human as input12. Following therapeutic dosing, plasma colchicine peaks at 16 nM or less and it is rapidly cleared from the circulation. Because at least 2% of total tubulin must bind to colchicine to destabilize microtubules21, we set the threshold for drug action at this level of site occupancy. A two-compartment PK-PD model suggested that the fraction of colchicine-bound tubulin in circulating myeloid cells falls short of this threshold (Extended Data Fig. 5), consistent with lack of effect on leukocyte microtubules in mice (Fig. 1h). These data argue against direct targeting of myeloid cells in vivo in the therapeutic regime. Most tests of colchicine action on cultured myeloid cells have used concentrations of 1–10 μM or more, which model the toxic regime11,22.
Colchicine activates Nrf2 in hepatocytes
Liver-selective action was surprising, since colchicine is not usually toxic in the liver even when taken daily for years in FMF7,23. We therefore hypothesized that hepatocytes actively defend themselves against the effects of microtubule disassembly by colchicine. Nrf2, encoded by nuclear factor erythroid-derived 2-like 2 gene (NFE2L2), is a master transcription factor that protects the liver from xenobiotics by induction of cytoprotective proteins24. Nrf2 is negatively regulated by the E3 ligase adaptor Keap1. Activation of SQSTM1/p62-dependent selective autophagy leads to degradation of Keap1 and hence activation of Nrf2 (refs. 25,26,27). Three- and 6-h treatment of colchicine decreased Keap1 and increased Nrf2 levels in mouse livers (Fig. 2a). We observed concomitant phosphorylation of SQSTM1/p62 at Ser349 (Fig. 2a), which enhances the binding affinity of SQSTM1/p62 to Keap1 (ref. 27), LC3 activation (measured by conversion of LC3-I to LC3-II (ref. 28); Fig. 2b), increased interaction of SQSTM1/p62 with Keap1 in the co-immunoprecipitation analysis (Fig. 2c) and increase in SQSTM1/p62 puncta by immunofluorescence (Fig. 2d). These data suggest that colchicine activates SQSTM1/p62-dependent selective autophagy in hepatocytes, leading to Keap1 degradation and Nrf2 activation.

a, Colchicine (colc.) activated the Nrf2–Keap1 pathway in the liver. Immunoblotting analysis was performed with indicated antibodies to identify proteins in this pathway. Colchicine-treated livers were collected at the indicated times. Colchicine triggered degradation of Keap1, stabilization of Nrf2 and phosphorylation of the selective autophagy receptor SQSTM1/p62. b, Colchicine-activated LC3 as measured by conversion of the cytosolic LC3-I isoform to the membrane-bound lapidated LC3-II isoform. c, Colchicine promoted physical interaction between SQSTM1/p62 and Keap1 as measured by co-immunoprecipitation analysis. d, SQSTM1/p62 puncta in the liver from mice with vehicle (veh.) or colchicine treatment were detected by immunofluorescence analysis. The number of SQSTM1/p62 puncta per nucleus was measured. Data are represented as mean ± s.d. Two-sided t-tests were used for statistical analysis; three biological replicates. Scale bar, 10 μm.
To test whether Nrf2 activation protects hepatocytes against colchicine, we treated wild-type and Nrf2 germline knockout (Nrf2−/−) mice with our safe dose. We observed hepatocyte injury using standard serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) assays in Nrf2−/− mice, but no liver damage in wild-type mice (Fig. 3). Thus, colchicine at safe doses has the potential for liver injury but normal hepatocytes are protected from the consequences of microtubule disassembly by Nrf2 activation.

Colchicine (colc.)-induced liver toxicity was prevented by Nrf2. Wild-type and Nrf2−/− mice were treated with either vehicle or a safe dose of colchicine as indicated. Serum levels of AST (left) and ALT (right) were measured as standard clinical biomarkers to infer hepatocyte cell death. Each dot represents one mouse; five and seven mice were used for vehicle and colchicine treatment, respectively. Data are presented as mean ± s.d. Two-sided t-tests were used for statistical analysis.
GDF15 is a colchicine-induced anti-inflammatory hepatokine
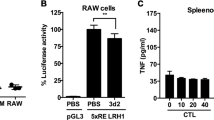
To identify proteins secreted by the liver in response to colchicine we queried a rat toxicogenomic database29, looking for messenger RNAs whose induction and decay kinetics matched those of the colchicine-induced anti-inflammatory factors we found in blood (Fig. 1). GDF15, a divergent TGF-β member also known as MIC-1 or NAG-1, met these criteria (Fig. 4a and Extended Data Fig. 6a,b). GDF15 regulates feeding and energy homeostasis by binding to the brainstem-restricted receptor, GFRAL30. It has been linked to inflammation biology and liver health, but the underlying mechanisms are unclear31,32,33,34. Plasma GDF15 is a biomarker of various diseases including cancer cachexia, mitochondrial disorders and cardiovascular diseases, as well as normal ageing and pregnancy35,36,37,38,39. Plasma GDF15 is increased by colchicine in humans40.

a, Gene expression profile of rat livers comparing vehicle to colchicine (colc.) treatment for 3 h (x axis) and 6 h (y axis). Each dot represents one gene. Red and grey dots denote genes encoding secreted and non-secreted proteins respectively, and GDF15 is highlighted in blue. Data are from the Open TG-GATEs toxicogenomic database. All data points were derived from biological triplicates. b, Induction of GDF15 in mouse livers as detected by immunoblotting. Livers were collected at the indicated times. c, Plasma GDF15 as measured by ELISA analysis. Red dots show induction kinetics in mice treated with 0.4 mg kg–1 colchicine; blue dots show the dose response after 6-h treatment; five and three mice were used for induction kinetics and dose response, respectively. d,e, Colchicine-induced GDF15 was mainly secreted by hepatocytes. d, Immunoblotting analysis revealed that induction of GDF15 in livers was blocked by treatment of hepatocyte-specific siRNA (GalNAc-siRNA) against GDF15. Luc, luciferase. e, ELISA analysis showed that hepatocyte-specific knockdown of GDF15 reduced colchicine-induced GDF15 in plasma. Remaining colchicine-induced plasma GDF15 was probably derived from the liver due to incomplete knockdown. At least four mice were used per condition. f, Nrf2 was required for colchicine-induced GDF15 expression in liver hepatocytes. g,h, In vivo peritonitis induced by i.p. zymosan challenge and assayed by neutrophil recruitment (g) and plasma IL-1β levels (h). Both Nrf2 and GDF15 were required for in vivo anti-inflammatory activity of colchicine using germline knockouts, hepatocyte-specific siRNA knockdowns and a GDF15-neutralizing antibody (anti-GDF15). Each dot represents one mouse, at least six mice per condition and three biological replicates. Data are represented as mean ± s.d. Two-sided t-tests were used for statistical analysis. Veh., vehicle.
The presence of GDF15 protein in liver and plasma was strongly induced by colchicine in a time- and dose-dependent manner, in both sexes (Fig. 4b,c and Extended Data Fig. 6c,d). Its temporal induction pattern (Fig. 4b) matched that of JNK phosphorylation in liver and the anti-inflammatory kinetics of colchicine measured in vivo and ex vivo (Fig. 1). To test the role of hepatocytes in GDF15 secretion after colchicine dosing, we developed a hepatocyte-specific knockdown reagent, triantennary N-acetylgalactosamine (GalNAc), that conjugated small interfering RNAs targeting GDF15 (GalNAc-siGDF15). Triantennary GalNAc confers hepatocyte-specific siRNA uptake41. GalNAc-siGDF15 treatment markedly attenuated the induction of GDF15 by colchicine in both liver (Fig. 4d) and plasma (Fig. 4e), indicating that hepatocytes are the source of most colchicine-induced GDF15. To model the clinical dosing route, we treated mice with colchicine by oral gavage. GDF15 was induced in liver and plasma, but not in the intestine or colon (Supplementary Fig. 3).
To test whether the Nrf2–Keap1 pathway is involved in GDF15 induction, we used either Nrf2 germline knockout (Nrf2−/−) or hepatocyte-specific knockdown (GalNAc-siNrf2). Both significantly reduced the expression of colchicine-induced GDF15 in liver and plasma (Fig. 4f and Extended Data Fig. 7a,b). Furthermore, we overexpressed Nrf2 in HEK 293T cells containing a GDF15 promoter upstream of luciferase. The luciferase gene was induced by Nrf2 (Extended Data Fig. 7c). CHOP, a key regulator of the cellular integrated stress response (ISR), is involved in metformin-induced GDF15 expression42. However, CHOP was not induced by colchicine in the liver, and the expression pattern of ISR regulators did not match that of colchicine-induced GDF15 (Extended Data Fig. 7d,e). Thus, colchicine causes an increase in GDF15 levels in plasma by activation of Nrf2 in hepatocytes.
We next tested the role of GDF15, and its upstream regulator Nrf2, in the anti-inflammatory action of colchicine. We developed peritonitis assays to measure neutrophil recruitment in response to i.p. challenge with zymosan to model infection, or monosodium urate crystals (MSU) to model gout. In wild-type mice, with or without control siRNA (GalNAc-siLuc) treatment, colchicine reduced neutrophil infiltration by 80 and 70% in zymosan and MSU models, respectively (Fig. 4g and Extended Data Fig. 7f). This anti-inflammatory activity was completely lost when GDF15 or Nrf2 was ablated in the whole mouse by using germline knockouts, or specifically in hepatocytes by using hepatocyte-targeting siRNAs (Fig. 4g and Extended Data Fig. 7f). We also measured circulating IL-1β following i.p. zymosan challenge. Colchicine reduced circulating IL-1β as expected (Fig. 4h). Systemic inhibition of GDF15 in mice by using a GDF15-neutralizing antibody blocked this anti-inflammatory activity of colchicine (Fig. 4h). We concluded that colchicine inhibits inflammation indirectly by activation of Nrf2 in hepatocytes, triggering the secretion of anti-inflammatory factors that include GDF15.
GDF15 is required for anti-inflammatory effects on myeloid cells
To test whether the anti-inflammatory actions of post-colchicine plasma on myeloid cells depend on GDF15, we returned to our ex vivo activation assays and tested both neutrophils and macrophages (Fig. 5 and Extended Data Figs. 8 and 9). Four groups of mice (wild type, Gdf15−/− and wild type pretreated with GalNAc-siLuc or GalNAc-siGDF15) were dosed with vehicle or colchicine, and plasma was collected 6 h later. Neutrophils from untreated wild-type mice were pretreated with these plasma samples and then challenged with PMA. Plasma from colchicine-treated wild-type or GalNAc-siLuc mice, but not Gdf15−/− or GalNAc-siGDF15 mice, strongly inhibited the activation of adhesion (Fig. 5b) and pro-IL-1β induction (Fig. 5d and Extended Data Fig. 8a). For independent validation of GDF15 dependence, we tested the effect by three different neutralizing antibodies. All of these robustly blocked the ex vivo anti-inflammatory effects of post-colchicine plasma (Fig. 5c,e and Extended Data Fig. 8b).

a, Ex vivo myeloid cell activation assays. Neutrophils from untreated mice or immortalized BMDMs were incubated in plasma collected from mice (wild type, Gdf15−/− or hepatocyte-specific siRNA knockdowns) following vehicle (veh.) or colchicine (colc.) treatment, then challenged with PMA (neutrophils) or Pam3CSK4 (BMDMs). Anti-GDF15, GDF15-neutralizing antibody; TPI-1, a SHP-1 inhibitor. b, PMA-stimulated adhesion of neutrophils to coverslips was inhibited by plasma from colchicine-treated wild-type mice, but not by Gdf15−/−. Co-treatment with TPI-1 blocked the anti-adhesive activity of post-colchicine plasma. c, Inhibition of GDF15 using a GDF15-neutralizing antibody blocked the anti-inflammatory activity of post-colchicine plasma. Veh. plasma, plasma from vehicle-treated mice; colc. plasma, plasma from colchicine-treated mice. Two-sided t-tests were used for statistical analysis. d, Induction of pro-IL-1β was blocked by post-colchicine plasma from wild-type mice, but not from Gdf15−/− mice. e, Co-treatment with a GDF15-neutralizing antibody blocked plasma activity. f–h, SHP-1 was activated by colchicine-induced GDF15. Phosphorylation of SHP-1 at Y564 marks the active form of SHP-1, while phosphorylation at S591 marks the inactive form. i, TPI-1 rescued pro-IL-1β induction, which was blocked by post-colchicine plasma. Post-colchicine plasma increased the active form (f) and decreased the inactive form (g) of SHP-1. h, The anti-inflammatory activity of post-colchicine plasma was blocked by inhibition of GDF15. j, SHP-1 inhibition by TPI-1 blocked the in vivo anti-inflammatory effect of colchicine in the zymosan peritonitis assay. Each dot represents one mouse. Data are presented as mean ± s.d. k, Model for indirect anti-inflammatory action of colchicine. Colchicine activates Nrf2–Keap1 signalling in hepatocytes, which protects them from damage and induction of a novel anti-inflammatory hepatokine, GDF15, into plasma. Colchicine-induced GDF15 then acts on circulating myeloid cells by supporting the active state of SHP-1.
We next tested immortalized mouse bone-marrow-derived macrophages (BMDMs). When activated by the TLR2/TLR1 ligand Pam3CSK4 followed by the microbial toxin nigericin, immortalized BMDMs expressed pro-IL-1β and secreted mature IL-1β. Post-colchicine plasma reduced both expression and secretion in a GDF15-dependent manner (Extended Data Fig. 9). Collectively, our data show that GDF15 polypeptide is required for the anti-inflammatory action of post-colchicine plasma on myeloid cells, consistent with our in vivo data.
SHP-1 phosphatase mediates anti-inflammatory effects on myeloid cells
Myeloid cell activation is controlled by activating and inhibitory surface receptors whose signals are integrated by the protein tyrosine phosphatase SHP-1 (PTPN6)43,44. The activity state of SHP-1 can be conveniently assayed by measurement of activating (pY564) and inhibitory (pS591) phosphorylation sites45. When neutrophils were treated with PMA in control plasma, pY564-SHP-1 levels decreased and pS591-SHP-1 levels increased, showing SHP-1 inactivation as expected. When neutrophils were incubated in GDF15-containing, post-colchicine plasma, PMA no longer caused SHP-1 inactivation (Fig. 5f,g). Neutralization of GDF15 by three different clones of anti-GDF15 antibodies robustly blocked the ability of post-colchicine plasma to maintain the active state of SHP-1 after PMA challenge ex vivo (Fig. 5h and Extended Data Fig. 8c). Moreover, plasma from GalNAc-siGDF15 mice lacked this activity (Extended Data Fig. 8d). Thus, the ability of post-colchicine plasma to positively regulate SHP-1 depends on GDF15.
To test whether SHP-1 is causally involved in the myeloid-inhibitory effects of colchicine, we used the SHP-1 inhibitor TPI-1 (ref. 46). TPI-1 treatment counteracted the ability of post-colchicine plasma to prevent PMA-induced neutrophil adhesion (Fig. 5b) and pro-IL-1β induction (Fig. 5i). Furthermore, TPI-1 blocked the anti-inflammatory effect of colchicine in vivo in the zymosan-induced peritonitis model (Fig. 5j). These data suggest that SHP-1 transduces the GDF15-dependent, anti-inflammatory activity of colchicine.
Discussion
Our work shows that colchicine acts therapeutically in mice by selectively targeting hepatocytes and inducing anti-inflammatory hepatokines, including GDF15, which inhibit myeloid cell activation by positive regulation of SHP-1. These results reveal an unprecedented therapeutic mechanism and a new pathway for communication between hepatocytes and myeloid cells (Fig. 5k). Hepatocytes specialize in clearance of xenobiotic compounds, so this kind of indirect drug action might play an unappreciated role in the pharmacology of other traditional medicines.
Selective accumulation of colchicine in the liver was already known from PK experiments14. Our work demonstrates selective action in hepatocytes by using several PD biomarkers, including microtubules, JNK, Nrf2 and GDF15. Importantly, the microtubule biomarker showed that circulating myeloid cells were not responsive at the safe dose while hepatocytes were, consistent with our PK-PD model, which showed that colchicine can act only in cells where it is actively concentrated. We propose that selective action in hepatocytes arises from concentration of free colchicine across the sinusoidal plasma membrane by transporters not expressed in other cell types. In hepatocytes, microtubule depolymerization by colchicine-activated SQSTM1/p62-dependent selective autophagy, leading to Keap1 degradation, Nrf2 activation and extensive changes in gene expression. Nrf2 activation had at least two effects: it protected hepatocytes from colchicine toxicity and induced the secretion of hepatokines including GDF15. Circulating plasma GDF15 was required for the anti-inflammatory action of colchicine measured in both in vivo and ex vivo assays.
Our work reveals a new role of GDF15 in communication between hepatocytes and myeloid cells, which further complicates the biology of this interesting signalling protein. Induction of GDF15 in the liver by toxins was reported previously47. GDF15 contributes to liver health31 and systemic triglyceride metabolism in response to infection34. The best-characterized function of GDF15 is inhibition of food intake via binding to the receptor GFRAL, leading to recruitment of the co-receptor RET and activation of a signalling cascade involving ERK phosphorylation. GFRAL is expressed in only a small fraction of neurons in the hindbrain, and not in blood cells30,48. The effects of colchicine-induced plasma GDF15 on myeloid cells reported here were almost certainly not mediated by GFRAL, since GFRAL is not expressed in myeloid cells. We performed multiple ex vivo and in vivo experiments and confirmed the absence of GFRAL dependence (Extended Data Fig. 10a–d). Modification, maturation, secretion, chaperoning and ligand presentation in blood are very complex for TGF-β itself49,50, and knowledge of other family members is limited51. It is therefore plausible that more than one molecular form of GDF15 has biological activity. We tested recombinant mature GDF15, which is a disulfide-linked dimer, from multiple sources in our ex vivo and in vivo assays, and found that it lacked anti-inflammatory activity although it efficiently activated ERK phosphorylation via GFRAL-RET in artificial models (Extended Data Fig. 10e–g). We therefore suspect that the molecular form of plasma GDF15 induced by colchicine, and the receptor on myeloid cells through which it acts, are new and require further characterization.
The tyrosine phosphatase SHP-1 plays a central role in regulation of myeloid cell activation, by coordinating signals from multiple activating and inhibitory receptors45. Our work implicates SHP-1 activation in the therapeutic activity of colchicine. Our proposal that colchicine-induced hepatokines prevent SHP-1 inactivation by inflammatory signals is consistent with clinical data. For example, a single dose of colchicine in humans reduces the adhesion of circulating neutrophils to nylon fibres for several days52, and SHP-1 is known to inhibit integrin activation45. SHP-1 also negatively regulates the expression of pro-inflammatory cytokines, consistent with the clinical effects of colchicine53.
Our work has notable implications for colchicine repurposing and new drug development. Colchicine is under investigation for repurposing in cardiovascular disease4,5, auto-inflammatory dermatitis54 and COVID-19 infection6. The mechanism we report may be useful for guiding patient selection and providing PD biomarkers. The molecular action of colchicine on microtubules is intrinsically toxic, which limits its therapeutic application. Drugs that mimic colchicine by less toxic mechanisms—for example, by activation of Nrf2 in hepatocytes or SHP-1 in myeloid cells—might provide safer and more effective treatments for inflammatory diseases.
Methods
Antibodies and reagents
Colchicine (no. CO115) at USP grade was obtained from Spectrum Chemical. TPI-1 (no. HY-100463) and BBT594 (no. HY-18840) were acquired from MedChemExpress. MSU crystals (no. tlrl-msu), nigericin (no. tlrl-nig), Pam3CSK4 (no. tlrl-pms) and LPS (no. tlrl-pb5lps) were obtained from Invivogen; 3-desmethylcolchicine (no. D230885) was acquired from Toronto Research Chemicals. Antibodies against GAPDH (no. 2118; 5,000× dilution), Nrf2 (no. 12721; 1,000× dilution), phospho-JNK (no. 9255; 3,000× dilution), SQSTM1/p62 (no. 23214; 1,000× dilution), phospho-S349-SQSTM1/p62 (no. 16177; 1,000× dilution), phospho-Y564-SHP-1 (no. 8849; 2,000× dilution), LC3B (no. 2775; 1,000× dilution) and mouse anti-rabbit IgG conformation specific (no. 3678) were all from Cell Signaling Technology. Tunicamycin (no. 3516) was from Tocris Bioscience. Antibodies against mouse IL-1β (no. AF-401; 2,000× dilution) and GDF15 enzyme-linked immunosorbent assay (ELISA) (no. DY-6385) for plasma GDF15 measurement were from R&D Systems. Antibodies against HNF4 (no. ab181604) and GDF15 (nos. ab189358 and ab105738) were from Abcam. The polyclonal GDF15 antibody was generated using the N terminus of mouse GDF15 as the antigen. The monoclonal GDF15-blocking antibody (patent ID: WO2014100689A1) was a kind gift of H. Tian. The antagonistic monoclonal GFRAL antibody (no. 3P10) was generated by immunization of mice with recombinant GFRAL extracellular domain (no. D1-D3). PMA (no. P1585), Zymosan (no. z4250), SPP86 (no. SML1435) and antibodies against tubulin (no. T6199) and Keap1 (no. MABS514; 2,000× dilution) were from Millipore Sigma. Antibodies against SHP-1 (no. sc-7289; 100× dilution), JNK (no. sc-7345; 1,000× dilution) and actin (no. sc-47778; 5,000× dilution) were from Santa Cruz. The anti-S591-SHP-1 antibody (no. SP1531; 2,000× dilution) was from ECM Biosciences. Anti-F4/80 antibody (no. MCA497GA) was from Bio-Rad. Anti-Ly-6G antibodies for fluorescent activated cell sorter (FACS) analysis (nos. 127614 and 127608) were from Biolegend. For immunoblotting, anti-rabbit IgG (no. 535571) and anti-mouse IgG (no. 35518) were from Invitrogen and anti-rat IgG (no. 926-68076) was from LI-COR. Dihydrorhodamine 123 (no. D23806), DAPI (no. D1306) and Lipofectamine 3000 Transfection Reagent (no. L3000008) were from ThermoFisher. The Dual-Glo Luciferase Assay System (no. E2920) was from Promega. Recombinant mouse GDF15, as the disulfide cross-linked, mature homodimer, was purified from conditioned medium collected from Expi-293F cells (Invitrogen), which express the core peptides of mouse GDF15.
Mouse maintenance
Gdf15−/− mice were a kind gift of S.-J. Lee47. Gdf15−/− mice were generated on a hybrid C57BL/6/129/SvJ background and back-crossed to C57BL/6J47. Following arrival at the Harvard Medical School animal facility, mice were bred to C57BL/6J for three generations. Nrf2−/− (no. 017009) and wild-type C57BL/6J (no. 000664) mice at 7–8 weeks of age were purchased from Jackson Laboratories and kept for at least 1 week before commencement of experiments. GFRAL knockout mice were purchased from Taconic, back-crossed for seven generations to a >98% C57BL/6 background and kept at NGM Biopharmaceuticals animal facility30. Mice were maintained on a 12/12-h light/dark cycle under specific-pathogen-free conditions. Eight- to 11-week-old male mice were used for all animal experiments in this study and wild-type C57BL/6J mice were used as control. To examine GDF15 induction, both male and female mice were used. The animal protocol was approved by the Harvard Medical Area Standing Committee on Animals, and mice were housed and handled in accordance with accreditation guidelines. Studies with GFRAL knockout mice were performed at NGM Biopharmaceuticals with NGM Institutional Animal Care and Use Committee-approved protocols, and all relevant ethical regulations were complied with.
Depleting genes of hepatocytes
N-acetylgalactosamine-conjugated siRNAs were synthesized by Alnylam Pharmaceuticals. To identify those GalNAc-siRNAs that deplete GDF15 and Nrf2 efficiently, we initially tested 40 GalNAc-siRNAs against different regions of the genes in primary mouse hepatocytes, and then picked five for follow-up testing in wild-type mice to identify those most efficient. To deplete colchicine-induced expression of GDF15 or Nrf2, mice were subcutaneously injected with GalNAc-siRNA at 10 mg kg–1. Seven days after GalNAc-siRNA injection, mice were treated with colchicine and levels of GDF15 or Nrf2 in livers were measured by immunoblotting. The GalNAc-siRNA against luciferase was used as a control.
Zymosan- or MSU-induced peritonitis
Murine peritonitis was induced by intraperitoneal injection of either 0.1 mg zymosan or 1 mg MSU, suspended in PBS. Three hours after challenge, peritoneal exudate cells were collected and stained with the antibody against Ly-6G for detection of neutrophils. The samples were then processed on an LSRII flow cytometry analyser. FACS files were analysed using FlowJo (Becton, Dickinson & Company). For measurement of plasma IL-1β levels, plasma samples were collected 5 h after challenge. For neutralization of GDF15, 10 mg kg–1 GDF15-blocking antibodies were used. For testing of mature GDF15 effects, recombinant mouse GDF15 was injected subcutaneously at 1 mg kg–1 one hour before zymosan challenge. For blocking of GFRAL, 3 mg kg–1 GFRAL antagonistic antibodies were used.
Conditioned plasma collection, GDF15 ELISA and liver function assessment
After vehicle or colchicine treatment, mice were euthanized with isoflurane and cardiac puncture was performed to collect blood samples. Plasma was separated with heparinized tubes (BD Pharmingen). The concentration of GDF15 in conditioned plasma samples was measured with a mouse GDF15 ELISA kit (R&D Systems) in accordance with the manufacturer’s instructions. For measurement of ALT and AST, serum samples were collected with Microtainer tubes (BD Pharmingen) and analysed on a Beckman AU680 Clinical Chemistry Analyzer.
Ex vivo inflammation assays on ROS production, adherence and cytokine induction
Bone marrow cells were flushed out from femurs and tibias of wild-type male mice, and red blood cells were removed using RBC Lysis solution (Miltenyi Biotec) in accordance with the manufacturer’s protocol. Neutrophils were labelled with Ly-6G antibodies at 4 °C. Then, blood cells were incubated with 50% conditioned plasma from vehicle- or colchicine-treated mice for 30 min at 37 °C. To measure ROS production in neutrophils, cells were treated with vehicle or 200 nM PMA for 15 min, followed by treatment with 5 μM dihydrorhodamine 123 for 15 min to detect ROS production. The staining reaction was stopped by placing the samples on ice and washing cells with cold PBS three times. Subsequently, cells with both Ly-6G and activated rhodamine 123 signals were detected with an LSRII flow cytometry analyser. To detect neutrophil adhesion to the walls of plastic tubes, cells were activated with PMA for 30 min, suspended cells were collected and Ly-6G-expressing cells were measured with the LSRII analyser. To measure neutrophil adhesion to glass coverslips, primary neutrophils were isolated from bone marrow cells according to the instructions provided in the Neutrophil Isolation Kit (Miltenyi Biotec), and were cultured with 50% conditioned plasma for 30 min at 37 °C. Then, primary neutrophils were treated with PMA, seeded on glass coverslips and cultured for 30 min at 37 °C. Adherent cells were fixed with 4% paraformaldehyde (PFA) in PBS buffer, and stained with DAPI. Images were acquired with a Nikon Ti fluorescence microscope. Cell numbers per field were quantified by ImageJ software (National Institutes of Health). To analyse induction of the pro-inflammatory cytokine, IL-1β, purified neutrophils or immortalized BMDM cells were co-treated with conditioned plasma and PMA (neutrophils) or Pam3CSK4 (immortalized BMDM) for 3 h. Expression of pro-IL-1β was measured by immunoblotting. To measure the secretion of mature IL-1β, Pam3CSK4-primed BMDM cells were activated with nigericin.
Immunocytochemistry and microscopy
Livers were rinsed with PBS and fixed with 4% PFA in PEM buffer (100 mM PIPES pH 6.9, 5 mM MgCl2, 5 mM EGTA). After fixation, the fixative buffer was replaced by 15–30% sucrose series, embedded in Tissue-Tek optimal cutting temperature (Sakura Finetek) and stored at −80 °C. OCT-embedded livers were cut to a thickness of 10 μm. Whole-blood samples were fixed in PEM buffer with acid citrate-dextrose. Liver sections were stained with antibodies against tubulin, HNF4 and F4/80. Blood cells were stained with antibodies against tubulin. Nuclei were labelled with DAPI. Images were acquired on a Zeiss Axio Observer Z1 equipped with a Plan Apo ×63/1.4 numerical aperture DIC objective and an LSM780 single-point scanning confocal scan head. Images were collected using ZEN black SP5.
Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblotting were performed as described previously55. Briefly, liver tissues were lysed with lysis buffer (50 mM Tris-HCl pH 7.5, 5 mM EDTA, 300 mM NaCl, 1% Triton X-100) for 30 min on ice, and the cell lysate was incubated with antibodies against SQSTM1/p62. After incubation for 2 h at 4 °C, Protein G affinity beads (Millipore Sigma) were added and samples were incubated for an additional 1 h. Proteins were eluted from beads with gel sample buffer (50 mM Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 0.1% bromophenol blue, 100 mM DTT) and separated by NuPAGE 4–12% Bis-Tris Protein Gels (ThermoFisher). Image Studio was used for immunoblot quantification.
Luciferase reporter assay
HEK 293T cells in 24-well plates were transfected with 100 ng of luciferase reporter plasmids, 200- or 1,000-ng transcription factor Nrf2 plasmids as indicated and 10 ng of Renilla luciferase internal control plasmids using Lipofectamine 3000 Transfection Reagent (ThermoFisher) in accordance with the manufacturer’s instructions. Two days post transfection, luciferase assays were performed according instructions provided with the Dual-Glo Luciferase Assay System (Promega).
TG-GATEs analysis
Biological triplicates of liver gene expression data were acquired from the Open TG-GATEs database (https://toxico.nibiohn.go.jp/open-tggates/english/search.html). The CEL files were normalized and log-transformed using the packages affy, affyio, BufferedMatrix, BufferedMatrixMethods and rat2302.db. Two-sided t-testing was adapted for computation of the significance of gene expression change. The collection of genes encoding secreted proteins was obtained from the Human Protein Atlas (https://www.proteinatlas.org/) and Plasma Proteome Database (http://plasmaproteomedatabase.org/).
Human PK-PD simulation
A two-compartment PK-PD model, based on ref. 12, was reconstructed in the SimBiology package in MATLAB (MathWorks). The central and peripheral compartments in the model were built to represent the plasma and myeloid cell, respectively. All parameters, including absorption, absorption time lag, duration of absorption, urinary elimination, central elimination and transfer between compartments, were taken from ref. 12. The fraction of colchicine-bound tubulin was calculated from the following equations (1)–(3) (variables described in Extended Data Fig. 5):
To simulate concentration of unbound colchicine across the plasma membrane, kaPlasma was increased by a factor of 10 or 100, respectively. All simulations were performed in SimBiology, and results were exported for post-processing in Prism v.7 (GraphPad).
Image quantification
Quantification of microtubule intensity was done using CellProfiler 3.1.8 (The Broad Institute). For hepatocytes, the nuclei were first segmented using the HNF4 stain. Hepatic cytoplasmic areas were then determined by expansion of a bubble outside the nucleus and subtraction of the nuclear area. Microtubule intensities were then calculated in this area. Kupffer cells were segmented using the F4/80 stain, and microtubule intensity in each cellular area was then calculated. The minimum cross-entropy method was used for cell segmentations, along with diameter constraints to exclude objects that were too small. All data, at single-cell level, were then exported to MATLAB (MathWorks) and Prism 7 (GraphPad) for post-processing. For quantification of SQSTM1/p62 puncta, images were first smoothened with a Gaussian filter in CellProfiler and puncta were then segmented using the minimum cross-entropy method. The total number of puncta, as well as the intensity values of individual puncta, were then calculated and exported for post-processing.
Data analysis
Quantification results were analysed in Excel software (Microsoft) and post-processed in Prism v.7 (GraphPad). All results are shown as mean ± s.d., and two-sided Student’s t-tests were used for statistical analysis.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding authors on reasonable request. Source data are provided with this paper.
Change history
05 May 2021
A Correction to this paper has been published: https://doi.org/10.1038/s42255-021-00397-5
References
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Dinarello, C. A. Anti-inflammatory agents: present and future. Cell 140, 935–950 (2010).
Dasgeb, B. et al. Colchicine: an ancient drug with novel applications. Br. J. Dermatol. 178, 350–356 (2018).
Nidorf, S. M., Eikelboom, J. W., Budgeon, C. A. & Thompson, P. L. Low-dose colchicine for secondary prevention of cardiovascular disease. J. Am. Coll. Cardiol. 61, 404–410 (2013).
Tardif, J. C. et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 381, 2497–2505 (2019).
Deftereos, S. G. et al. Effect of colchicine vs standard care on cardiac and inflammatory biomarkers and clinical outcomes in patients hospitalized with coronavirus disease 2019: the GRECCO-19 randomized clinical trial. JAMA Netw. Open 3, e2013136 (2020).
Finkelstein, Y. et al. Colchicine poisoning: the dark side of an ancient drug. Clin. Toxicol. (Phila.) 48, 407–414 (2010).
Weisenberg, R. C., Borisy, G. G. & Taylor, E. W. The colchicine-binding protein of mammalian brain and its relation to microtubules. Biochemistry 7, 4466–4479 (1968).
Dinarello, C. A. et al. Effect of prophylactic colchicine therapy on leukocyte function in patients with familial Mediterranean fever. Arthritis Rheumatol. 19, 618–622 (1976).
Caner, J. E. Colchicine inhibition of chemotaxis. Arthritis Rheumatol. 8, 757–764 (1965).
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006).
Thomas, G., Girre, C., Scherrmann, J. M., Francheteau, P. & Steimer, J. L. Zero-order absorption and linear disposition of oral colchicine in healthy volunteers. Eur. J. Clin. Pharm. 37, 79–84 (1989).
Florian, S. & Mitchison, T. J. Anti-microtubule drugs. Methods Mol. Biol. 1413, 403–421 (2016).
Hunter, A. L. & Klaassen, C. D. Biliary excretion of colchicine. J. Pharmacol. Exp. Ther. 192, 605–617 (1975).
Ghorpade, D. S. et al. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 555, 673–677 (2018).
Ferguson, F. C. Jr. Colchicine. I. General pharmacology. J. Pharmacol. Exp. Ther. 106, 261–270 (1952).
Terkeltaub, R. A. et al. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheumatol. 62, 1060–1068 (2010).
Wang, T. H. et al. Microtubule-interfering agents activate c-Jun N-terminal kinase/stress-activated protein kinase through both Ras and apoptosis signal-regulating kinase pathways. J. Biol. Chem. 273, 4928–4936 (1998).
Cereghini, S. Liver-enriched transcription factors and hepatocyte differentiation. FASEB J. 10, 267–282 (1996).
Faber, K. N., Muller, M. & Jansen, P. L. Drug transport proteins in the liver. Adv. Drug Deliv. Rev. 55, 107–124 (2003).
Bergen, L. G. & Borisy, G. G. Tubulin-colchicine complex inhibits microtubule elongation at both plus and minus ends. J. Biol. Chem. 258, 4190–4194 (1983).
Magupalli, V. G. et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369, eaas8995 (2020).
Ozen, S. et al. EULAR recommendations for the management of familial Mediterranean fever. Ann. Rheum. Dis. 75, 644–651 (2016).
Cuadrado, A. et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 18, 295–317 (2019).
Komatsu, M. et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223 (2010).
Bae, S. H. et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 17, 73–84 (2013).
Ichimura, Y. et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 51, 618–631 (2013).
Johansen, T. & Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296 (2011).
Igarashi, Y. et al. Open TG-GATEs: a large-scale toxicogenomics database. Nucleic Acids Res. 43, D921–D927 (2015).
Hsu, J. Y. et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 550, 255–259 (2017).
Kim, K. H. et al. Growth differentiation factor 15 ameliorates nonalcoholic steatohepatitis and related metabolic disorders in mice. Sci. Rep. 8, 6789 (2018).
de Jager, S. C. et al. Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J. Exp. Med. 208, 217–225 (2011).
Kempf, T. et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ. Res. 98, 351–360 (2006).
Luan, H. H. et al. GDF15 Is an Inflammation-Induced central mediator of tissue tolerance. Cell 178, 1231–1244 e1211 (2019).
Yatsuga, S. et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 78, 814–823 (2015).
Wollert, K. C., Kempf, T. & Wallentin, L. Growth differentiation factor 15 as a biomarker in cardiovascular disease. Clin. Chem. 63, 140–151 (2017).
Johnen, H. et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 13, 1333–1340 (2007).
Tanaka, T. et al. Plasma proteomic signature of age in healthy humans. Aging Cell 17, e12799 (2018).
Fejzo, M. S. et al. Placenta and appetite genes GDF15 and IGFBP7 are associated with hyperemesis gravidarum. Nat. Commun. 9, 1178 (2018).
Demidowich, A. P. et al. Colchicine’s effects on metabolic and inflammatory molecules in adults with obesity and metabolic syndrome: results from a pilot randomized controlled trial. Int. J. Obes. 44, 1793–1799 (2020).
Nair, J. K. et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 136, 16958–16961 (2014).
Coll, A. P. et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature 578, 444–448 (2020).
Mocsai, A., Ruland, J. & Tybulewicz, V. L. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 10, 387–402 (2010).
Veillette, A., Latour, S. & Davidson, D. Negative regulation of immunoreceptor signaling. Annu. Rev. Immunol. 20, 669–707 (2002).
Abram, C. L. & Lowell, C. A. Shp1 function in myeloid cells. J. Leukoc. Biol. 102, 657–675 (2017).
Kundu, S. et al. Novel SHP-1 inhibitors tyrosine phosphatase inhibitor-1 and analogs with preclinical anti-tumor activities as tolerated oral agents. J. Immunol. 184, 6529–6536 (2010).
Hsiao, E. C. et al. Characterization of growth-differentiation factor 15, a transforming growth factor beta superfamily member induced following liver injury. Mol. Cell. Biol. 20, 3742–3751 (2000).
Yang, L. et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 23, 1158–1166 (2017).
Constam, D. B. Regulation of TGFbeta and related signals by precursor processing. Semin. Cell Dev. Biol. 32, 85–97 (2014).
Derynck, R. & Budi, E. H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 12, eaav5183 (2019).
Hinck, A. P., Mueller, T. D. & Springer, T. A. Structural biology and evolution of the TGF-beta family. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a022103 (2016).
Fordham, J. N., Kirwan, J., Cason, J. & Currey, H. L. Prolonged reduction in polymorphonuclear adhesion following oral colchicine. Ann. Rheum. Dis. 40, 605–608 (1981).
Martinez, G. J. et al. Colchicine acutely suppresses local cardiac production of inflammatory cytokines in patients with an acute coronary syndrome. J. Am. Heart Assoc. 4, e002128 (2015).
Robinson, K. P. & Chan, J. J. Colchicine in dermatology: a review. Australas. J. Dermatol. 59, 278–285 (2018).
Weng, J. H. et al. Pregnenolone activates CLIP-170 to promote microtubule growth and cell migration. Nat. Chem. Biol. 9, 636–642 (2013).
Acknowledgements
We thank S.-J. Lee for the Gdf15−/− mice and J. Kagan for immortalized BMDM cells. The GDF15-blocking antibody was a kind gift from H. Tian of NGM Biopharmaceuticals. We thank W.-T. Kuo of Harvard Medical School for assistance with tissue immunostaining. We thank M. Zhou of NGM Biopharmaceuticals for assistance with animal experiments. This work was supported by grants from the National Institutes of Health (no. GM131753 to T.J.M) and the American Heart Association (no. 8POST34080251 to J.-H.W.).
Author information
Authors and Affiliations
Contributions
J.-H.W. designed and conducted the experiments and wrote the manuscript. P.D.K. performed PK-PD modelling and image quantification. H.H.L. and I.N. examined GFRAL neutralization with the peritonitis assay. H.-C.T. generated hepatocyte-specific siRNAs. K.S. and R.J. examined the Open TG-GATEs database. R.V. tested the activity of recombinant GDF15. T.J.M. oversaw the execution of the project and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
H.-C.T. was an employee of Alnylam Pharmaceuticals when the work was done. H.H.L., I.N. and R.V. were employees of NGM Biopharmaceuticals when the work was done. The other authors declare no competing interests.
Additional information
Peer review information Primary Handling Editor: Christoph Schmitt.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Selection of colchicine doses for mouse treatment.
a, Dose translation from humans to animal studies based on clinical guidelines. The colchicine doses for mouse via the oral or i.p. routes were calculated according to the guidelines from the Food and Drug Administration and the European League Against Rheumatism, and were listed in the table. i.p., intraperitoneal. i.v., intravascular. b-f, Safety analysis of colchicine based on measurement of gut toxicity. Diarrhea is the dose-limiting toxicity in man. Mice received either vehicle, colchicine at 0.4 mg/kg, or 2.4 mg/kg. b, Intestines were harvested 6 hours after vehicle or colchicine treatment. Scale bars, 1 cm. c, The physiological severity score for diarrhea. d, Colchicine at 0.4 mg/kg did not affect the region of intestine with feces, but 2.4 mg/kg reduced it. Each dot represents one mouse. e, Detection of fecal water content by measuring weight of feces before and after drying. f, Colchicine at 0.4 mg/kg did not affect the fecal water content. Data are represented as mean ± s.d. Two-sided t-tests were used for statistical analysis. A single colchicine dose of 0.4mg/mg i.p. was used in all subsequent experiments unless otherwise indicated.
Extended Data Fig. 2 Colchicine at a safe dose selectively targets hepatocytes.
Additional images related to Fig. 1g. Microtubules in the livers of mice treated with a, vehicle or b, 0.4 mg/kg colchicine. Livers were collected 6 hours after treatment. Colchicine selectively depolymerized microtubules in hepatocytes, identified by HNF4+. The boxed areas are magnified and shown at the bottom. Liver cells from 4 livers with vehicle or colchicine treatment respectively. Scale bars, 10 μm.
Extended Data Fig. 3 Colchicine at a safe dose does not damage microtubules in circulating blood cells.
Six hours after treatment of vehicle or colchicine (0.4 mg/kg or 2.4 mg/kg), the whole blood samples were collected and fixed directly. Microtubules were visualized by immunofluorescence and imaged by confocal microscopy. a, Representative images of microtubules in circulating blood cells. b, Quantification of microtubule staining intensity. Individual cells exhibited a wide range of microtubule intensities that were similar following vehicle or 0.4 mg/kg colchicine (the safe dose). Microtubules were depolymerized in cells from mice receiving 2.4 mg/kg (the toxic dose). AU, arbitrary unit. Scale bar, 5 μm.
Extended Data Fig. 4 Colchicine at a toxic dose targets all cell types in the liver.
Microtubules in a, vehicle- or b, 2.4 mg/kg colchicine-treated livers were visualized, and c, intensities of microtubules were quantified. Microtubules were depolymerized in all cell types after treatment of colchicine at 2.4 mg/kg. The boxed areas are magnified and shown at the bottom. AU, arbitrary unit. Scale bars, 10 μm.
Extended Data Fig. 5 Modeling of human pharmacokinetics-pharmacodynamics (PK-PD) of colchicine.
a, Simulation diagram of two-compartment PK-PD model, based on human pharmacology data from Thomas, G. et al. Two blue boxes represent the central and peripheral compartments, which correspond to plasma and myeloid cell respectively. Light green boxes correspond to reaction species (colchicine concentration in plasma or myeloid cell), and yellow circles correspond to reactions (absorption, urinary elimination, central elimination, and transfer between plasma and myeloid cell). Colchicine absorption takes place from t = 0.22 to t = 1 hour. Tubulin-colchicine binding biochemistry was ignored and all colchicine in the myeloid cells was assumed to bind to tubulin. Fraction bound was calculated based on equations listed in Methods. b, Constants for modeling. c, Human PK-PD modeling of colchicine supported the indirect action of colchicine. The blue line represents colchicine concentrations in plasma over time after colchicine dosing. Magenta lines show the fraction of colchicine-bound tubulin in myeloid cells. Dashed and dotted magenta lines show unbound colchicine concentrated across the cell membrane by a factor of 10 and 100 respectively. The green line marks the 2% threshold of site occupancy for colchicine action.
Extended Data Fig. 6 GDF15 is a novel hepatokine induced by colchicine.
a, Volcano plot for the gene expression changes of rat livers comparing 3 hours after vehicle to colchicine treatment. Biological triplicate data are from the Open TG-GATEs database. Red and gray dots represent genes encoding secreted and non-secreted proteins respectively. GDF15 is highlighted in blue. P, probability value. b, Time course of GDF15 mRNA expression in the rat liver. Data are from Open TG-GATEs. 3 Mice per condition. c, GDF15 was induced in the liver but not kidney collected from colchicine-treated mice. d, ELISA analysis for measurement of plasma GDF15 in both mouse sexes. Induction was reproducibly stronger in males in the C57BL/6J mouse strain. Each dot represents one mouse. 5 Mice per condition. 3 Biological replicates. Data are represented as mean ± s.d. Two-sided t-tests were used for statistical analysis.
Extended Data Fig. 7 GDF15 is a novel anti-inflammatory hepatokine induced by Nrf2 signaling triggered by colchicine.
a, ELISA analysis for measurement of plasma GDF15 in wild-type and Nrf2 germline knockout (Nrf2−/−) mice. Each dot represents one mouse. b, Immunoblotting analysis revealed that induction of GDF15 in livers was blocked by treatment of hepatocyte-specific siRNA (GalNAc-siRNA) against Nrf2. Luc, luciferase. 3 Biological replicates. c, Activation of GDF15 promoter activity by Nrf2. HEK 293T cells were co-transfected with the luciferase reporter driven by the GDF15 promoter and the Nrf2 plasmid. Luciferase activities were determined from three samples. Data from three independent experiments. d, Immunoblotting analysis for detection of CHOP expression in livers collected at the indicated times after colchicine or Tunicamycin treatment. e, The temporal mRNA expression pattern of the integrated stress response regulators in the rat liver. Data are from Open TG-GATEs. 3 Mice per condition. f, The anti-inflammatory effect of colchicine in the MSU (monosodium urate crystals) peritonitis model depended on GDF15 induction in hepatocytes. Mice were challenged with 1 mg MSU. GalNAc-siRNA, hepatocyte-specific siRNA. Luc, luciferase. Each dot represents one mouse. Data are represented as mean ± s.d. Two-sided t-tests were used for statistical analysis.
Extended Data Fig. 8 Colchicine-induced GDF15 inhibits pro-IL-1β expression by activating the immuno-inhibitory signal of SHP-1 in primary neutrophils.
Ex vivo activation of neutrophils evaluated by the levels of (a-b) pro-IL-1β induction or (c-d) phosphorylated SHP-1. Cells were pre-incubated with plasma from the indicated mice and then activated with PMA. a, Induction of GDF15 specifically in hepatocytes was required for the anti-inflammatory activity of post-colchicine plasma, as assayed by pro-IL-1β induction. GalNAc-siLuc, hepatocyte-specific siRNA against luciferase. GalNAc-siGDF15, hepatocyte-specific siRNA against GDF15. b, The anti-inflammatory activity of post-colchicine plasma was blocked by two different clones of antibodies (#2 and #3) against GDF15. ctr, control antibody. veh plasma, plasma from vehicle-treated mice. Colc plasma, plasma from colchicine-treated mice. c, GDF15 neutralization by two clones of anti-GDF15 antibodies (#2 and #3) blocked plasma activity on stabilizing SHP-1 activation. d, Expression of colchicine-induced GDF15 specifically in hepatocytes was required for activating SHP-1. 3 Biological replicates.
Extended Data Fig. 9 Colchicine-induced GDF15 inhibits pro-IL-1β expression and mature IL-1β secretion from macrophages.
Ex vivo activation of immortalized BMDMs evaluated by the levels of (a-b) pro-IL-1β expression and (d-e) IL-1β secretion. a-b, Cells were pre-incubated with plasma from the indicated mice and then activated with Pam3CSK4. Expression of colchicine-induced GDF15 in hepatocytes was required for plasma activity on blocking pro-IL-1β expression. GalNAc-siLuc, hepatocyte-specific siRNA against luciferase. GalNAc-siGDF15, hepatocyte-specific siRNA against GDF15. c, Activation methods for measuring IL-1β production. d, Colchicine did not alter IL-1β production assayed with two activation methods. 5 Samples per condition over 3 biological replicates e, Colchicine-induced GDF15 was required for plasma activity on reducing IL-1β production. 4 Samples per condition over 3 biological replicates Data are represented as mean ± s.d. Two-sided t-tests were used for statistical analysis.
Extended Data Fig. 10 Canonical mature GDF15-GFRAL-RET signaling is not involved in the anti-inflammatory actions of colchicine.
a-c, and f, Ex vivo activation of neutrophils evaluated by the levels of pro-IL-1β expression. Inhibition of the GFRAL-RET signaling by a, RET inhibitor, SPP86 (SPP) and BBT594 (BTT), or b, a GFRAL neutralizing antibody (α-GFRAL) did not alter post-colchicine plasma activity on blocking pro-IL-1β expression. c, Post-colchicine plasma from wild-type and GFRAL germline knockout mice blocked pro-IL-1β expression. d, GFRAL inhibition by a GFRAL neutralizing antibody did not affect the in vivo anti-inflammatory effect of colchicine in the zymosan peritonitis assay. e, Efficacy of recombinant mature GDF15 (rGDF15) measured by the levels of phosphorylated ERK in a GFRAL-RET reporter cell model. 3 Samples per condition. Recombinant GDF15 was a disulfide-linked mature homodimer. Recombinant mature GDF15 did not affect f, ex vivo pro-IL-1β expression, and did not inhibit g, in vivo zymosan-induced neutrophil recruitment as colchicine. Each dot represents one mouse. Data are represented as mean ± s.d. Two-sided t-tests were used for statistical analysis.
Supplementary information
Source data
Source Data Fig. 1
Unprocessed immunoblots.
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Unprocessed immunoblots.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Source data.
Source Data Fig. 4
Unprocessed immunoblots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Unprocessed immunoblots.
Source Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed immunoblots.
Source Data Extended Data Fig. 7
Unprocessed immunoblots.
Source Data Extended Data Fig. 8
Unprocessed immunoblots.
Source Data Extended Data Fig. 9
Unprocessed immunoblots.
Source Data Extended Data Fig. 10
Unprocessed western blots.
Rights and permissions
About this article

Cite this article
Weng, JH., Koch, P.D., Luan, H.H. et al. Colchicine acts selectively in the liver to induce hepatokines that inhibit myeloid cell activation. Nat Metab 3, 513–522 (2021). https://doi.org/10.1038/s42255-021-00366-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-021-00366-y
This article is cited by
-
Colchicine Inhibits NETs and Alleviates Cardiac Remodeling after Acute Myocardial Infarction
Cardiovascular Drugs and Therapy (2024)
-
Docking experiments suggest that gloriosine has microtubule-targeting properties similar to colchicine
Scientific Reports (2023)
-
Macrophage fumarate hydratase restrains mtRNA-mediated interferon production
Nature (2023)
-
GDF15 promotes weight loss by enhancing energy expenditure in muscle
Nature (2023)
-
Tumor-derived GDF-15 blocks LFA-1 dependent T cell recruitment and suppresses responses to anti-PD-1 treatment
Nature Communications (2023)
