
Abstract
Ischaemic heart disease and stroke are the most common causes of death worldwide. Anoxia, defined as the lack of oxygen, is commonly seen in both these pathologies and triggers profound metabolic and cellular changes. Sphingolipids have been implicated in anoxia injury, but the pathomechanism is unknown. Here we show that anoxia-associated injury causes accumulation of the non-canonical sphingolipid 1-deoxydihydroceramide (DoxDHCer). Anoxia causes an imbalance between serine and alanine resulting in a switch from normal serine-derived sphinganine biosynthesis to non-canonical alanine-derived 1-deoxysphinganine. 1-Deoxysphinganine is incorporated into DoxDHCer, which impairs actin folding via the cytosolic chaperonin TRiC, leading to growth arrest in yeast, increased cell death upon anoxia–reoxygenation in worms and ischaemia–reperfusion injury in mouse hearts. Prevention of DoxDHCer accumulation in worms and in mouse hearts resulted in decreased anoxia-induced injury. These findings unravel key metabolic changes during oxygen deprivation and point to novel strategies to avoid tissue damage and death.
Similar content being viewed by others


Main
Oxygen is essential for the survival of most animals1. In humans, tissue damage in response to oxygen deprivation is the number-one cause of death worldwide2. While our brain cells and cardiomyocytes can only survive a few minutes without oxygen, other animals have developed adaptations to live through longer periods of anoxia3. The nematode Caenorhabditis elegans (C. elegans) can enter into a hypometabolic state of suspended animation and survive for days in anoxic conditions by slowing down its metabolism, development and energy consumption3. This makes the worm an ideal model to study metabolic and cell-biology changes in response to oxygen deprivation.
Lethal or debilitating ischaemia–reperfusion injury is the result of inadequate blood supply to a tissue, leading to oxygen deprivation (ischaemia) that causes profound metabolic and cell-biology changes1. When blood flow and oxygen supply is restored (reperfusion), these initial alterations trigger subsequent cellular damage, leading to tissue injury and death1,4.
The membrane-lipid class of sphingolipids encompasses several bioactive small molecules that are involved in growth control, cell migration, inflammation and many stress responses5. De novo sphingolipid biosynthesis starts with serine palmitoyl-CoA transferase (SPT) condensing serine with acyl-CoA to produce 3-ketosphinganine, which is subsequently reduced to the sphingoid base sphinganine. Ceramide synthases (CerSs) use different acyl-CoA species to N-acylate sphingoid bases producing dihydroceramide (DHCer) from sphinganine, which is desaturated to form ceramide. DHCer and ceramide serve as building blocks for head-group-bearing sphingolipids like sphingomyelin or hexosylceramide (HexCer) by transfer of different head groups onto their 1-hydroxyl group. CerSs can also use other sphingoid bases as substrates, like sphingosine, which is generated when existing sphingolipids are recycled6 (Fig. 1i). The first steps of sphingolipid biosynthesis and many sphingolipid functions are conserved in a number of species, ranging from yeast to humans. These species include the nematode C. elegans7, which has a structural particularity in that worms produce iso-branched C17 sphingoid bases instead of the C18 straight-chain sphingoid bases found in yeast and humans8,9.

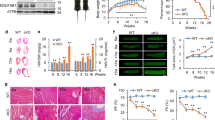
a, Volcano plot of sphingolipid changes in hyl-2(gnv1) worms compared with wild-type worms, as determined by non-targeted lipid analysis. Non-canonical DoxSa-derived C16 DoxDHCer and its isotopic peak (M + 1) (red dots) are upregulated in hyl-2(gnv1). Blue dots are sphingolipids that are downregulated in hyl-2(gnv1) and correspond to canonical sphinganine-derived C19–C23 sphingomyelins and HexCers, including C22 sphingomyelin and its isotopic peak (M + 1); yellow dots are not statistically significant. n = 6 (wild type) and n = 3 (hyl-2) independent biological replicates. b,c, Graphs representing the abundance of C16 DoxDHCer (b) and C22 sphingomyelin (c) in both wild-type (WT(N2)) and hyl-2(gnv1) mutants. Molecular structures of C16 DoxDHCer and C22 sphingomyelin highlight non-canonical DoxSa (red) and canonical sphingosine (blue). Replicates are as in a. Arbitrary units (a.u.), thick lines show the median, boxes are upper and lower quartiles, whiskers are maximum and minimum, and circles are outliers. d, SPTLC1 homologue multiple-sequence alignment using human (Hs), fish (Dr), fly (Dm), worm (Ce) and yeast (Sc). Coloured areas correspond to similar (grey) and identical (orange) residues. Conserved cysteines are highlighted in red. Mutations in this cysteine (C133W or C133Y) cause hereditary sensory and autonomic neuropathy type 1A (HSAN1A)16. C121W is the corresponding worm mutation. e, A schematic of anoxia-reoxygenation survival experiments. Worms are grown for 3 days in normoxia, exposed to anoxia for either 28, 42 or 48 h (X h), reoxygenated and grown for 1 d in normoxia before survival is determined. f, Survival of wild-type worms and wild-type worms expressing the sptl-1C121W allele after 42 h anoxia and reoxygenation; n = 3 independent biological replicates. g, Survival of hyl-2(tm2031) worms and hyl-2(tm2031) worms expressing the sptl-1C121W allele after 28 h anoxia and reoxygenation. n = 10 independent biological replicates. h, Abundance of DoxDHCer relative to ceramide (Cer) in worms that were wild type, wild type expressing the sptl-1C121W allele, hyl-2(tm2031) and hyl-2(tm2031) expressing the sptl-1C121W allele, both under normoxia (solid) and directly after 20 h anoxia (shaded). n = 4 independent biological replicates. For f–h, bars are means with s.e.m. Unpaired two-sided Student’s t-test P (a,f–h) and their FDR-corrected q values (b,c) are given. i, Illustrations of canonical and non-canonical sphingolipid biosynthesis pathways: SPT, serine palmitoyl-CoA transferase; 3-KDR, 3-keto dihydrosphingosine reductase; DHS1P, sphinganine-1-phosphate; S1P, sphingosine-1-phosphate.
Several studies have implicated sphingolipids in the pathogenesis of myocardial infarction and stroke10,11 but the exact molecular species involved in anoxic death and its pathomechanism are not known. We have previously shown that in C. elegans the ceramide synthase HYL-2 plays a protective role in anoxia-reoxygenation. While wild-type animals can survive in anoxic conditions for up to 3 days12, hyl-2-mutant worms are hypersensitive to lack of oxygen and die of massive necrosis already after 2 days of anoxia followed by one day of reoxygenation13. Our previous targeted lipidomic analysis revealed that much like mammalian CerSs14 the worm CerSs have acyl-CoA substrate specificity. HYL-2 mainly incorporates C19–C23 fatty acids into ceramides that are further metabolized to head-group-bearing sphingolipids like sphingomyelin or HexCer13. How the lack of a specific subset of ceramides would translate into tissue injury in response to anoxia-reoxygenation is unknown.
Results
Anoxia-hypersensitive mutants accumulate 1-deoxydihydroceramide
We first identified the relevant sphingolipid species that cause anoxia-reoxygenation hypersensitivity in hyl-2 mutants. We used an untargeted lipidomics approach to compare the sphingolipidomes of wild type and hyl-2 strains in normoxia and found that non-canonical DoxDHCer was strongly upregulated in hyl-2 mutants (Fig. 1a,b). As previously shown, hyl-2 mutants also showed a significant decrease in sphingomyelins and HexCers containing C19 to C23 acyl chains13 (Fig. 1a,c and Extended Data Fig. 7).
DoxDHCer differs from canonical DHCer in that it forms from the non-canonical sphingoid base 1-deoxysphinganine6 (DoxSa, red in molecular structure in Fig. 1b). DoxSa is produced when serine palmitoyl-CoA transferase (SPT), which has a strong preference for serine as a substrate, uses alanine instead for condensation with acyl-CoA15,16. In contrast to DHCer, DoxDHCer is neither degraded by the canonical pathway nor further metabolized to head-group-bearing sphingolipids, leading to accumulation of it6,16 (see scheme in Fig. 1i).
We next investigated whether accumulation of DoxDHCer causes hypersensitivity to anoxia. To increase the levels of DoxDHCer, we introduced two additional mutated alleles of one of the SPT subunits (sptl-1C121W) into the worm genome. This mutation alters SPT specificity from serine to alanine, resulting in an increase in the levels of DoxSa16 (Fig. 1d). We then assessed the survival of the mutant worms after anoxia-reoxygenation exposure (Fig. 1e). Wild-type worms carrying the sptl-1C121W alleles showed a significant decrease in survival after anoxia (89.4 ± 3.9% versus 65.7 ± 5.2%; Fig. 1f). A strong synergistic effect was observed when the sptl-1C121W alleles were expressed in anoxia-hypersensitive worms with mutated hyl-2 (88.9 ± 1.8% versus 7.2 ± 0.7%; Fig. 1g), suggesting a role for DoxDHCer in anoxia-induced death.
Consistently, the levels of DoxDHCer increased compared with those of canonical ceramide in wild-type worms, even after sublethal exposure to anoxia. Levels of DoxDHCer were higher in both worms expressing the sptl-1C121W alleles and in those with mutated hyl-2 (Fig. 1h and Extended Data Fig. 1), indicating that anoxia induces an increase in DoxDHCer levels that correlates with a higher mortality rate.
Anoxic metabolism increases DoxDHCer precursors
We next tested how the accumulation of DoxDHCer in response to anoxia is produced. Time-course experiments in wild-type worms that were increasingly exposed to anoxia showed a rapid accumulation of the DoxDHCer precursor DoxSa (Fig. 2a). In contrast, the levels of the canonical sphinganine were reduced (Fig. 2b), resulting in a relative increase in DoxSa compared with sphinganine upon anoxia exposure (Fig. 2c).

a–f, Wild-type young adult worms were grown under normoxia or different durations of anoxia (striped), and the following compounds were measured: concentration of the worm DoxSa (iso-branched C17 DoxSa) (a), worm sphinganine (iso-branched C17 sphinganine) (b) and their relative abundance, given as the DoxSa/sphinganine ratio (c); and the amounts of alanine (d), serine (e) given as the percentage of total amino acid signals and their relative abundance, given as the alanine/serine ratio (f). For a–f, n = 4 independent biological replicates, and bars are means with s.e.m. g,h, Two-day-old larvae were grown for 24 h on either DMSO control or 5 µM rotenone, and the following compounds were measured: relative amounts of alanine and serine (g) and relative abundance of DoxSa and sphinganine (h); n = 3 independent biological replicates, and bars are means with s.e.m. i, Normoxia survival of wild-type worms and hyl-2(tm2031) mutants with knockdown of de novo sphingolipid biosynthesis (sptl-1 RNAi) and treated with 25 µM exogenous worm DoxSa (iso-branched C17 DoxSa); n = 3 (wild type) and n = 4 (hyl-2) independent biological replicates, and bars are means with s.e.m. For c and f–i, an unpaired two-sided Student’s t-test was used for statistical analysis. j, Simplified scheme of anoxic metabolism leading to non-canonical alanine-derived DoxDHCer production. TCA, tricarboxcylic acid cycle; ETC, electron transport chain.
This change in DoxSa/sphinganine ratio correlated with a change in the levels of alanine and serine. Increasing anoxia exposure resulted in a rise in alanine levels, with a concomitant decrease in the levels of serine (Fig. 2d,e), resulting in an increasing alanine/serine ratio over the time course (Fig. 2f).
In the absence of oxygen, cells can no longer generate ATP via oxidative phosphorylation. Elevated glycolytic activity results in a depletion of serine and an accumulation of pyruvate which, in part, is transformed into alanine17 (scheme in Fig. 2j). Mimicking anoxic metabolism by mitochondrial complex I inhibition with rotenone led to an increase in the ratio of alanine versus serine (Fig. 2g) and an increase in the DoxSa/sphinganine ratio (Fig. 2h). Taken together, this indicates that oxygen deprivation in C. elegans leads to an imbalance of alanine over serine that results in an increase in the production of DoxSa and a decrease in the production of sphinganine. DoxSa is further metabolized, resulting in the accumulation of DoxDHCer, while the levels of canonical sphinganine-derived ceramides are reduced (Fig. 1h and Extended Data Fig. 1, scheme in Fig. 2j). Both conditions (increase in DoxSa and decrease in sphinganine) are needed to cause anoxic death.
Indeed, specifically lowering the levels of sphinganine in normoxia does not result in a high mortality rate (97% survival; Fig. 2i). Only when the levels of sphinganine are reduced and the levels of DoxSa are increased at the same time, the survival rate drops to 75% (Fig. 2i), suggesting that the relative amount of DoxSa compared to sphinganine determines survival versus anoxia-induced mortality.
Ceramide synthases show sphingoid base specificity
The CerS HYL-2 has a protective role in anoxia: the rate of survival in hyl-2 mutants is lower after anoxia-reoxygenation treatment13 (Fig. 3e). In normoxia, hyl-2 mutants exhibit higher mortality when the levels of DoxSa are increased and the levels of sphinganine are decreased (Fig. 2i). Therefore, we conclude that hyl-2 is hypersensitive to anoxia-reoxygenation because these mutants are hypersensitive to the non-canonical DoxSa produced during anoxia.

a, Dilution growth assay of wild-type yeast expressing individual mammalian CerS1–CerS6 on rich medium (YPD) containing 0.05% tergitol (T) as well as vehicle (EtOH) or 5, 10 or 20 µM C18 DoxSa. This experiment was repeated at least five times with similar results. b, Representative structure of DoxDHCer from S. cerevisiae containing non-canonical alanine-derived C18 DoxSa (red). c, Heat map of increase in specific DoxDHCer species levels in wild-type yeast expressing individual mammalian CerSs when grown in rich medium with C18 DoxSa (+2.5 µM DoxSa) instead of vehicle. Concentration is given as nM per 25 optical density at 600 nm (OD600). d, Schematic of worm anoxia-reoxygenation survival experiments to identify suppressor mutant in an ethylmethansulfonat (EMS) screen33. e, Survival after 48 h of anoxia and 1 d reoxygenation for the following groups of worms: wild type, ttc-17(gnv3), CerS mutant hyl-2(gnv1), rescued ttc-17(gnv3);hyl-2(gnv1) double mutant and reverted ttc-17(gnv3);hyl-2(gnv1) double-mutant expressing TTC-17::GFP; n = 9 independent biological replicates, and bars are means with s.e.m. f, Schematic of TTC-17 protein (T19A5.1) with its tetratricopeptide repeat (TPR1) homology domain; amino acid changes due to suppressor point mutations are indicated. g,h, Targeted lipid analysis of wild-type, ttc-17(gnv3), hyl-2(gnv1) and rescued ttc-17(gnv3);hyl-2(gnv1) double-mutant worms. Shown are log2-transformed sum values normalized to inorganic phosphate (Pi). Box-and-whisker graphs (bottom) with general molecular structures (top) are shown. In g, depletion in canonical serine-derived sphingoid base-containing (blue) C20-23 sphingomyelin (C20–C23 sphingomyelin) is not rescued, while in h, elevated non-canonical 1-deoxysphingoid base-containing (red) C24–C26 DoxDHCer (C24–C26 DoxDHCer) levels are lowered to wild-type levels in rescued double mutants. n = 6 independent biological replicates; the thick line shows the median, the box shows upper and lower quartiles, and the whiskers are maximum and minimum. P values are from unpaired two-sided Student’s t-tests. ns, nonsignificant.
CerSs catalyse the conversion of canonical and non-canonical sphingoid bases (sphinganine and DoxSa) into both DHCer and DoxDHCer6. Therefore, hyl-2 mutants should show an increase in sphingoid bases. However, hyl-2 mutants show an increase in DoxDHCer, while canonical sphinganine derivatives are downregulated (Fig. 1a–c,h), raising the possibility that in the absence of HYL-2, another CerS with higher affinity for DoxSa takes over and catalyses DoxDHCer production.
To test whether CerSs show specificity for DoxSa, we expressed the six mammalian CerSs in Saccharomyces cerevisiae yeast cells and assessed their specificity to sphingoid base substrates by exposing them to increasing levels of DoxSa and scoring for DoxDHCer-dependent growth arrest (Fig. 3a–c). We found that wild-type yeast as well as yeast cells expressing CerS1, CerS2 and CerS4 grew even when exposed to high levels of DoxSa. However, CerS3, CerS5 and CerS6 arrested growth at 5 µM DoxSa (CerS3) and at 10–20 µM (CerS5 and CerS6) (Fig. 3a) due to an increase in DoxDHCer (Fig. 3b,c). This indicates that even though all six mammalian CerSs are active when expressed in yeast cells and maintain their known acyl-CoA substrate specificity14 (shown in Extended data Fig. 2), CerSs also exhibit enzymatic specificity for sphingoid base substrates (DoxSa versus sphinganine). Therefore, different CerSs have different capacity to convert DoxSa into DoxDHCer, which explains why hyl-2 mutants exhibit higher levels of DoxDHCer.
Reducing DoxDHCer levels is sufficient to revert anoxic death
Expressing the mammalian CerSs in yeast cells not only uncovered a sphingoid base substrate specificity of CerSs, but also showed that growth arrest is induced specifically by the increase in the levels of DoxDHCer (Fig. 3a–c). Accumulation of DoxSa by itself is not sufficient to cause growth arrest. DoxSa needs to be converted to DoxDHCer to stop the growth of yeast cells (Fig. 3a–c). Indeed, DoxDHCer-induced growth arrest in yeast cells expressing CerS3 can be prevented by inhibition of CerS3 using Fumonisin B1 (Extended Data Fig. 2).
We then tested whether decreasing the levels of DoxDHCer also prevents mortality in worms exposed to anoxia-reoxygenation. In a screen looking for suppressors of the anoxia hypersensitivity observed in hyl-2(gnv1) mutants (Fig. 3d), we isolated and identified the ttc-17(gnv3) mutant (for details see Fig. 3f and Extended Data Fig. 2). The ttc-17 mutation promoted survival of hyl-2(gnv1) mutants after anoxia-reoxygenation exposure (Fig. 3e). Lipidomic analysis revealed that the ttc-17(gnv3) mutation does not restore hyl-2 activity because the levels of canonical sphinganine-derived sphingolipids normally produced by HYL-2 (C20-C23 sphingomyelin) remained low in the double mutant (Fig. 3g). However, DoxDHCer was reduced to wild-type levels in the double mutant (Fig. 3h), indicating (1) that accumulation of DoxDHCer rather than the reduction of canonical sphinganine-derived ceramides caused anoxia hypersensitivity in hyl-2 mutants and (2) that reducing the levels of DoxDHCer is sufficient to revert anoxic death.
DoxDHCer is sufficient to cause ischaemia–reperfusion injury in mouse hearts
Pathological conditions, such as ischaemic heart disease, are characterized by oxygen deprivation1. Using an in vivo mouse model of ischaemia–reperfusion heart injury (Fig. 4a and Extended Data Fig. 3), we first analysed changes in sphingolipid levels of mouse hearts in normoxia (sham) and after ischaemia–reperfusion and found that the levels of sphingolipids, including DoxSa and DoxDHCer, were upregulated after ischaemia and correlated with increased infarct formation18 (Extended Data Fig. 3).

a, Schematic of mouse heart, showing occlusion (blue) in the left anterior descending coronary artery and cut sections (dashed black lines) for quantification of tissue injury. b, Timeline of ischaemia–reperfusion experiment with preconditioning. c, Illustration of a cut section showing the area at risk of necrosis (AR), which is devoid of blue dye as it was cut off from blood supply, and the area of necrosis (AN) which is pale in contrast to the viable myocardial tissue that is stained red. d–f, Quantification of metabolite levels after ischaemia–reperfusion in vehicle (Veh)-, myriocin (Myr)- and myriocin plus DoxSa (Myr+DoxSa)-treated hearts. d, Quantification of alanine/serine levels. e, Quantification of DoxSa. f, Quantification of non-canonical DoxDHCer normalized to inorganic phosphate (Pi). g, Quantification of infarct size, measured as the area of necrosis/area at risk ratio, shows cardioprotection by myriocin preconditioning and partial reversion of this upon Myr+DoxSa exposure. h, Plot representation of area of necrosis relative to area at risk shows significant cardioprotection following myriocin treatment, which is reverted upon addition of DoxSa (Myr+DoxSa). The coefficient of determination (r2) is given for each linear regression, and P was determined by analysis of covariance (ANCOVA). For d,e, n = 11 (Veh and Myr) and n = 10 (Myr+DoxSa), for f, n = 10 (Veh), n = 12 (Myr) and n = 9 (Myr+DoxSa); and for g,h, n = 14 (Veh), n = 12 (Myr) and n = 10 (Myr+DoxSa) are heart-tissue samples from individual animals. Statistics in d–g are unpaired two-sided Student’s t-test P values, and bars are mean values with s.d.
We then analysed whether interfering with the production of DoxDHCer during ischaemia has any effect on cardiac injury caused by lack of oxygen (Fig. 4a–c). Specific inhibition of SPT by myriocin16 did not have any influence on alanine versus serine levels because myriocin does not affect amino acid metabolism upstream of SPT (Fig. 4d). However, myriocin treatment resulted in downregulation of all sphingolipids tested (Extended Data Fig. 4), including DoxSa and DoxDHCer (Fig. 4e,f), leading to a decrease in the area of necrosis compared to the area at risk (Fig. 4c,g,h and Extended Data Fig. 4 for controls).
While several sphingolipids are increased in ischaemia–reperfusion (Extended Data Fig. 3), an increase in DoxDHCer is sufficient to revert the protective effects of myriocin. Simultaneous myriocin plus DoxSa treatment increased DoxSa and DoxDHCer in mouse hearts (Fig. 4e,f) without affecting the levels of canonical sphinganine-derived sphingolipids (Extended Data Fig. 4) and resulted in a partial reversion of myriocin cardioprotection (Fig. 4g,h). Indeed, throughout the whole dataset, area of necrosis shows a low positive correlation with amounts of DoxDHCer in mouse hearts (Extended data Fig. 4). Also, ceramides show a similar positive correlation, but not DHCers (Extended data Fig. 4) indicating that other ceramides might also play a role in ischaemia–reperfusion injury in mice, as suggested previously18,19,20. We conclude that (1) ischaemia–reperfusion leads to a general upregulation of sphingolipid production, (2) SPT inhibition during ischaemia is cardioprotective; and (3) specifically increasing the levels of DoxDHCer is sufficient to aggravate ischaemia–reperfusion injury in the mouse heart.
Cytosolic chaperonin TRiC is a target of DoxDHCer
To investigate the DoxDHCer-related cell biological processes that are affected during anoxia, we first performed quantitative proteomic analysis in which we compared worms under normoxic conditions and after 24 h of exposure to anoxia (Fig. 5a). Consistent with our metabolomic data (Fig. 2), many of the electron transport chain and tricarboxylic acid cycle proteins were downregulated in anoxia, while glycolysis-related proteins were unchanged (Fig. 5a, left). We also observed downregulation of microtubule-associated proteins, tubulin monomers and F-actin-associated factors. Actin monomers themselves remained unchanged (Fig. 5a, middle). Tubulin downregulation, F-actin disruption and actin monomer aggregation have been associated with alteration of the cytosolic chaperonin TRiC in C. elegans21 (Fig. 5b). TRiC (TCP-1 ring complex, also called CCT for chaperonin containing TCP-1) is a multi-subunit complex that folds globular proteins, such as actin and tubulin, and is required for skeletal muscle biogenesis in vertebrates22 and in cardiac physiology in flies23,24 and humans25. TRiC-impaired cells exhibit an accumulation of actin aggregates21. We found that all eight TRiC subunits were downregulated in worms after anoxia (Fig. 5a, right), raising the possibility that DoxDHCer produced during anoxia impairs TRiC function, leading to defects in the actin cytoskeleton.

a, Stable-isotope-mediated quantitative proteomic analysis shows proteome differences upon 24 h anoxia treatment. Shown are foward ratios of light (L) anoxic proteome (Anox) over heavy-labelled (H) normoxic proteome (Normox) on the x axis against the reverse ratios of heavy-labelled (H) anoxic proteome (Anox) over light (L) normoxic proteome (Normox) on the y axis. Proteins with certain GO terms are highlighted including glycolysis, tricarboxylic acid cycle, electron transport chain, microtubule, actin binding, actin, unfolded protein binding and the cytosolic chaperonin TRiC. b–d, mCherry::ACT-5 (mC::ACT-5) intestinal signals of wild-type animals in control conditions and upon RNA interference (RNAi)-mediated knockdown of the chaperonin subunit gene cct-5 (b), under normoxia or after 24- and 48-h anoxia and 24-h reoxygenation treatment (c) and after treatment with exogenous worm DoxSa (iso-branched C17 DoxSa) (d). Scale bars, 10 µm. The experiments in b–d were repeated three times with similar results. e, Yeast cells expressing mammalian CerS3 showing F-actin staining with phalloidin grown in rich medium (YPD) and upon DoxSa treatment. Scale bar, 2 µm. Yeast phalloidin stainings have been repeated at least five times with similar results. f, Dilution growth assay of wild-type yeast and thermo-sensitive TRiC chaperonin mutants expressing control or mammalian CerS3 constructs on rich medium containing 0.05% tergitol, as well as vehicle (EtOH) or 4 and 8 µM DoxSa at 24 °C. Experiments examining the hypersensitivity of yeast TRiC mutants were repeated three times with similar results.
As in TRiC impaired worms (Fig. 5b and Extended Data Fig. 5), apical F-actin localization associated with intestinal microvilli was also disrupted, and the amount of cytoplasmic actin aggregates increased in worms after anoxia, after an additional 24-h reoxygenation (Fig. 5c and Extended Data Fig. 5), and in normoxia upon addition of exogenous DoxSa (Fig. 5d and Extended data Fig. 5).
Like in worms, addition of DoxSa to yeast cells expressing CerS3 to produce DoxDHCer results in disruption of actin cables and actin patches, with a concomitant increase in actin aggregates (Fig. 5e and Extended Data Fig. 5), raising the possibility that TRiC is the essential factor inhibited during DoxDHCer-dependent growth arrest in yeast.
Haploid yeast carrying a temperature-sensitive TRiC mutation and TRiC haploinsufficient yeast strains, both expressing CerS3 and exposed to increasing amounts of DoxSa, arrested growth at a lower concentration of DoxSa than control yeast cells (Fig. 5f and Extended Data Fig. 5), consistent with DoxDHCer acting on TRiC. In accordance with this, overexpression of a gene encoding one of the TRiC subunits, tailless complex polypeptide (TCP1, also known as CCT1), partially restores growth of yeast cells expressing CerS3 and exposed to DoxSa (Extended Data Fig. 5).
DoxDHCer-dependent impairment of TRiC in ischaemia–reperfusion
To address whether TRiC is also a target of DoxDHCer during ischaemia–reperfusion injury, we first looked at TRiC (CCT5) expression in mouse hearts that underwent ischaemia–reperfusion (Fig. 6 and Extended Data Fig. 6). CCT5 was found in a regular pattern in non-ischaemic parts of the heart, in irregular patterns in ischaemic tissue that did not undergo necrosis, and was absent in the necrotic area itself (Extended Data Fig. 6). In non-ischaemic parts of the hearts, CCT5 colocalized with striated actin (Fig. 6b,c and Extended Data Fig. 6). This colocalization was mainly lost in non-necrotic ischaemic tissue (Fig. 6a and Extended Data Fig. 6). In this tissue, actin lost its striated pattern and appeared as cytosolic aggregates, as seen in worms and yeast (Fig. 5b–e), indicating that ischemia-reperfusion impairs TRiC function, leading to the formation of actin aggregates and eventually cell death.

a, Immunofluorescence images of myofibrils in cardiomyocytes of the left ventricle upon ischaemia–reperfusion injury after preconditioning with vehicle, myriocin or myriocin plus DoxSa. Anti-CCT5 is shown in cyan, and anti-actin in magenta; scale bars, 5 µm. b, Immunofluorescence of myofibrils in cardiomyocytes of the right non-ischaemic ventricle; scale bars, 5 µm. c, Pearson’s coefficient quantification of co-localization between TRiC chaperonin (anti-CCT5) and actin in non-ischaemic and ischaemic cardiomyocytes of hearts preconditioned with vehicle, myriocin or myriocin plus DoxSa. n = 10 independent images from the different heart regions for each treatment. Bars are means with s.e.m. For c, statistics were done with an unpaired two-sided Student’s t-test.
To correlate the increase in DoxDHCer during ischaemia–reperfusion with impaired function of TRiC, we treated the hearts with myriocin and DoxSa, as in Fig. 4, and looked at CCT5 and actin. Addition of DoxSa resulted in a more prominent irregular pattern of CCT5 and an increase in actin aggregate formation, even in the non-ischaemic tissue (Fig. 6b,c and Extended Data Fig. 6).
Other sphingolipid species, including ceramides, are also upregulated in ischaemia–reperfusion (Extended Data Fig. 3), decrease in response to cardioprotective myriocin treatment, and show similar positive correlation with necrosis as DoxDHCer (Extended Data Fig. 4). Therefore, we cannot exclude that ceramide, or its precursor DHCer, also have a role during ischaemia–reperfusion injury in mice18,20. In worms and yeast, we can exclude this possibility, as we can clearly separate the detrimental effects of non-canonical DoxDHCer from canonical sphingolipids in anoxia-reoxygenation (Fig. 3) and because the observed actin phenotype in yeast cells (Fig. 5e) is clearly distinct from the actin phenotype caused by ceramide accumulation in response to aureobasidin treatment26,27. If ceramides contribute to ischaemia–reperfusion injury in mice, they probably do so through a distinct mechanism from the DoxDHCer-mediated chaperonin impairment that we observe in yeast, worms and mouse hearts.
Discussion
In this study, we have unravelled the molecular basis of tissue injury and death in response to oxygen deprivation. We have found that lack of oxygen, which also occurs during ischaemic heart disease, changes energy metabolism, leading to an increase in the alanine/serine ratio (Fig. 2). This causes the upregulation of the non-canonical alanine-derived DoxSa and DoxDHCer (Fig. 1h and Fig. 2). Suppression of DoxDHCer formation protects from tissue injury caused by oxygen deprivation (Figs. 3 and 4). DoxDHCer impairs the function of the cytosolic chaperonin TRiC (Fig. 5), resulting in protein misfolding and the formation of actin aggregates, which ultimately leads to cell death (Figs. 5 and 6). This is consistent with previous work showing that: (1) non-canonical DoxSa has an effect on the organization of the cytoskeleton in worms9 and human cells16,28; (2) protein aggregates are formed in cerebral ischaemia29; and (3) mutations in the TRiC subunit CCT7 are associated with higher risk of myocardial infarction25.
We have identified a critical sphingolipid species responsible for anoxic death. While myriocin treatment improves tissue survival in ischaemia–reperfusion18 (Fig. 4 and Extended Data Fig. 3), it does so by downregulating all sphingolipids, most of which are essential for many cellular processes. Therefore, a more targeted treatment against DoxDHCer should be a more suitable therapy against ischaemia–reperfusion injury. This therapeutic strategy could also be used in other DoxDHCer-related pathologies, such as hereditary sensory and autonomic neuropathy type 1A16 and diabetic neuropathy30, which are both associated with late-onset peripheral loss of temperature and pain sensation, as well as skin ulcerations. Very similar symptoms with onset already at birth have been described for TRiC mutations in rats31 and humans32, in accordance with our hypothesis that TRiC function is the target of DoxDHCer.
Methods
Detailed information on experimental design and reagents can be found on the Reporting Summary.
Material
Commercial C18 DoxSa was purchased from Avanti Polar Lipids (Alabaster, Alabama, USA); all other compounds and solvents at highest purity were obtained from Sigma-Aldrich (Buchs, Switzerland) or Acros Organics/Chemie Brunschwig (Basel, Switzerland). Iso-branched C17 DoxSa was synthesized previously9.
C. elegans wild-type N2 (var. Bristol) and E. coli strains OP50 and SLE1 were received from the Caenorhabditis Genetics Centre (University of Michigan, Michigan, USA); the hyl-2(tm2031) and oatr-1(tm5454) strains were from the National BioResource Project (Tokyo Women’s Medical University, Tokyo, Japan); strain EI4348 was from NemaGENETAG34; and GK454 was a kind gift from K. Sato21. A complete list of yeast and nematode strains used in this study can be found in Supplementary Tables 1 and 2. Unless stated otherwise, bacteria were grown on LB medium, yeast on rich medium (YPUATD: 1% yeast extract, 2% Bacto peptone, each 40 mg L–1 uracil, adenine, tryptophan, 2% glucose, 2% bacto agar), and worms on NGM medium seeded with OP50 bacteria33.
C. elegans codon-optimized wild-type sptl-1 and mutant sptl-1C121W as well as the S. cerevisiae codon-optimized mammalian CerSs were synthetized by GENEART (Regensburg, Germany). Adeno-associated virus vectors for wild-type SPTLC1 (AAV-WT) and the hereditary sensory and autonomic neuropathy type 1A mutant SPTLC1 (AAV-C133W) expression were custom prepared by Vector Biolabs (Philadelphia, PA) from previously published pcDNA3.1 vectors16.
Construction of worm line carrying sptl-1 C121W allele
Single-gene insertion was performed by the Mos1-mediated single-copy insertion (MosSCI) method, as described before35 with minor modifications: the background strain was IE4348, which carries an inserted Mos1 transposon in chromosome I between nucleotides 2850991 and 2851093. The strain was out-crossed with N2 for 4 rounds to eliminate secondary transposon insertion. The strain was maintained on NGM plates seeded with E. coli HB101. A targeting plasmid was constructed by replacing cbunc-119-positive selection marker of pCFJ352 with Peft-3::GFP::H2B and inserting a codon-optimized sptl-1C121W allele including a C-terminal triple flag-tag with the endogenous promoter and terminator. The injection mix consisted of 50 ng μl–1 of pCFJ601 (Peft-3::transposase), 22.5 ng μl–1 of the targeting plasmid, 10 ng μl–1 of pMA122 (Phsp::peel-1), 10 ng μl–1 of pGH8 (Prab-3::mCherry), 2.5 ng μl–1 of pCFJ90 (Pmyo-2::mCherry), and 5 ng μl–1 of pCFJ104 (Pmyo-3::mCherry). The injection mix was centrifuged at 20,800g for 1 min to pellet impurities. Young adult hermaphrodites were injected with the injection mix using the standard worm injection setup, and were placed at room temperature for 1 h to recover. Injected worms were placed on individual NGM plates seeded with E. coli OP50 and incubated at 20 °C for 7 d, until all adult worms had died off after starvation. Next, the worms were heat treated by placing the plate inside an air incubator at 34 °C for 2 h. The worms were visually screened for putative gene insertion at least 6 h after the heat treatment. The worms were L1 larvae with green fluorescent protein (GFP)-labelled nuclei but devoid of mCherry signal. Gene insertion was verified by single-worm PCR and sequencing. Next, the worms were out-crossed with N2 for four rounds to eliminate secondary mutations before being used in an experiment.
Isolation and identification of C. elegans suppressor mutant
Ethylmethanesulfonate (EMS) mutagenesis of hyl-2(gnv1) single mutants was performed following the Aroian lab protocol: L4 larvae were agitated for 4 h in M9 buffer containing 50 mM EMS. Mutagenized worms were then plated onto seeded 100 mm ENG plates (0.5% peptone 0.1% yeast extract, 50 mM sodium chloride, 2% agar plus 5 µg cholesterol, 1 mM calcium chloride, 1 mM magnesium sulfate, 1 mM potassium phosphate at pH6) and left to become gravid adults overnight. Gravid P0 adults were harvested and bleached, and the F1 embryos were plated. Gravid F1 adults were bleached again, F2 embryos were left to hatch in M9 buffer, and L0 larvae were plated and grown to obtain young adult F2 worms that were placed in anoxia for selection. Isolated suppressor mutations were identified using rapid single polymorphism (RSNP) mapping, as described36. Whole-genome sequencing (WGS) was performed at the genomic platform of the Centre Médical Universitaire, Geneva. Genomic DNA from pooled recombinant worms from the RSNP mapping was extracted and analysed. Data analysis was performed using the free software IGV (Integrative Genomic Viewer) from the Broad Institute37. The suppressor mutants were outcrossed with N2 animals four times and ttc-17(gnv3) SNPs were followed by PCR and DNA sequencing using the following primer sets: For the SNP 1 (GGA to GAA), forward 5′–3′: GTGTCCCGAGTTCCATGAGT, and reverse 5′–3′: AGCTCTCCAGTAGGCAGCAG, with a PCR fragment of 451 base pairs (bp). For the SNP 2 (CAG to TAG), forward 5′–3′: GCTGCCATGCTTCACAGTAA and reverse 5′–3′: ACTCGAATTCTTTCCCAGCA, with a PCR fragment of 463 bp.
RNA interference experiments
RNAi experiments were performed on solid NGM plates containing 25 µg ml–1 carbenicillin and 1 mM IPTG. Worms were fed with HT115 strains containing either the empty vector L4440 as a control or L4440 containing T19A5.1 (ttc-17), mre-11 from C. elegans RNAi library obtained at Source Bioscience LifeScience, or daf-2, which was obtained from the Kenyon lab (University of California at San Francisco, CA)38. Ahringer RNAi Feeding Library L4440 constructs containing fragments of sptl-1, cct-1, cct-2, cct-3, cct-4, cct-5, cct-6, cct-7 and cct-8 were a kind gift from M. Gotta’s lab (Centre Médical Universitaire, Geneva). Every clone was verified by sequencing before use. All the experiments were performed at 20 °C following the Ahringer lab RNAi Feeding Protocol (Version 11.04.01)39, and the downregulation of ttc-17 was verified by semi-quantitative PCR.
Worm anoxia-reoxygenation survival assay
C. elegans strains were tested in anoxia for different amounts of time as described previously13. Anoxia experiments were always performed at 20 °C, and a maximum of 60 strains were tested at the same time. All tested animals were synchronized young adults (72 h post L0 stage), and around 30 worms per strain were tested in each well of a 12-well plate containing 1.5 ml NGM agar seeded with 5× concentrated bacteria. Survivors were scored using a binocular (Leica MZ6) after recovering for a period of 24 h in a normoxic environment.
Other worm survival and feeding assays
To determine the lethal toxicity effects of exogenous synthetic worm DoxSa (iso-branched C17 DoxSa) on C. elegans strains in normoxia, starved L0 larvae were grown on RNAi NGM plates containing 0 or 25 µM worm DoxSa and having been seeded with HT115 bacteria expressing either an empty vector (L4440) or an sptl-1 RNAi knockdown construct. Read-out was survival after 3 days of development. For inhibition of mitochondrial oxidative phosphorylation, L0 larvae were grown for 2 days on normal NGM plates seeded with 5× OP50 bacteria, and then switched for 24 hours on NGM plates containing either DMSO control or 5 µM rotenone (mitochondrial complex I inhibitor, Sigma Aldrich R8875).
Construction of transgenic strains containing ttc-17 transcriptional and translational fusions
Transgenic strains were obtained by gene bombardment using the Particle Delivery System PDS-1000/He Biolistic (BIO-RAD). For the transcriptional fusion reporter strain, a Pttc-17::GFP construct was cloned into pBluescript II KS+ plasmid containing a wild-type copy of Cbunc-119(+) at the SacI site. The resulting construct gnvEx2 was introduced into unc-119(ed3) worms by bombardment to give rise to strain JCM2, while for the translational fusion Pttc-17::GFP::ttc-17;Cbunc-119(+) was cloned into the same vector, resulting in gnvEx3, which was introduced into unc-119(ed3);ttc-17(gnv3);hyl-2(gnv1) to give rise to JCM3. Non-unc worms were isolated and tested for their GFP expression and/or resistance to anoxia.
Microscopy
Observations were performed using confocal Zeiss LSM700 microscope at the bioimaging centre of the Faculty of Science (University of Geneva). For each GFP worm observation, the setup of the gain was always done with the wild-type animal N2 for maximal reduction of the background GFP signal from C. elegans autofluorescence. In heart immunohistochemistry and immunofluorescence, antibody specificities of immunostainings were tested with control samples missing the primary antibody.
Samples for yeast F-actin imaging were obtained from exponentially growing cells (OD600 of 0.05, or 1 million cells per ml) in YPD liquid, which were treated with control vehicle or 4 µM DoxSa and incubated at 30 °C for 3 h. Next, the cells were fixed by directly adding paraformaldehyde to the culture at a final concentration of 4%. Then, 1 OD600 value of cells were washed with 3 ml of washing buffer (0.1 M of Potassium phosphate pH 7.5, 1.2 M of sorbitol), stained with 50 μl of 1 μM of phalloidin-Atto488 (Sigma-Aldrich) at 4 °C for 1 h in the dark, and observed by confocal microscopy.
Worm lipid extraction
Cryolysis was performed with a ‘Precellys 24, lysis & homogenization’ machine (Bertin Technologies). About 8,000 synchronized young adults per strain were collected with double-distilled H2O (without embryos). Bacteria were washed away, and worms were resuspended in 1 ml and transferred into a Cryolysis tube, spun down at 1,000g for 2 min and then washed once with 1 ml LC/MS grade water (Fisher Scientific). As much supernatant as possible was removed before the pelleted worms were frozen in liquid nitrogen and stored at −80 °C.
Worms were broken at 3–4 °C with 100 µl 1.4-mm zirconium oxide beads (Bertin Technologies) in 800 µl MS-grade H2O in the Precellys machine: 3 bursts of 45 s at 6,200 r.p.m. with 45-s interruptions. Lysates were eluted into a glass tube containing lipid standards, spun down at 600g and eluted again with 200 µl MS-grade H2O.
Lipid extraction was done with 3.6 ml chloroform:methanol (1:2). After vortexing and centrifugation at 800g for 5 min, single-phase supernatant was transferred to a new glass tube. Phase separation was induced by adding 0.5 ml of MS-grade H2O plus 0.5 ml of chloroform. After centrifugation at 800g for 5 min, the lower organic phase was collected. The sphingolipid fraction was obtained by treatment with methylamine followed by a butanol extraction.
Untargeted lipidomic analysis
In order to compare the sphingolipidome of worm CerS mutant hyl-2(gnv1) with the wild type in an unbiased way, we analysed sphingolipid fractions on an LTQ Orbitrap (ThermoFisher, Bremen, Germany) Fourier Transform mass spectrometer using direct infusion via a TriVersa NanoMate (Advion, Ithaca, NY, USA), collecting full scans for 3 min without a priori assumptions as to which lipid species might be present in a given solution. For each biological replicate (n = 6 for WT and n = 3 for mutants), three technical replicates were acquired. To facilitate the discovery process when using the Orbitrap, an automated pipeline that goes from raw mass spectrometry files to statistical evaluation was put in place and was executed on the Vital-IT high performance computing cluster (Lausanne, Switzerland). In short, one run of Orbitrap analysis consists of hundreds of scans, and the first step of the pipeline was to put them together to produce a single spectrum for each sample. Experiments aimed at discovering lipid composition differences between wild type and mutants. Our pipeline aligned all spectra against each other and identified peaks, given a user-specified signal threshold. Each peak was then quantified in each spectrum, and peaks were statistically evaluated concerning differential abundance in distinct strains based on a series of unpaired two-sided Student’s t tests. P values were then corrected using the R package for estimating local and tail area-based false discovery rates (FDRs)40, and hits were determined on the basis of a q < 0.01 cut-off. Hits were annotated using our lipid database and relying on the high mass precision of the Orbitrap, which is below 2 ppm. All results were sorted by significance, and graphic summaries were obtained, such as heat maps, volcano plots and box-and-whisker plots. Volcano plot shows –log10 of P against log2-transformed fold-change, and the dotted line indicates a significance cut-off of P = 0.01.
Targeted lipidomic analysis
For targeted analysis of candidate lipid species, we followed a previously published protocol41 based on multiple reaction monitoring. Briefly, a Triversa NanoMate (Advion, Ithaca, NY, USA) was used to infuse samples onto a TSQ Vantage triple quadrupole mass spectrometer (ThermoFisher Scientific, Waltham, USA). MRM-MS was used to identify and quantify lipid species as previously described42. Data were converted and quantified relative to standard curves of internal standards that had been spiked in before extraction. At least two independent biological replicates were analysed, each of which comprised up to six technical replicates.
Targeted metabolomics analysis
Pyridine extraction of metabolites and liquid chromatography–mass spectrometry (LC–MS) analysis was done according to Höglinger et al.43. In short, 6,000 worms and internal standards were extracted twice using pyridine extraction buffer (ethanol:water:diethylether:pyridine = 15:15:5:1, 2.1 10−3 M ammonium hydroxide), cryolysis and incubation on ice for 20 min. Extracts were centrifuged, separated from cell debris and dried. Metabolites were re-suspended in borate buffer (200 mM boric acid pH 8.8, 10 mM tris(2-carboxyethyl)phosphine, 10 mM ascorbic acid and 33.7 mM 13C15N-valine), derivatized with 6-aminoquinolyl-hydroxysuccinimidyl carbamate (AQC, 2.85 mg ml–1 in acetonitrile) at 55 °C for 15 min and left at 24 °C overnight. Samples were analysed using a reverse-phase C18 column (HPLC EC 100/2 Nucleoshell RP-18 2.7 mm) on an Accela high-performance liquid chromatography system (ThermoFisher Scientific, Waltham, MA), coupled to a TSQ Vantage triple quadrupole mass spectrometer (ThermoFisher Scientific, Waltham, USA) using multiple reaction monitoring.
Yeast strain construction
Yeast strains expressing mammalian CerSs were constructed as follows: yeast codon-optimized genes of Mus musculus CerSs 1, 2, 4, 5 and 6 and Homo sapiens CerS 3 were synthesized by GENEART (Regensburg, Germany) containing an amino-terminal flag-tag, subcloned first into pRS424 (BamHI/EcoRI) then together with the Tdh3 promoter and Cyc1 terminator (SacI/KpnI) into integrative vector Yiplac204. For CerS double-mutant complementation strain RH7165 (lag1Δlac1Δ) was first transformed with pRS416-LAG1, mammalian CerSs were introduced through PmlI-linearized Yiplac204 constructs into the Trp1 locus, and pRS416-LAG1 was removed on 5-FOA plates. For the wild-type strains expressing mammalian CerSs strain RH2881 (wild type) was transformed with the PmlI-linearized Yiplac204 constructs directly.
EUROSCARF BY4741- and BY4743-derived CerS3 expressing strains were generated similarly. The synthetic codon-optimized gene of N-terminally flag-tagged CerS3 was inserted into the pRS424-GPD plasmid at the BamHI and EcoRI sites. Then, the pTDH3::CERS3::Cyc1T fragment, obtained by cutting the plasmid with SacI and KpnI, was inserted into the YIplac211 plasmid. Next, 70 bp of the left homology region of the URA3 locus was introduced into the plasmid at the AatII site, while the 70 bp of the right homology region was introduced into the plasmid at the KpnI and AflII sites. Then, the CerS3 URA3 cassette amplified with forward (CCATGAAGCTTTTTCTTTC) and reverse (CATATTTATGGTGAAGGATAAGTT) oligos was integrated into the genome of BY4741, BY4743, the related temperature-sensitive strains and the haploid insufficient strains.
To generate strains overexpressing individual TRiC subunits, LEU2 was amplified from the pRS415-LEU2 plasmid using the following oligonucleotides: forward (CAGTGGATCCGCGCCTGATTCAAGAAATA), reverse (CTGTGAGCTCCTAACTTTTTGTGTGGTGCC). The resulting LEU2 fragment with the BamHI and SacI restriction sites was introduced into the pFA6a-KanMX6 plasmid at the BamHI and SacI sites, replacing the KanMX6 gene. Next, the promoter of Tdh3 obtained by cutting the pRS424-GPD plasmid with SacI and SpeI was introduced into the pFA6a-LEU2 at the SacI and SpeI sites. Then, the LEU2::pTDH3 promoter cassette was amplified and inserted just upstream of the start codon of individual CCT genes.
Yeast growth assays
Yeast strains were grown at 30 °C to saturation in liquid YPD with additional uracil, adenine and tryptophan (YPUATD). Stationary yeast cultures were diluted to OD600 = 1.5 and were subjected to consecutive ten-fold dilutions with double-distilled water. About 3 µl of the dilutions were pinned either onto YPUATD agar plates in the case of complementation of lag1Δlac1Δ or on YPAUTD containing 0.05% tergitol NP-40, ethanol vehicle and 0, 5, 10 and 20 µM DoxSa or 5 µM DoxSa and 100 µM Fumonisin B1 (Sigma Aldrich F1147) for DoxSa sensitivity assays. Plates were normally incubated for 2 days at 30 °C, and in the case of temperature sensitive mutants, for 3 days at 24 °C before monitoring growth.
Yeast lipid analysis
Yeast lipid extraction and analysis was performed as previously described41,42. Complemented lag1Δlac1Δ strains expressing mammalian CerSs were grown in liquid YPUATD until early log phase before stopping growth with 5% trichloroacetic acid (TCA), while wild-type yeast strains expressing the mammalian CerSs were grown to OD600 = 1.0 in simple YPUATD and then grown for 90 minutes in YPUATD with ethanol vehicle control or 2.5 µM DoxSa before metabolism was stopped with 5% TCA.
Myriocin preparation for in vivo mouse experiments
Stock solution of myriocin (Sigma M1177) was prepared at 2.5 mg ml–1 in a mixture of Cremophor and DMSO (3:1 vol/vol). On the day of the experiment, a working solution at 0.05 mg ml–1 was made by a dilution 1:50 in 0.9% NaCl. Mixture of myriocin and DoxSa (Avanti Polar Lipids) was prepared similarly.
Mouse in vivo model of acute myocardial ischaemia–reperfusion injury, infarct size measurement and intramyocardial adenovirus injection
All procedures conformed to European Parliament Directive 2010/63/EU and the September 22, 2010 Council on the Protection of Animals and were approved by the local institutional animal research committees (no. BH2012-64 and no. 17627-2018112210138309). C57Bl6 mice (male, 8–12 weeks, Charles River Laboratories) were anesthetized by intraperitoneal injection of xylazine (5 mg per kg (body weight), Rompun), ketamine (100 mg per kg (body weight), Imalgen) and were supplemented subcutaneously with buprenorphine (0.075 mg per kg (body weight)). The animals were orally intubated with a 22-gauge metallic cannula and ventilated via a rodent ventilator according to their body weight (Physiosuite MouseVent, Kent Scientific). Body temperature was maintained at 37 °C using a heating pad. Mice were randomized to blindly receive either the vehicle (mixture of Cremophor and DMSO (3:1 vol/vol) in 0.9% saline), 0.3 mg per kg (body weight) myriocin (Sigma M1177) or each 0.3 mg per kg (body weight)myriocin and DoxSa 10 min before the start of ischaemia by intravenous injection in the jugular vein (n = 10–16 per group). A left thoracotomy was performed in the fourth left intercostal space, and the pericardium was opened to expose the heart. An 8-0 polypropylene suture attached to a small curved needle was passed around the left anterior descending coronary artery. Successful left anterior descending coronary artery occlusion was confirmed by ST-segment shift on the ECG (EMKA) and the appearance of myocardial pallor. Reperfusion was performed at the end of the 45-min of ischaemia by reopening of the suture. The mouse chests were then closed, and once spontaneous breathing resumed, the endotracheal tube was removed and the animals were allowed to recover in a temperature- and oxygen-controlled intensive-care unit. They were then returned to the animal facilities until the end of the reperfusion period. After 24 h of reperfusion, infarct size was assessed, or the heart was fixed for histology. To this end, mice were anesthetized; the coronary artery was briefly reoccluded followed by an intravenous injection of 0.5 mg per kg (body weight) Uniperse blue pigment to delineate the in vivo area at risk. For infarct size measurement, the heart was removed and cut into 5 1-mm-thick transverse slices parallel to the atrioventricular groove. All slices were incubated for 15 min in a 1% solution of triphenyltetrazolium chloride at 37 °C to differentiate infarcted (pale) from viable (brick red) myocardial area. Extent of the area at risk and area of necrosis was quantified by computerized planimetry using SigmaScanPro5 (version 5.0.0) and corrected for the weight of the tissue slices.
For the intramyocardial adenovirus injection, C57Bl6 mice (male, 8–12 weeks) were anesthetized under 3.5% Sevoflurane and supplemented subcutaneously with buprenorphine (0.075 mg per kg (body weight)). The chest cavity was opened for access to the heart similarly to the ischaemia procedure described above. 5 × 108 PFU of adenovirus in 20 µl NaCl were injected into the left ventricular wall in 7 different points. The chest cavity was closed, and the mice were allowed to recover for 1 week prior to ischaemia–reperfusion surgery, as described above, with no additional injections during the sequence.
Echocardiography
Cardiac contractile function was assessed by echocardiography before the ischaemia and at 24 h of reperfusion. Echocardiography was performed under slight sedation (5 mg per kg (body weight) butorphanol sc). Images were acquired with a 13-MHz linear-array transducer with a digital ultrasound system (Vivid 7, GE Medical Systems, Waukesha, Wis). Conventional measurements were performed: left ventricle (LV) end-diastolic diameter, LV end-systolic diameter, LV fractional shortening, interventricular thickness, and posterior wall thickness.
Mouse heart histology
Following ischaemia–reperfusion, mice were perfused with PBS followed by 4% paraformaldehyde through the right carotid. The heart was then removed and placed in 4% paraformaldehyde at 4 °C. At 24 h after fixation of the mouse heart in 4% paraformaldehyde, the heart was washed with PBS and embedded in paraffin by the automatic tissue processor STP120 (Thermo Fisher 813160). Ultrasectioning, haematoxylin and eosin (H&E) staining, phosphotungstic acid haematoxylin (PTAH) staining, anti-CCT5 immunohistochemical, as well as anti-CCT5 and anti-actin immunofluorescence staining were performed by the EPFL Histology Core Facility (Lausanne, Switzerland) using standard procedures and utilizing 1/100-diluted primary antibodies anti-CCT5 (abcam rabbit ab129016) and anti-pan actin (Thermo mouse MA5-11869) as well as 1/1,000-diluted secondary antibodies donkey anti-mouse Alexa568 and donkey anti-rabbit Alexa647.
Mouse heart lipid and metabolite analysis
After planimetric analysis slices were transferred to Eppendorf tubes, frozen in liquid nitrogen, lyophilized and ground to powder. Lipids were extracted from powder following the methyl tertiary butyl ester (MTBE) method44. Briefly, 4 mg dry powder was resuspended in 100 µl MS-grade water. 50 µl zirconium beads, 340 µl methanol and internal standards were added and tissue was disrupted using cryolysis at 4 °C before adding 1.2 ml of MTBE. After 1-h incubation at room temperature on a shaker, phase separation of the extract was induced with 200 µl MS-grade water, and the upper organic phase was collected after centrifugation. Aqueous phase was extracted one more time with artificial upper phase (MTBE: methanol:water = 20:6:3, vol/vol) and after centrifugation, organic phases were combined and dried. The sphingolipid fraction was prepared as described earlier, and lipid analysis was performed as described above. Metabolite analysis was performed from 5 mg of dried tissue powder using the pyridine extraction as described for worm metabolomic analysis.
In order to distinguish non-canonical 1-deoxyceramides from canonical DHCers, we followed an established protocol15. Briefly, we separated ceramides using normal phase LC–MS (Uptisphere 5 µm NH2 150 × 2.1 mm column) at a flow rate of 0.375 ml min–1 and an isocratic solvent system (CH3CN/CH3OH/CH3COOH/CH3(CH2)3OH/H2O, 95/3/1/0.4/0.3, vol/vol with 5 mM ammonium acetate) delivered by an Accela autosampler and HPLC system coupled to a TSQ Vantage triple quadrupole mass spectrometer (ThermoFisher Scientific, Waltham, USA). MRM-MS was used to identify and quantify the following ceramide species: at retention time 1.1 min internal standard C12 DoxDHCer (Q1: 468.48 and Q3: 268.3), C16-26 DoxDHCer (Q1: 524.54, 552.57, 580.60, 608.63, 636.66, 664.69 and Q3: 268.3) and C16-26 1-deoxyceramides (Q1: 522.54, 550.57, 578.60, 606.63, 634.66, 662.69 and Q3: 266.3) and at retention time 1.4 min C17 ceramide internal standard (Q1: 534.52 and Q3: 264.3) as well as C16-26 DHCers (Q1: 522.54, 550.57, 578.60, 606.63, 634.66, 662.69 and Q3: 266.3). Areas under the curve were quantified using Thermo’s Xcalibur Quan browser software, and normalization was performed relative to the respective internal standards.
Sample preparation for worm proteome analysis by LC–MS/MS
Ornithine amino transferase mutant oatr-1(tm5454) strain was grown on NGM plates without peptone seeded with SLE1 bacteria45 grown in M63 medium containing 80 µg ml–1 l-proline as well as either 40 µg ml–1 each of light l-lysine and l-arginine or 41.44 µg–1 heavy [13C6,15N2]l-lysine (CNLM-291 Cambridge Isotope Laboratories, Andover, MA, USA) and 41.88 µg ml–1 heavy [13C6, 15N4]l-arginine (CNLM-539 Cambridge Isotope Laboratories, Andover, MA, USA). After four generations, heavy-isotope-labelled worms showed heavy/light incorporations of more than 99%. For each condition, two biological replicates of worms each of heavy and light cultures were grown to young adulthood and were either harvested directly or exposed to 24 h of anoxia. Worms were harvested in double-distilled water and washed with 10 mM NH4HCO3, and worm pellets were frozen. Lysates were prepared in 10 mM NH4HCO3 using the cryolysis and zirconium beads.
Protein concentration was determined using the Bradford assay (Bio-Rad), and then light and heavy proteomes were mixed together at a 1:1 ratio. Proteomes (40 µg) were denatured with 6 M urea in 50 mM NH4HCO3, reduced with 10 mM TCEP for 30 min and alkylated with 25 mM iodoacetamide for 30 min in the dark. Samples were diluted to 2 M urea with 50 mM NH4HCO3 and digested with trypsin (Thermo Scientific, 2 μL of 0.5 μg μl–1) in the presence of 1 mM CaCl2 for 12 hours at 37 °C. Samples were acidified to a final concentration of 5% acetic acid, desalted over a self-packed C18 spin column and dried. Samples were analysed by LC-MS/MS (see below) and the MS data was processed with MaxQuant (see below).
LC–MS/MS proteomics analysis
Peptides were resuspended in water with 0.1% formic acid (FA) and analysed using Proxeon EASY-nLC 1000 nano-UHPLC coupled to QExactive Plus Quadrupole-Orbitrap mass spectrometer (Thermo Scientific). The chromatography column consisted of a 30-cm long, 75-μm internal diameter microcapillary capped by a 5-μm tip and packed with ReproSil-Pur 120 C18-AQ 2.4 μm beads (Dr. Maisch GmbH). LC solvents were 0.1% TFA in water (Buffer A) and 0.1% TFA in acetonitrile (Buffer B). Peptides were eluted into the mass spectrometer at a flow rate of 300 nl min–1 over a 240-min linear gradient (3–35% Buffer B) at 65 °C. Data were acquired in data-dependent mode (top-20, NCE 30, R = 17’500) after full MS scan (R = 70’000, m/z 400–1’300). Dynamic exclusion was set to 10 s and peptide match to prefer, and isotope exclusion was enabled.
The MS data were analysed with MaxQuant1 (V1.5.2.8) and searched against the C. elegans proteome (UniProt) and a common list of contaminants (included in MaxQuant)46. The first peptide search tolerance was set to 20 ppm; 10 ppm was used for the main peptide search, and fragment mass tolerance was set to 0.02 Da. The FDR for peptides, proteins and sites identification was set to 1%. The minimum peptide length was set to six amino acids, and peptide re-quantification, label-free quantification (MaxLFQ) and ‘match between runs’ were enabled. The minimal number of peptides per protein was set to two. l-lysine and l-arginine were set as light isotope labels and [13C6,15N2]l -lysine and [13C6,15N4]l -arginine were set as heavy isotope labels. Oxidized methionine was searched as variable modifications and carbamidomethylation of cysteines was searched as a fixed modification.
1,270 proteins were identified, and of those, 1,197 proteins were found at least once in both forward (light anoxia versus heavy normoxia) and reverse (heavy anoxia versus light normoxia) labelling. 867 proteins with a coefficient of variation below 25% between forward and reverse labelling were selected and Gene Ontology (GO) terms were searched with Generic Gene Ontology (GO) Term Finder47 (https://go.princeton.edu).
Statistical analysis
Unless otherwise stated, statistical analysis including unpaired two-sided Student’s t test, two-tailed analysis of variance (ANOVA) and outlier detection was performed using the GraphPad Prism 8 software. Slopes of linear regressions were compared using ANCOVA.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Kalogeris, T., Baines, C. P., Krenz, M. & Korthuis, R. J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 298, 229–317 (2012).
World Health Organization. WHO Methods and Data Sources for Country-level Causes of Death 2000–2015 (Geneva, 2017).
Padilla, P. A. & Ladage, M. L. Suspended animation, diapause and quiescence: arresting the cell cycle in C. elegans. Cell Cycle 11, 1672–1679 (2012).
Ganote, C. & Armstrong, S. Ischaemia and the myocyte cytoskeleton: review and speculation. Cardiovasc Res. 27, 1387–1403 (1993).
Hannun, Y. A. & Obeid, L. M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 19, 175–191 (2018).
Duan, J. & Merrill, A. H. Jr. 1-Deoxysphingolipids encountered exogenously and made de Novo: Dangerous mysteries inside an enigma. J. Biol. Chem. 290, 15380–15389 (2015).
Hannich, J. T., Umebayashi, K. & Riezman, H. Distribution and functions of sterols and Sphingolipids. Cold Spring Harb. Perspect. Biol. 3, 189–202 (2011).
Chitwood, D. J., Lusby, W. R., Thompson, M. J., Kochansky, J. P. & Howarth, O. W. The glycosylceramides of the nematode Caenorhabditis elegans contain an unusual, branched-chain sphingoid base. Lipids 30, 567–573 (1995).
Hannich, J. T., Mellal, D., Feng, S. H., Zumbuehl, A. & Riezman, H. Structure and conserved function of iso-branched sphingoid bases from the nematode Caenorhabditis elegans. Chem. Sci. 8, 3676–3686 (2017).
Novgorodov, S. A. & Gudz, T. I. Ceramide and mitochondria in ischemia/reperfusion. J Cardiovasc. Pharmacol. 53, 198–208 (2009).
Siddique, M. M., Li, Y., Chaurasia, B., Kaddai, V. A. & Summers, S. A. Dihydroceramides: from bit players to lead actors. J. Biol. Chem. 290, 15371–15379 (2015).
Padilla, P. A., Nystul, T. G., Zager, R. A., Johnson, A. C. & Roth, M. B. Dephosphorylation of cell cycle-regulated proteins correlates with anoxia-induced suspended animation in Caenorhabditis elegans. Mol. Biol. Cell. 13, 1473–1483 (2002).
Menuz, V. et al. Protection of C. elegans from anoxia by HYL-2 ceramide synthase. Science 324, 381–384 (2009).
Mullen, T. D., Hannun, Y. A. & Obeid, L. M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 441, 789–802 (2012).
Zitomer, N. C. et al. Ceramide synthase inhibition by fumonisin B1 causes accumulation of 1-deoxysphinganine: a novel category of bioactive 1-deoxysphingoid bases and 1-deoxydihydroceramides biosynthesized by mammalian cell lines and animals. J. Biol. Chem. 284, 4786–4795 (2009).
Penno, A. et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J. Biol. Chem. 285, 11178–11187 (2010).
Butler, J. A. et al. Profiling the anaerobic response of C. elegans using GC-MS. PLoS One. 7, e46140 (2012).
Reforgiato, M. R. et al. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res. Cardiol. 111, 12 (2016).
Devlin, C. M. et al. Dihydroceramide-based response to hypoxia. J. Biol. Chem. 286, 38069–38078 (2011).
Testai, F. D. et al. Multiple sphingolipid abnormalities following cerebral microendothelial hypoxia. J. Neurochem. 131, 530–540 (2014).
Saegusa, K. et al. Caenorhabditis elegans chaperonin CCT/TRiC is required for actin and tubulin biogenesis and microvillus formation in intestinal epithelial cells. Mol. Biol. Cell 25, 3095–3104 (2014).
Berger, J. et al. In Vivo Function of the Chaperonin TRiC in alpha-Actin Folding during Sarcomere Assembly. Cell Rep. 22, 313–322 (2018).
Gill, S., Le, H. D., Melkani, G. C. & Panda, S. Time-restricted feeding attenuates age-related cardiac decline in Drosophila. Science 347, 1265–1269 (2015).
Melkani, G. C. et al. TRiC/CCT chaperonins are essential for maintaining myofibril organization, cardiac physiological rhythm, and lifespan. FEBS Lett. 591, 3447–3458 (2017).
Erdmann, J. et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 504, 432–436 (2013).
Berchtold, D. et al. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat. Cell Biol. 14, 542–547 (2012).
Rispal, D. et al. Target of rapamycin complex 2 regulates actin polarization and endocytosis via multiple pathways. J. Biol. Chem. 290, 14963–14978 (2015).
Guntert, T. et al. 1-Deoxysphingolipid-induced neurotoxicity involves N-methyl-d-aspartate receptor signaling. Neuropharmacology 110, 211–222 (2016).
Kahl, A. et al. Cerebral ischemia induces the aggregation of proteins linked to neurodegenerative diseases. Sci. Rep. 8, 2701 (2018).
Othman, A. et al. Plasma deoxysphingolipids: a novel class of biomarkers for the metabolic syndrome? Diabetologia 55, 421–431 (2012).
Lee, M. J. et al. Hereditary sensory neuropathy is caused by a mutation in the delta subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct4) gene. Hum. Mol. Genet. 12, 1917–1925 (2003).
Bouhouche, A., Benomar, A., Bouslam, N., Chkili, T. & Yahyaoui, M. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 43, 441–443 (2006).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Vallin, E. et al. A genome-wide collection of Mos1 transposon insertion mutants for the C. elegans research community. PLoS One. 7, e30482 (2012).
Frokjaer-Jensen, C. et al. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375–1383 (2008).
Davis, M. W. et al. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics 6, 118 (2005).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Dillin, A., Crawford, D. K. & Kenyon, C. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science 298, 830–834 (2002).
Kamath, R. S., Martinez-Campos, M., Zipperlen, P., Fraser, A. G. & Ahringer, J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2, research0002.1 (2001).
Strimmer, K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462 (2008).
da Silveira Dos Santos, A. X. et al. Systematic lipidomic analysis of yeast protein kinase and phosphatase mutants reveals novel insights into regulation of lipid homeostasis. Mol. Biol. Cell 25, 3234–3246 (2014).
Guan, X. L., Riezman, I., Wenk, M. R. & Riezman, H. Yeast lipid analysis and quantification by mass spectrometry. Methods Enzymol. 470, 369–391 (2010).
Hoglinger, D. et al. Intracellular sphingosine releases calcium from lysosomes. eLife 4, e10616 (2015).
Matyash, V., Liebisch, G., Kurzchalia, T. V., Shevchenko, A. & Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 49, 1137–1146 (2008).
Larance, M. et al. Stable-isotope labeling with amino acids in nematodes. Nat. Methods 8, 849–851 (2011).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Boyle, E. I. et al. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20, (3710–3715 (2004).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Acknowledgements
We would like to thank L. Pineau for construction of yeast strains, I. Riezman for help with LC–MS analysis, V. Nesatyy and F. David for help with lipidomics analysis, M. Müller for help with statistical analysis, J. Dessimoz for histological support and I. Castanon for helpful comments on the manuscript. Worms and bacterial strains were received from the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440), from National BioResource Project (Tokyo Women’s Medical University, Tokyo, Japan), from NemaGENETAG. Worm strain GK454 was a kind gift from K. Sato. J.T.H. was supported by an EMBO/Marie-Curie Long-Term Fellowship. This work was supported by grants from the Swiss National Science Foundation (SNSF) (to H.R. and J.-C.M.), the National Centre of Competence in Research (NCCR) Chemical Biology and SystemsX (evaluated by the SNSF) and the Canton of Geneva. A.Z. was supported by an SNSF assistant professorship.
Author information
Authors and Affiliations
Contributions
J.T.H. and H.R. designed the study, J.T.H. performed lipidomic and metabolomic analyses, as well as functional studies in worms and yeast. A.G.H. constructed the sptl-1C121W worm and performed yeast actin staining. S.G. isolated and characterized the ttc-17(gnv3) suppressor mutant under the guidance of J.-C.M. M.P. and L.G. designed, M.P., L.G., B.P. and H.T. performed, and M.P. and L.G. interpreted the mouse ischaemia–reperfusion experiments under the guidance of M.O. A.Z. synthesized iso-branched C17 DoxSa. D.A. performed proteomics analysis under the guidance of A.A. N.G. developed peak identification and quantification for non-targeted lipidomics. J.T.H. and H.R. wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Primary Handling Editor: Pooja Jha
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Non-canonical DoxDHCer increases during anoxia while normal ceramide is depleted.
a,b, Targeted lipid analysis of DoxDHC (a) and ceramide (b) species from wild-type and hyl-2(tm2031) worms, and the respective animals expressing the sptl-1C121W allele (+C121W) either after 3 d of normoxia (solid columns) or after an additional 20 h of anoxia (shaded columns); log2-transformed data from n = 4 independent biological replicates; bars are means with s.e.m. c,d, Correlations of DoxDHCer (c) and ceramide (d) amounts in pmole per nmoles inorganic phosphate between normoxic and anoxic conditions, for the same dataset as in in a and b; n = 4 independent biological replicates, points are means with s.e.m.; slopes of the linear regressions for each strain are given as m with a P value testing the difference to a line with the slope 1.
Extended Data Fig. 2 Activity and substrate specificity of mammalian CerS is conserved when expressed in yeast, and anoxia-reoxygenation survival of hyl-2(gnv1) mutant worms is extended by knock down of ttc-17, which is widely expressed in L4 larvae and young adults.
a, Yeast dilution growth assay of lag1Δlac1Δ CerS double mutants showing rescue with all six mammalian CerSs CerS1–CerS6 on rich medium. The experiment was repeated twice with similar results. b, Representative structure of an inositol phosphoceramide (IPC), which is the S. cerevisiae higher sphingolipid analogous to mammalian sphingomyelin and contains canonical serine-derived phytosphingosine (blue). c, Heat maps of specific IPC species levels in wild-type and CerS mutants rescued with individual mammalian CerSs showing IPC levels of the major molecular species produced as nM per 25 OD600 or percent of total. d, Yeast dilution growth assay of wild-type yeast expressing control (EV) and the six mammalian CerSs CerS1–CerS6 on rich medium (YPD) containing 0.05% tergitol and vehicle (EtOH), plus either 5 µM DoxSa or 5 µM and 100 µM fumonisin B1 (FB1). The experiment was repeated twice with similar results. e, Survival of hyl-2(gnv1) after 48 h of anoxia and 24 h reoxygenation following 3-d feeding on bacteria expressing empty vector (L4440) or RNAi vectors targeting ttc-17 or the positive control (daf-2) and the negative control (mre-11); n = 3 independent experiments with 2 independent biological replicates each; bars are means with s.e.m., P values are determined by unpaired two-sided Student’s t-test. f, cDNA quantification via PCR shows knock down of ttc-17 mRNA in response to ttc-17 RNAi compared with empty vector (L4440). The experiment was repeated three times with similar results. g–j, Confocal microscopy images of adults (g,i) and L4 larva (h) expressing a GFP reporter from a ttc-17 promoter showing widespread expression, especially in the hypodermis (arrow head), head neurons, the intestine and the developing vulva (arrow). j, Confocal microscopy image of a wild-type adult not expressing any fluorescent protein to show background signal mainly from gut granules in the intestine. Similar results for g–j were obtained in three independent experiments. Scale bars represent 100 μm.
Extended Data Fig. 3 Both non-canonical as well as canonical sphingolipids are upregulated upon ischaemia–reperfusion in mouse hearts overexpressing wild-type or mutant (C133W) SPTLC1 constructs.
a, Mouse hearts expressing wild-type or mutant SPTLC1 constructs for 1 week after adeno-associated viral (AAV) infection were analysed either following sham treatment or ischaemia–reperfusion. b–f, Both non-canonical (b,c) and canonical (d–g) sphingolipids are upregulated upon ischaemia–reperfusion, as can be seen for DoxSa (b), DoxDHCer (c), sphinganine (d), sphingosine (e), ceramide (f) and DHCer (g). While wild-type worms and C133W mutants are mostly similar upon ischaemia–reperfusion, C133W shows a weak increase in DoxDHCer in non-ischaemic sham (c) and lower (dihydro-)ceramide levels than WT after ischaemia–reperfusion (f,g), but surprisingly also significantly lower DoxDHCer levels than wild type after ischaemia–reperfusion (c). This shows that C133W mutation causes an increase of DoxDHCer relative to wild type only in sham conditions in which sufficient serine is available as substrate for the wild-type enzyme. Serine is not used as substrate by C133W that uses alanine instead. In ischaemic conditions, wild-type SPT readily uses alanine as a substrate, even more efficiently than C133W; n = 4 for sham and n = 8 for ischaemia–reperfusion are individual animals, bars are means with s.d. All statistical tests in b–g are unpaired two-sided Student’s t-tests. h, Plot representation of area of necrosis relative to area at risk for animals with AAV expression of wild-type and mutant (C133W) SPTLC1. Under these conditions, very little difference can be seen compared with the vehicle-treated control animals (grey line), but hearts with higher DoxDHCer levels do show a tendency of more necrosis. Vehicle linear regression from Fig. 4h; for AAV, n = 8 individual animals; coefficient of determination (r2) given for each linear regression and P as determined by ANCOVA.
Extended Data Fig. 4 Non-canonical DoxSa reverts myriocin cardioprotection by increasing levels of non-canonical 1-deoxy(dihydro)ceramide, but not canonical ceramide.
a–d, While animals treated with control vehicle or myriocin were comparable, mice involved in the group treated with myriocin plus DoxSa (Myr+DoxSa) by chance happened to show significantly increased body weight (a), left ventricle (LV) weight (b) and area at risk in left ventricle (c). While experiments were performed blinded, the batch of mice used for Myr+DoxSa was slightly larger at time of surgery. Still, relative area at risk (d) was comparable between all three conditions, n = 14 (Vehicle), n = 12 (Myriocin) and n = 10 (Myriocin+DoxSa) are individual animals; bars are means with s.d. e,f, LC–MS analysis of free sphingoid bases from mouse hearts after ischaemia–reperfusion treatment, whose hearts had been preconditioned either with vehicle, myriocin or myriocin and DoxSa (Myr+DoxSa), shows a significant decrease upon myriocin treatment for both sphinganine (e) and sphingosine (f); n = 10–11 animals. g,h, Direct infusion lipid analysis of ceramide levels from the same hearts as in e,f, showing downregulation of ceramide (g) and dihydroceramides (h) upon myriocin treatment, which is not reverted by myriocin plus DoxSa treatment (Myr+DoxSa) for ceramide (g). Apparent reversal of dihydroceramides (h) levels upon Myr+DoxSa treatment can be explained by LC–MS (i–k) as 1-deoxyceramides have the same m/z ratio as dihydroceramides and need to be separated by LC15. Indeed, only the reduced levels upon myriocin treatment in DoxDHC (i) and 1-deoxyceramides (j) but not in the dihydroceramides (k) are reverted by myriocin plus DoxSa treatment; n = 12 (Vehicle), n = 12 (Myriocin) and n = 10 (Myriocin+DoxSa) are individual animals, bars are means with s.d . All statistical tests are unpaired two-sided Student’s t-tests. l–n, Analyses of area of necrosis in mg relative to the amount of DoxDHCer (l) and ceramide (m) show a low positive correlation, while DHCer (n) shows only negligible correlation; r values are the correlation coefficients from the linear regressions (solid lines), discontinuous lines are 95% confidence intervals; n = 25 (l), n = 24 (m,n) individual animals.
Extended Data Fig. 5 Non-canonical 1-deoxysphingolipids impair protein folding and disrupt the cytoskeleton.
a–c, Larger images of mCherry::ACT-5 (mC::ACT-5) intestinal signals of wild-type animals (a) upon treatment with empty vector (L4440) or RNAi knock down feeding constructs targeting all eight subunits of the chaperonin TRiC complex under normoxia or after 24- and 48-h anoxia and 24-h reoxygenation treatment (b), and after treatment with exogenous worm DoxSa (iso-branched C17 DoxSa) (c); scale bars, 25 µm. Experiments were repeated three times with similar results. d, Yeast cells expressing mammalian CerS3 showing F-actin staining with phalloidin upon DoxSa treatment for 3 h; scale bar, 2 µm. Experiments were repeated five times with similar results. e, Yeast dilution growth assay of wild-type yeast and thermo-sensitive TRiC chaperonin mutants expressing control or mammalian CerS3 constructs on rich medium containing 0.05% tergitol as well as vehicle (EtOH) or 2, 4 and 8 µM DoxSa (DoxSa) at a permissive temperature of 24 °C. Experiments were repeated three times with similar results. f, Yeast dilution growth assay of diploid wild-type yeast and haplo-insufficient TRiC chaperonin mutants expressing control or mammalian CerS3 constructs on rich medium containing 0.05% tergitol as well as vehicle (EtOH) or 4, 8 and 10 µM DoxSa at 30 °C. Experiments were repeated twice with similar results. g, Yeast dilution growth assay of wild-type yeast (BY4741) expressing mammalian CerS3 as well as a control construct, or overexpressing all 8 chaperonin subunits from a genomic Tdh3 promoter on rich medium (YPD) and rich medium containing 0.05% tergitol as well as vehicle (EtOH) or 2.5, 5, 7.5, and 10 µM DoxSa at 30 °C. Experiments were repeated five times with similar results.
Extended Data Fig. 6 Ischaemia–reperfusion and non-canonical DoxSa treatments both damage the cytoskeleton by reducing TRiC chaperonin and actin colocalization in cardiomyocytes.
a, Immunohistochemical detection of the TRiC chaperonin complex CCT5 subunit after ischaemia–reperfusion injury showing irregular staining in the area at risk of the left ventricle; staining around the right ventricle (RV) is more regular. Scale bar, 1 mm. Stainings were repeated with similar results on hearts from nine individual animals, using staining without primary antibody as negative control. b, Close-up of H&E staining (upper), anti-CCT5 immunohistochemical staining (middle) and PTAH staining (lower) in area at risk in the left ventricle showing both necrotic (N) and healthy (H) cardiomyocyte fibres, as well as edema (E); scale bars, 100 µm. Stainings were repeated with similar results on hearts from at least two individual animals. c, Immunofluorescence imaging of cardiomyocytes treated with vehicle, myriocin or myriocin plus DoxSa after ischaemia–reperfusion in the left ventricle and in non-ischaemic right ventricle; anti-CCT5 staining is shown in cyan, anti-actin in magenta and DAPI in yellow. Upper panels show overviews with scale bars of 20 µm, while lower panels show close-ups with scale bars at 5 µm. Experiments were repeated four times with similar results.
Extended Data Fig. 7 Non-targeted high-mass-accuracy lipidomics screen identifies significantly altered sphingolipid species in hyl-2(gnv1) mutant animals.
Columns are as follows: measured mass per charge ratios, as detected with the LTQ orbitrap mass spectrometer and identified with in-house software; relative change between WT(N2) and hyl-2(gnv1) (increased species have red borders, and decreased species have blue borders); previously detected sphingolipid changes from Menuz et al.13 (colours as in the previous column); statistical significance as determined by unpaired two-sided Student’s t-test; FDR-corrected q-values; assigned lipid species with mass error below 2 ppm (most significantly increased assigned species are highlighted in red, and most significantly decreased assigned species are in blue); molecular formula of identified lipid species; theoretical mass per charge ratios; mass errors between measured and theoretical mass per charge ratio in parts per million. n = 6 (wild type) and 3 (hyl-2) independent biological replicates. Hex, hexose. Short hand for total back-bone carbons was used: AA:b, where AA is the total ceramide backbone carbons, and b is the number of double bonds. -OH, number of additional hydroxylations. Short hand for detailed species description was used: ihMM:n/X:Y:Z where I is iso-branched, h is sphingoid base hydroxyl groups (m, one; d, two; t, three), MM is the number of sphingoid base carbons, n is the number of sphingoid base double bonds, X is the number of fatty-acid carbons, Y is the number of fatty-acid double bonds and Z is the number of fatty acid hydroxylations.
Supplementary information
Source data
Source data Fig. 1
Statistical Source Data
Source data Fig. 2
Statistical Source Data
Source data Fig. 3
Statistical Source Data
Source data Fig. 4
Statistical Source Data
Source data Fig. 5
Statistical Source Data
Source data Fig. 6
Statistical Source Data
Source Data
Extended Data Fig. 1
Source Data
Extended Data Fig. 2
Source Data
Extended Data Fig. 3
Source Data
Extended Data Fig. 4
Rights and permissions
About this article

Cite this article
Hannich, J.T., Haribowo, A.G., Gentina, S. et al. 1-Deoxydihydroceramide causes anoxic death by impairing chaperonin-mediated protein folding. Nat Metab 1, 996–1008 (2019). https://doi.org/10.1038/s42255-019-0123-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-019-0123-y
This article is cited by
-
Imbalanced unfolded protein response signaling contributes to 1-deoxysphingolipid retinal toxicity
Nature Communications (2023)
-
Patched regulates lipid homeostasis by controlling cellular cholesterol levels
Nature Communications (2021)
-
Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia-reperfusion
Cell Death & Disease (2020)
