
Abstract
The nature and extent of mitochondrial DNA variation in a population and how it affects traits is poorly understood. Here we resequence the mitochondrial genomes of 169 Drosophila Genetic Reference Panel lines, identifying 231 variants that stratify along 12 mitochondrial haplotypes. We identify 1,845 cases of mitonuclear allelic imbalances, thus implying that mitochondrial haplotypes are reflected in the nuclear genome. However, no major fitness effects are associated with mitonuclear imbalance, suggesting that such imbalances reflect population structure at the mitochondrial level rather than genomic incompatibilities. Although mitochondrial haplotypes have no direct impact on mitochondrial respiration, some haplotypes are associated with stress- and metabolism-related phenotypes, including food intake in males. Finally, through reciprocal swapping of mitochondrial genomes, we demonstrate that a mitochondrial haplotype associated with high food intake can rescue a low food intake phenotype. Together, our findings provide new insight into population structure at the mitochondrial level and point to the importance of incorporating mitochondrial haplotypes in genotype–phenotype relationship studies.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
The sequencing data available at the NCBI Sequence Read Archive is currently submitted under accession no. SRP168326 (see also Supplementary Table 20). Source data for Fig. 3 is provided with the paper. All other data (that is, variant files, GRDs) can be found in the remaining Supplementary Tables.
Code availability
All code used for this study has been deposited with GitHub (https://github.com/DeplanckeLab/BeversLitovchenko2018).
Change history
06 April 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014).
Pesole, G. et al. The neglected genome. EMBO Rep. 13, 473–474 (2012).
Latorre-Pellicer, A. et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565 (2016).
Tranah, G. J. et al. Mitochondrial DNA sequence variation in multiple sclerosis. Neurology 85, 325–330 (2015).
Hudson, G., Gomez-Duran, A., Wilson, I. J. & Chinnery, P. F. Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet. 10, e1004369 (2014).
Marom, S., Friger, M. & Mishmar, D. MtDNA meta-analysis reveals both phenotype specificity and allele heterogeneity: a model for differential association. Sci. Rep. 7, 43449 (2017).
Wei, W. et al. Germline selection shapes human mitochondrial DNA diversity. Science 364, eaau6520 (2019).
Weerts, M. J. A. et al. Sensitive detection of mitochondrial DNA variants for analysis of mitochondrial DNA-enriched extracts from frozen tumor tissue. Sci. Rep. 8, 2261 (2018).
McCarthy, M. I. et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 9, 356–369 (2008).
Peirce, J. L., Lu, L., Gu, J., Silver, L. M. & Williams, R. W. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 5, 7 (2004).
Mackay, T. F. C. et al. The Drosophila melanogaster Genetic Reference Panel. Nature 482, 173–178 (2012).
Huang, W. et al. Natural variation in genome architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res. 24, 1193–1208 (2014).
Anholt, R. R. H. & Mackay, T. F. C. The road less traveled: from genotype to phenotype in flies and humans. Mamm. Genome 29, 5–23 (2018).
Richardson, M. F. et al. Population genomics of the Wolbachia endosymbiont in Drosophila melanogaster. PLoS Genet. 8, e1003129 (2012).
Cooper, B. S., Burrus, C. R., Ji, C., Hahn, M. W. & Montooth, K. L. Similar efficacies of selection shape mitochondrial and nuclear genes in both Drosophila melanogaster and Homo sapiens. G3 (Bethesda) 5, 2165–2176 (2015).
Salminen, T. S. et al. Mitochondrial genotype modulates mtDNA copy number and organismal phenotype in Drosophila. Mitochondrion 34, 75–83 (2017).
Zhu, C. T., Ingelmo, P. & Rand, D. M. G×G×E for lifespan in Drosophila: mitochondrial, nuclear, and dietary interactions that modify longevity. PLoS Genet. 10, e1004354 (2014).
Mossman, J. A., Biancani, L. M., Zhu, C. T. & Rand, D. M. Mitonuclear epistasis for development time and its modification by diet in Drosophila. Genetics 203, 463–484 (2016).
Hazkani-covo, E., Zeller, R. M. & Martin, W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 6, e1000834 (2010).
Rogers, H. H. & Griffiths-jones, S. Mitochondrial pseudogenes in the nuclear genomes of Drosophila. PLoS ONE 7, e32593 (2012).
Haag-Liautard, C. et al. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 6, e204 (2008).
Templeton, A. R., Crandall, K. A. & Sing, C. F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132, 619–633 (1992).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Kapun, M. et al. Genomic analysis of European Drosophila populations reveals major longitudinal structure, continent-wide selection, and unknown DNA viruses. Preprint at bioRxiv https://doi.org/10.1101/313759 (2019).
Burman, J. L. et al. A Drosophila model of mitochondrial disease caused by a complex I mutation that uncouples proton pumping from electron transfer. Dis. Model. Mech. 7, 1165–1174 (2014).
Swalwell, H. et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur. J. Hum. Genet. 19, 769–775 (2011).
Corbett-Detig, R. B., Zhou, J., Clark, A. G., Hartl, D. L. & Ayroles, J. F. Genetic incompatibilities are widespread within species. Nature 504, 135–137 (2013).
Greene, J. C. et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl Acad. Sci. USA 100, 4078–4083 (2003).
Matzkin, L. M., Johnson, S., Paight, C., Bozinovic, G. & Markow, T. A. Dietary protein and sugar differentially affect development and metabolic pools in ecologically diverse Drosophila. J. Nutr. 141, 1127–1133 (2011).
Castello, P. R., Drechsel, D. A. & Patel, M. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J. Biol. Chem. 282, 14186–14193 (2007).
Colinet, H., Renault, D. & Roussel, D. Cold acclimation allows Drosophila flies to maintain mitochondrial functioning under cold stress. Insect Biochem. Mol. Biol. 80, 52–60 (2017).
Lovero, D. et al. Characterization of Drosophila ATPsynC mutants as a new model of mitochondrial ATP synthase disorders. PLoS ONE 13, e0201811 (2018).
Kristensen, T. N., Loeschcke, V., Tan, Q., Pertoldi, C. & Mengel-From, J. Sex and age specific reduction in stress resistance and mitochondrial DNA copy number in Drosophila melanogaster. Sci. Rep. 9, 12305 (2019).
Garlapow, M. E., Huang, W., Yarboro, M. T., Peterson, K. R. & Mackay, T. F. C. Quantitative genetics of food intake in Drosophila melanogaster. PLoS ONE 10, e0138129 (2015).
Harbison, S. T., McCoy, L. J. & Mackay, T. F. C. Genome-wide association study of sleep in Drosophila melanogaster. BMC Genomics 14, 281 (2013).
Unckless, R. L., Rottschaefer, S. M. & Lazzaro, B. P. A genome-wide association study for nutritional indices in Drosophila. G3 (Bethesda) 5, 417–425 (2015).
Kaneko, M., Satta, Y., Matsuura, E. T. & Chigusa, S. I. Evolution of the mitochondrial ATPase 6 gene in Drosophila: unusually high level of polymorphism in D. melanogaster. Genet. Res. 61, 195–204 (1993).
Ballard, J. W. & Kreitman, M. Unraveling selection in the mitochondrial genome of Drosophila. Genetics 138, 757–772 (1994).
Rand, D. M. & Kann, L. M. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol. Biol. Evol. 13, 735–748 (1996).
Mishmar, D. et al. Natural selection shaped regional mtDNA variation in humans. Proc. Natl Acad. Sci. USA 100, 171–176 (2003).
Fiedorczuk, K. & Sazanov, L. A. Mammalian mitochondrial complex I structure and disease-causing mutations. Trends Cell Biol. 28, 835–867 (2018).
Salminen, T. S. et al. Lethal interaction of nuclear and mitochondrial genotypes in Drosophila melanogaster. G3 (Bethesda) 9, 2225–2234 (2019).
Rand, D. M., Fry, A. & Sheldahl, L. Nuclear-mitochondrial epistasis and drosophila aging: introgression of Drosophila simulans mtDNA modifies longevity in D. melanogaster nuclear backgrounds. Genetics 172, 329–341 (2006).
James, A. M., Collins, Y., Logan, A. & Murphy, M. P. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol. Metab. 23, 429–434 (2012).
Suomalainen, A. & Battersby, B. J. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 19, 77–92 (2018).
Sharpley, M. S. et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343 (2012).
Rand, D. M., Haney, R. A. & Fry, A. J. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol. Evol. 19, 645–653 (2004).
Moschall, R., Gaik, M. & Medenbach, J. Promiscuity in post-transcriptional control of gene expression: Drosophila sex-lethal and its regulatory partnerships. FEBS Lett. 591, 1471–1488 (2017).
Tower, J. Mitochondrial maintenance failure in aging and role of sexual dimorphism. Arch. Biochem. Biophys. 576, 17–31 (2015).
Schwarze, S., Weindruch, R. & Aiken, J. Oxidative stress and aging reduce COX I RNA and cytochrome oxidase activity in Drosophila. Free Radic. Biol. Med. 25, 740–747 (1998).
Picelli, S. et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040 (2014).
Alpern, D. et al. BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome Biol. 20, 71 (2019).
Krueger, F. Trim Galore!: a wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Rensch, T., Villar, D., Horvath, J., Odom, D. T. & Flicek, P. Mitochondrial heteroplasmy in vertebrates using ChIP-sequencing data. Genome Biol. 17, 139 (2016).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Katoh, K., Kuma, K. I., Toh, H. & Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (2005).
Jha, P., Wang, X. & Auwerx, J. Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 6, 1–14 (2016).
Garcia, C. J., Khajeh, J., Coulanges, E., Chen, E. I. J. & Owusu-Ansah, E. Regulation of mitochondrial complex I biogenesis in Drosophila flight muscles. Cell Rep. 20, 264–278 (2017).
Aw, W. C., Bajracharya, R., Towarnicki, S. G. & Ballard, J. W. O. Assessing bioenergetic functions from isolated mitochondria in Drosophila melanogaster. J. Biol. Methods 3, e42 (2016).
Iuso, A., Repp, B., Biagosch, C., Terrile, C. & Prokisch, H. Assessing mitochondrial bioenergetics in isolated mitochondria from various mouse tissues using Seahorse XF96 analyzer. Methods Mol. Biol. 1567, 217–230 (2017).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Zhao, H. et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007 (2014).
Benjamini, Y. & Bogomolov, M. Selective inference on multiple families of hypotheses. J. R. Stat. Soc. B 76, 297–318 (2014).
Yim, A. et al. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations. Preprint at bioRxiv https://doi.org/10.1101/641423 (2019).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Alexa, A. & Rahnenfuhrer, J. topGO: Enrichment analysis for gene ontology. R package version 2.32.0. https://bioconductor.org/packages/release/bioc/html/topGO.html (2016).
Acknowledgements
We thank the laboratory members of the Deplancke laboratory for helpful suggestions regarding the experiments and analyses and in particular D. Alpern, V. Gardeux and M. Frochaux. We are also grateful for the assistance from the Auwerx and Schoonjans laboratories and in particular from V. Lemos, A. Mottis and P. Luan. We thank the genomics core facilities at the Université de Lausanne and École Polytechnique Fédérale de Lausanne (EPFL) for sequencing the libraries, and B. Habermann for valuable suggestions on mitonuclear genes. We are also grateful for the computational infrastructure provided by the Swiss Institute of Bioinformatics and Vital-IT at EPFL. This project was funded by a grant from SystemsX.ch to B.D. (AgingX, 51RTP0_151019) and by institutional support from EPFL.
Author information
Authors and Affiliations
Contributions
R.P.J.B., M.L. and B.D. conceptualized the study. R.P.J.B., M.L. and V.S.B. performed the experiments. M.L., R.P.J.B. and A.K. performed the computational analyses. M.R.R., J.A. and B.H. provided critical suggestions and comments on the manuscript. R.P.J.B., M.L. and B.D. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Primary Handling Editors: Christoph Schmitt, Ana Mateus.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Quality assessment of mtDNA-enriched libraries.
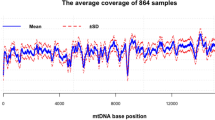
a) Each bar represents the coverage of a single DGRP line. Samples sequenced using a paired-end strategy are shown in grey and samples sequenced using a single-end strategy are shown in blue. See also Supplementary Table 1 for a detailed overview of sequence statistics per sample. b) Comparison of the normalized coverage between our mtDNA sequencing method (blue) and the regular sequencing profile of Mackay et al. 2012 [11] (pink) across the mitochondrial genome. Solid lines depict the coverage profile per DGRP line and dashed lines depict the overall coverage per bp achieved by each of the studies. GC-content is depicted with a grey solid line (200 bp bins). Light green blocks represent the mitochondrial genes excluding tRNAs. c) Dots represent the coverage per DGRP line in the coding region. The barplot represents the mean coverage over all DGRP lines in the coding region. Error bars represent the standard error of means. d) Number of DGRP lines with loci containing a coverage below 10× . e) Average number of loci with a coverage below 10 × . Dots represent number of loci below 10× per DGRP line, and the barplot represents the mean of all populations with loci below 10× coverage. Error bars represent the standard error of means of all represented populations.
Extended Data Fig. 2 NUMT analysis schematic.
A schematic representation for our newly developed computational strategy to detect NUMTs. The upper part shows the mapping of short Illumina reads solely to the mitochondrial genome and the detection of points that could be putative integration sites of mitochondrial fragments into the nuclear genome (breakpoints) as transitions from soft clipped bases to matching bases (SM) or the other way around (MS). All possible combinations of Illumina paired end reads in relation to their alignment to the breakpoints are depicted. The reads that could not be used in the breakpoint inference are shaded. Parts of the read matched to the mitochondrial genome is colored in orange, whereas soft clipped parts, putatively matching to the nuclear genome, are colored in green. The nuclear genome harboring the NUMT is shaded and represented with a dotted line to indicate that the reads were not aligned to it. A similar scenario would take place for the single end Illumina reads. For the second part, alignment of the 454 reads to SM and MS genomes is performed. SM reads are reads with the first part being soft clipped and the second part matching to the mitochondrial genome. Those reads mark the start of putative NUMTs. The consensus sequence of reads was inferred and was considered as an SM genome. Similarly, MS reads mark the end of putative NUMTs and thus the consensus of the reads presents an MS genome. Next, 454 reads were mapped to both SM and MS genomes and only reads aligned to both SM and MS ‘chromosomes’ were used to infer a consensus sequence of a putative NUMT.
Extended Data Fig. 3 Flow scheme of genotyping and variant calling.
Samples were first genotyped based on nuclear fragments that were sequenced by Mackay et al. 2012 [11]. Corrections for the genotype (where necessary) were applied prior to mitochondrial variant calling.
Extended Data Fig. 4 Variant distribution and heterozygosity within the DGRP.
a) The average number of mitochondrial variants per DGRP. b) Number of variants per basepair per gene per type corrected for gene length. Next to each bar, the number of variants and gene length are displayed. c) Flanking regions that were used to accurately retrieve reads containing the heteroplasmic repeat sequence. d) Sanger sequencing of randomly selected DGRP lines of the intergenic repeat region. e) Frequency of the dominant (maximum number of reads) types of the heteroplasmic intergenic repeat region in the DGRP. The reference (iso-1) type is in red. f) The frequency of all types of heteroplasmic intergenic repeat regions observed in all DGRP lines that are supported by 10 or more reads. The reference (iso-1) type is in red. g-j) Amplicon sequencing results for variant 3596_G/A for DGRP-528 (g), variant 10670_G/A for DGRP-136 (h) and variant 10868_G/A for DGRP-21 (i) and DGRP-373 (j). From left to right, results are depicted for eight males (upper), and females (lower) for amplicon sequencing of the respective DGRP line. Next, results are shown for the iso-1 reference strain followed by the initial sequencing results and variant calling from our mtDNA-enriched sequencing for the respective DGRP line and iso-1. Alternate alleles are displayed in light grey whereas reference alleles are dark grey. k) Heteroplasmy example. The upper panel shows amplicon sequencing results for male #1 for DGRP-21 versus male #1 for iso-1 (lower panel). In red, variants of the alternate allele are shown. l) Relationship between the number of mitochondrial variants in DGRP lines and Wolbachia infection. The lines within the violin represent the 25th (lower), 50th, and 75th (upper) percentile. Lower and upper end of the violin plot represent the bottom 25% and upper 75% of the data.
Extended Data Fig. 5 Population structure and metabolic effects of mitochondrial haplotypes.
a) Permutations of the genetic distance between haplogroups from Fig. 3a using the G′ST estimator. b) Outline of the analysis to quantify relative mitochondrial complex I levels. Relative intensity ratios between complex I of w1118 over complex I of a given DGRP line are calculated. c) Examples of DGRP lines displaying high, mid, and low relative levels of complex I. d) Oxygen consumption rates (OCR) in pMoles per minute for different mitochondrial respiration states. e) Oxygen consumption rates (OCR) for individual lines/genotypes per state. For each genotype, two biological replicates were used, except DGRP-235 for which only one replicate was available. For each biological replicate, four time-measurements were made for state II, five for state III, three for state IVo, and three for Rotenone. For the calculations, the individual measurements are presented as dots and the mean of the measurements are represented by the barplot. The error bars are the standard error of means. f) Respiratory control ratio (RCR) as calculated from a ratio of state III over state IVo. Statistical tests in d, e, and f were calculated using ANOVA. For the boxplots in d and f boxes represent the 25th (lower) to 75th (upper) percentile. Whiskers correspond to the bottom 25% and upper 75% of the data with the median indicated by a thick black line.
Extended Data Fig. 6 Variation within the sex-lethal gene region.
The location of nuclear genomic variants in the DGRP is shown above the genes.
Extended Data Fig. 7 Crossing scheme for genotype ratio distortion (GRD).
Detailed overview of the crossing scheme that was used to construct the ACI and ACR conplastic populations.
Extended Data Fig. 8 Supporting figures related to genotype ratio distortion (GRD) analysis and experiments.
a) Climbing activity of imposed (ACI) and rescue (ACR) allelic-combination populations (ANOVA, p-value = 0.7). Dots represent individual (n = 9) ACI or ACR populations (n = 3 biological replicates per group). Red dots represent the mean over all populations. Error bars represent standard error of means. For genotype w1118 n = 6 biological replicates were used. b) Epistatic interactions between the mitochondrial variant chrM 791 and nuclear variant located at position 3 L 10690916. The variants influence starvation resistance in males. The p-value is adjusted for multiple testing (p-value = 0.00022, χ2-test as implemented in PLINK). c) Epistatic interactions between the mitochondrial variant chrM 791 and nuclear variant located at position 3 L 10687695. The p-value is adjusted for multiple testing (p-value = 0. 00075, χ2-test as implemented in PLINK). Notably, there is a 3 kb distance between the epistatic nuclear variants that associated with starvation resistance in males versus females. For boxplots on b,c boxes represent the 25th (lower) to 75th (upper) percentile. Whiskers correspond to the bottom 25% and upper 75% of the data with the median indicated by a thick black line.
Extended Data Fig. 9 Food intake of males for individual F1 populations.
In blue, populations are shown with the mitochondrial haplotype 5 (MH5; high food intake), and in red, populations with the mitochondrial haplotype 1 (MH1; low food intake). Mat = maternal, pat = paternal. Boxes represent the 25th (lower) to 75th (upper) percentile. Whiskers correspond to the bottom 25% and upper 75% of the data with the median indicated by a thick black line.
Supplementary information
Supplementary Information
Supplementary Notes 1–7
Supplementary Tables
Supplementary Tables 1–24
Source data
Source Data Fig. 3
Unprocessed BN–PAGE gels.
Rights and permissions
About this article

Cite this article
Bevers, R.P.J., Litovchenko, M., Kapopoulou, A. et al. Mitochondrial haplotypes affect metabolic phenotypes in the Drosophila Genetic Reference Panel. Nat Metab 1, 1226–1242 (2019). https://doi.org/10.1038/s42255-019-0147-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-019-0147-3
This article is cited by
-
Two mitochondrial DNA polymorphisms modulate cardiolipin binding and lead to synthetic lethality
Nature Communications (2024)
