
Abstract
Understanding the photodamage mechanism underlying the highly nonlinear dynamic of femtosecond laser pulses at the second transparent window of tissue is crucial for label-free microscopy. Here, we report the identification of two cavitation regimes from 1030 nm pulses when interacting with the central nervous system in zebrafish. We show that at low repetition rates, the damage is confined due to plasma-based ablation and sudden local temperature rise. At high repetition rates, the damage becomes collateral due to plasma-mediated photochemistry. Furthermore, we investigate the role of fluorescence labels with linear and nonlinear absorption pathways in optical breakdown. To verify our findings, we examined cell death and cellular responses to tissue damage, including the recruitment of fibroblasts and immune cells after irradiation. These findings contribute to advancing the emerging nonlinear optical microscopy techniques and provide a strategy for inducing precise, and localized injuries using near-infrared femtosecond laser pulses.
Similar content being viewed by others


Introduction
Light microscopy has revolutionized our ability to observe and understand biological processes. However, does the use of intense light irreversibly perturb cellular processes? What is the most accurate definition of biological damage caused by our observation? Does the instantaneous death of irradiated cells, as noted by1, constitute damage? Or should any reversible cellular impairment in a living organism already be classified as damage2? What if the illumination leads to cell apoptosis by slowing down vital metabolic processes by degrading intracellular organic compounds3?
Life on Earth depends on light and is adapted to the solar flux of less than 1.4 kW/m2. It has been argued that this value should be used as a safe irradiance reference when observing dynamic biological processes4. However, the sun is a continuous source of incoherent radiation with a non-uniform multi-octave spectral coverage. While the organisms are adopted to harvest the energy of frequencies with the higher spectral density of the sun for their development through photosynthesis, they stayed ignorant of other lower spectral density components present in the solar spectrum5. Furthermore, the photodamage effect of continuous light on biological specimens differs from that of pulsed lasers at similar photon flux6,7,8. Therefore, it is crucial to carefully consider these factors when light microscopy is used for imaging and in medical applications. In the past few decades, two significant types of microscopy have gained widespread usage: fluorescence microscopy, which utilizes the emission of linearly or non-linearly excited fluorophores to capture microscopic images of the sample9,10,11,12,13,14; and label-free microscopy, which relies on the emission of the molecular composition of the sample caused by an induced nonlinear polarization at high laser peak intensities15,16,17,18,19,20,21,22,23. Despite the distinct imaging methodologies utilized in these two categories, they share a common mechanism of photodamage.
When a pulsed laser interacts with tissue, the molecules in the sample get excited to a higher energy state through single-photon or multiphoton absorption, depending on the illumination peak intensity and optical frequency. The molecules can then return to the ground state either non-radiatively, leading to thermal damage, or via de-excitation processes. De-excitation involves dissociation or changes in the redox state of the absorbing molecule, energy transfer to a dioxygen molecule, or formation of reactive oxygen species (ROS), resulting in localized chemical perturbations and cell death24,25,26,27. At extremely high peak intensities, ionization of molecules and formation of a low-density plasma takes place, leading to photochemical damage or ablation and production of supersonic shock waves, which likewise lead to tissue damage28,29,30,31. In both of these categories of microscopy, the interaction between the laser pulses and tissue involves absorbers, predominantly consisting of water molecules32 or other tissue components depending on the excitation wavelength33,34,35,36,37 in label-free microscopy, or fluorescence labels in fluorescence microscopy38,39. While ROS generation significantly impacts cell damage in fluorescence microscopy, its contribution in label-free microscopy is negligible. Therefore, there has been significant attention on the use of different label-free microscopy techniques in the recent years12,16,40,41,42,43. Moreover, due to its large penetration depth and image contrast, label-free microscopy has become a crucial tool for deep-tissue and long-range imaging. The complementary information provided by both categories of microscopy has led to the development of more advanced techniques, such as multimodal nonlinear microscopy44,45,46,47.
Over the last decades, a significant body of research has been conducted on the effects of short laser pulses on thin in vitro and ex vivo samples in nonlinear fluorescence microscopy1,19,48,49,50,51. Despite these numerous investigations, a comprehensive study on how laser irradiation affects cell viability in a long-range imaging setting in vivo, specifically for label-free and multimodal nonlinear microscopy, has not been fully explored. This study leverages the vertebrate species zebrafish (Danio rerio) to delve into the mechanisms of photodamage in tissue at a cellular level triggered by femtosecond excitation pulses. Our objectives revolve around elucidating several key questions: How do the dynamics of different photodamage mechanisms unfold across a spectrum of femtosecond pulses, particularly at near-infrared (NIR)? What are the dynamics behind the photodamage at high peak intensities at NIR, and does the repetition rate of the laser pulses influence the driving mechanism? Is it feasible to reach plasma formation without inducing gradual damage to the sample? What is the role of thermal effects in photodamage at high peak intensities? To what extent does the ROS generation due to fluorescence proteins cause acceleration in photodamage? How does the damage threshold scale relative to the numerical aperture of focusing optics?
Results
Evaluating in vivo phototoxicity in tissues is considerably more intricate than in cultured cells, where probes for ROS production or observation of cell death through carbonization are common. In this study, we used zebrafish larvae at three days post-fertilization (dpf) to investigate interactions between femtosecond pulses and tissues in a living organism. We employed two complementary criteria to assess photodamage at the level of the spinal cord and surrounding trunk tissue. Short-term damage was evaluated by monitoring loss of tissue integrity at the whole tissue level and by assessing specific cell types (neurons, glial cells, keratinocytes), which were monitored through fluorescence labels in specific transgenic zebrafish lines. Long-term damage was assessed by observing axon regeneration over a period of two days, histological stains to detect cell death, and injury-associated recruitment of reactive fibroblasts and immune cells (macrophages) within 24 hours post-irradiation (hpi)52,53.
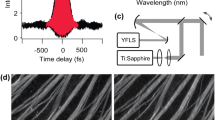
Figure 1a and b present a visual summary of the experiments conducted to investigate the damage mechanism upon the interaction of femtosecond laser pulses with the central nervous system (CNS) of a zebrafish larva. To generate the required femtosecond laser pulses at various wavelengths for this study, the output of a ytterbium amplifier delivering 250 fs pulses at 1030 nm was up-converted to 515 nm and 343 nm through second-harmonic and third-harmonic generation, respectively. A Pockels cell was utilized for pulse picking and reducing the laser pulse train’s repetition rate from 2 MHz to 127 kHz, enabling the differentiation of thermal damage contributions across various irradiation regimes. The generated femtosecond laser pulses were focused on the spinal cord using a reflective microscope objective. An imaging system was developed to monitor and adjust the focus position across different sections of the zebrafish. To monitor tissue damage following the different irradiation paradigms, we employed different transgenic fluorescence reporter zebrafish lines to examine the photodamage of the skin and spinal cord.

a Zebrafish larvae were used as a model organism to investigate the in vivo interaction of femtosecond pulses with tissues. Precisely targeted femtosecond laser pulses were focused on the central nervous system (CNS) under various irradiation settings. The cellular dynamics resulting from this pulse-tissue interaction were observed over time, focusing on neural cells and gauging the responsiveness of macrophages and fibroblasts. b Illumination setup for studying the interaction of femtosecond pulses at various wavelengths with the zebrafish CNS. HWP Half Wave Plate, TFP Thin Film Polarizer, SH-BS Second Harmonic Beam Splitter, TH-BS Third Harmonic Beam Splitter, BBO Beta Barium Borate crystal; BS Beam Splitter.
Two measurements were conducted to differentiate photodamage from linear processes, such as one-photon absorption and thermal damage (which should scale linearly with dwell time and photon flux), from nonlinear processes triggered by laser peak intensity. In the first set of measurements, samples were exposed to varying peak intensities while maintaining fixed dwell times. In the second series, laser pulses with varying dwell times and tissue at constant pulse energy were examined. Here, the peak intensity threshold was kept below the average damage intensity threshold measured in the previous experiment. Additionally, these measurements were carried out at two distinct repetition rates to discern the underlying photodamage processes further.
In the first series of measurements, a total number of 180 zebrafish larvae were individually irradiated for 30 s at various peak intensities, repetition rates, and irradiation wavelengths. Glial cells and neurons of the spinal cord were labeled with the fluorescence proteins GFP and mKate2, respectively (assessed in her4.3:GFP-F; elavl3:rasmKate2 double transgenic animals). Figure 2a displays the spinal cord damage probability versus peak intensity, while Fig. 2b presents the average damage intensity threshold for different parameters. Our data indicate the nonlinear scaling of the damage threshold versus irradiation wavelength. Furthermore, irradiation at a lower repetition rate across all wavelengths leads to a higher damage threshold than irradiation at higher repetition rates within the same dwell time. It has been observed that the damage behavior for the 1030 nm irradiation has a different qualitative behavior compared to 515 nm and 343 nm. The lesion’s dimension expands as the peak intensity increases for the two shorter irradiation wavelengths. However, damage occurs abruptly for the 1030 nm wavelength, immediately generating visible mechanical shockwaves. Figure 2c shows the corresponding average power threshold for damage at various wavelengths and repetition rates. Figure 2d summarizes the photons per area related to two irradiation regimes (see Supplementary Fig. 1 for more information).

a The damage probability of the zebrafish spinal cord versus peak intensity of 250 fs pulses at various irradiation wavelengths and laser repetition rates. Irradiation at 343 nm, 515 nm, and 1030 nm are represented by blue, green, and red curves, respectively. The circle indicates irradiation at 127 kHz repetition rates, while the triangle shows irradiation at 2 MHz repetition rates. b The average values of the peak intensity threshold at various wavelengths and laser repetition rates. c The average power damage threshold at various wavelengths and laser repetition rates. d The comparison of the average photons per area threshold for different parameters. Dwell time was fixed to 30 s. e The damage of the spinal cord was observed when the samples were irradiated at 343 nm and 515 nm below the peak intensity damage threshold and at longer dwell times of 30 s and 60 s respectively. For below-threshold irradiation at 1030 nm, no damage was observed by increasing the irradiation beyond 900 s. The thickness of the curves for 343 nm, and 515 nm illustrates the severity of the damage qualitatively. f The average values of the peak intensity threshold at 1030 nm and various laser repetition rates for wild-type (non-labeled) zebrafish and zebrafish in which neurons were labeled by either mKate2 (assessed in elavl3:rasmKate2 transgenic animals) or GFP (assessed in elavl3:GFP-F transgenic animals). b–d, f The error bars show standard deviation.
The dwell time in the second series of measurements increased while the peak intensity was below the damage threshold. Our study on photon flux revealed that, for irradiation at 343 nm and 515 nm, the lesion expands with increased dwell time, showing gradual damage. However, the response is entirely different for 1030 nm irradiation. No damage was observed by increasing the dwell time at a peak intensity below the damage threshold, as judged by loss of fluorescence signal or compromised tissue integrity. Figure 2e shows photon flux versus dwell time for three different wavelengths. At 343 nm, photo damage is visible following 30 s of irradiation, signifying that linear mechanisms predominantly drive tissue damage at this wavelength due to absorption by DNA54,55,56 (see Supplementary Fig. 2). In contrast, at 515 nm irradiation wavelength, the damage was observed at one order of magnitude higher photon flux threshold, hinting at the involvement of nonlinear processes. At 1030 nm, it is possible to extend the irradiation duration to several minutes.
Further measurements were performed on animals subjected to irradiation at 1030 nm as they showed distinct behaviors compared to irradiation at 343 nm and 515 nm. The mKate2 fluorophore has a high absorption cross-section for two-photon absorption at 1030 nm, while the nonlinear absorption cross-section of the GFP fluorophore at 1030 nm is weaker57,58. Therefore, we used transgenic lines in which all neurons were either labeled with GFP (assessed in elavl3:GFP-F transgenic zebrafish) or mKate2 (assessed in elavl3:rasmKate2 transgenic zebrafish) to isolate the additional role of the fluorescent labels in photodamage. Figure 2f compares the averaged peak intensity threshold for the two scenarios in addition to the wild-type fish. The damage threshold of samples labeled by GFP is higher than mKate2 labeled samples for irradiation at 127 kHz, indicating the role of the two-photon absorption pathway and ROS generation of mKate2. However, at 2 MHz repetition rates, the difference between the damage threshold at both labels becomes marginal, indicating that at these regimes, the role of the plasma-mediated chemical effects and nonlinear photochemistry becomes prominent (see Supplementary Fig. 1).
For the samples deemed healthy upon irradiation of intense femtosecond pulses at 1030 nm, we examined potential damage to the skin using krtt1c19e:GFP transgenic zebrafish with fluorescently labeled basal keratinocytes of the bilayered epidermis. Figure 3 depicts the brightfield, GFP-labeled basal keratinocytes and mKate2-labeled neurons (visualized in elavl3:rasmKate2 transgenic animals) following irradiation above the damage intensity threshold at 1030 nm. After irradiation, we observed a sharply declined fluorescence of the neuronal marker but not the epidermal marker, indicating that tissue damage was largely limited to the focal area. Similar results are observed for irradiation at the other two wavelengths, where no apparent damage was detected on the epidermis (see Supplementary Fig. 3 for more information).

Trunk of an elavl3:rasmKate2;krtt1c19e:EGFP double transgenic zebrafish larva with the fluorescently labeled spinal cord (neurons; mKate2) and epidermis (basal keratinocytes; GFP) after 30 s irradiation with 1030 nm pulses above the damage threshold. Arrowheads indicate the focus of irradiation. While damage is visible at the level of the spinal cord, the superficially located epidermis remains intact. Images shown are brightfield (BF) recordings and orthogonal projections of the confocal image (xy or yz view). Scale bars: 50 μm.
To exclude the possibility that the reduction of fluorescence signal in the spinal cord is a consequence of bleaching of the fluorophore and not due to tissue damage, we reassessed the irradiated animals at 24 hpi and 48 hpi. This showed an imperfect fluorescence pattern of the white matter tracts, supporting the regrowth of irradiation-damaged axonal fibers rather than recovery of fluorescence, which occurs in larval zebrafish within 48 hours after mechanical transection as previously reported59 (Fig. 4a, b). To provide further evidence for tissue damage or its absence in the respective conditions, we assessed cellular apoptosis by TUNEL assay and monitored the recruitment of immune cells and fibroblasts (Fig. 4c–e) to the irradiated site52,53. Consistent with tissue damage, we observed the emergence of TUNEL+ cells as well as the recruitment of reactive fibroblasts (visualized with pdgfrb:GFP transgenic animals) and macrophages (visualized with mpeg1:mCherry transgenic animals) in samples irradiated at 1030 nm for 30 s above the damage threshold. In contrast, for samples irradiated just below the damage threshold, we did not detect TUNEL signal, fibroblasts, or macrophages accumulating at the irradiation site. This supports our observation of the highly nonlinear scaling of the damage at NIR and the absence of any gradual damage at this wavelength.

a The timeline of experiments. Timepoints of irradiation and analysis are indicated. b Spinal cord lesion induced by 30 mW pulses at 2 MHz (arrowhead in overview). The same elavl3:rasmKate2 transgenic animal is shown at 2 hpi (\({b}^{{\prime} }\)) and 48 hpi (b″). Note regrowth of white matter tracts at 48 hpi (arrowhead in b). The neurons of the zebrafish spinal cord are genetically labeled with mKate2 fluorescent protein. c Cell death (arrowheads), as detected by the TUNEL assay, is observed in the irradiation site at above-damage threshold peak intensities but not below the damage threshold intensities. d Recruitment of macrophages (arrowheads) to the irradiation site is observed at 16 mW at 2 MHz and 1.6 mW at 127 kHz; however, not at below damage threshold irradiations at the average powers of 5.8 mW at 2 MHz and 0.7 mW at 127 kHz (visualized with mpeg1:Cherry transgenic animals). e Irradiation at above damage threshold intensity leads to the recruitment of reactive fibroblasts in the irradiation site (arrowhead; visualized with pdgfrb:GFP transgenic animals). b-e Scale bars: 50 μm.
Discussion
The photodamage peak intensity threshold for CNS tissue of zebrafish when irradiated by femtosecond pulses at 1030 nm is higher compared to irradiation at its harmonics. It was observed that when samples are subjected to femtosecond pulses at 343 nm and 515 nm, the extent of the photodamage is gradual and scales proportional to irradiation photon flux. A lower ablation threshold for irradiation at shorter wavelengths is expected due to the more efficient photoionization28. However, in our study, the gradual linear damage at shorter wavelengths, owing to linear absorption in tissue, prevents the irradiated samples from reaching the cavitation regime. In contrast, for irradiation at 1030 nm in both the absence and presence of a two-photon absorption path of fluorescence proteins, the damage appears abruptly. The cavitation was observed at an average fluence of 1.2 J/cm2 at 2 MHz operation and an average fluence of 2 J/cm2 for 127 kHz operation, which is consistent with previous observations on ablation threshold in porcine corneal stroma28. The severity of cavitation escalated when the larvae were irradiated at 2 MHz compared to 127 kHz. No luminous plasma was observed at these regimes, resembling the optical breakdown in aqueous media when irradiated with femtosecond pulses60,61,62,63.
Water can be treated as an amorphous semiconductor with a bandgap energy of 6.5 eV. For a transition from the molecular 1b1 orbital into an excitation band, the energy of six photons at 1030 nm is required64,65. In our study, we reach the critical peak power of 94 GW at 127 kHz and 57 GW at 2 MHz for irradiation at 1030 nm, which are clearly beyond the 6.2 MW of water critical power, considering the nonlinear index of 1.9 × 10−20 m2/W66. The energy thresholds for optical breakdown at 50% breakdown probability for the targeted CNS cells at z ≈ 75 μm are 7.8 nJ at 127 kHz and 5 nJ at 2 MHz extracted from Fig. 2a. The sharpness of the nonlinearity of the damage, which is defined by the ratio of the threshold energy to the energy difference between a 10% and a 90% breakdown probability, is calculated to be 2.7 at 127 kHz and 1.8 at 2 MHz67, emphasizing on the steeper break down at lower repetition rates.
Assuming pure water and according to simulated plasma dynamics and electron energy spectrum using the multi-rate-equation model68, a 1030 nm, 127 kHz, 250 fs laser pulse at 8.1 TW/cm2 peak intensity produces an electron density of the order of 1021 cm−3, which is similar to the free electron density at the optical breakdown threshold of water32,69,70. This regime of nonlinearity leads to 103 K temperature rise by a single pulse at 1030 nm, where only heating by nonlinear absorption is considered68. For femtosecond laser pulses, heat accumulates if the heat diffusion time is longer than the inter-pulse interval. For samples irradiated at 4.9 TW/cm2 at 2 MHz repetition rate, the generated free electron density is reaching the low plasma density. The inter-pulse interval at this repetition rate is 500 ns, which is considerably longer than the heat dissipation time. For focusing at a numerical aperture lower than 0.9, the steady-state temperature distribution is broader than the generated heat due to the single pulse distribution. However, at repetition rates lower than 2 MHz, temperature accumulation and broadening of the temperature distribution are negligible69 and can not cause the observed decrease in peak intensity threshold for optical breakdown and cell death at 2 MHz repetition rates71. The lowered cavitation threshold could be due to the low-density plasma generation, which could trigger plasma-mediated chemical effects and nonlinear photochemistry72. At 1030 nm, four photons are required for DNA excitation with a low cross-section. However, in the presence of free electrons, many different DNA damage mechanisms can be initiated by capturing the electron via DNA base73,74. Moreover, free-electron-mediated effects become possible once seed electrons in the conduction band of water are created, scaling with the irradiation wavelength32. Therefore, it can be concluded that the generated low-density plasma in samples irradiated with 1030 nm pulses at 2 MHz leads to the creation of ROS by the dissociation of water molecules25,74,75 and DNA damage, lowering the ablation threshold at higher repetition rates69,74,76.
The radius of the damage size for the CNS of the samples irradiated at 1030 nm is 40 μm for irradiation at 2 MHz and 13 μm for irradiation at 127 kHz at z ≈ 75 μm. The irradiation peak intensity at 127 kHz is above the required threshold for bubble formation by a single laser pulse69. Therefore, no collateral damage was observed, and the damage was confined to the region around the laser-induced plasma due to mechanical forces arising from bubble formation and shockwave emission77,78,79. For samples irradiated at 2 MHz repetition rates, collateral damage is observed due to bubble formation based on plasma-mediated accumulative chemical disintegration of biomolecules. Free electrons have picoseconds lifetime and nanometers traveling range and cannot have a seminal contribution to the size of the observed damage site80,81. However, the generated damaging “agents" at the focal spot, such as ROS, have a high diffusivity and can travel over 25 μm within less than a second75,82. Moreover, due to our relatively low numerical aperture, the size of the damage could be affected by the self-focusing and aberration of the irradiation beam83, while its expansion is restricted by the surrounding matrix84. Therefore, at the peak intensities reported in this work, the generated free electron also contributes to the damage size before reaching the beam waist85,86.
Conclusion
For the successful implementation of cutting-edge nonlinear microscopy techniques for deep-tissue, in vivo imaging and designing their use for medical applications, a comprehensive understanding of the non-invasive, optimal operational parameters and constraints of the imaging system is essential. This entails fine-tuning variables like the laser pulses’ average power, peak intensity, wavelength, pulse duration, and other factors such as imaging dwell time. In addition, a rigorous assessment of thermal or photon-based damage mechanisms across various irradiation wavelengths is required. Equally important is the distinction between the impact of peak intensity, average power, and dwell time in nonlinear microscopy, as the nonlinear signal scales with peak intensity. At the same time, the signal-to-noise ratio is proportional to the average power of the laser pulse and dwell time, while an in-depth assessment of biological tissue requires longer penetration of light and employment of the focusing optics with a long working distance. Though interconnected, these parameters may initiate various photodamage mechanisms, as shown in this study.
Water, a major component of biological systems, exhibits five main resonances in the near-infrared at 0.76 μm, 0.97 μm, 1.19 μm, 1.45 μm, and 1.94 μm, with an absorption cross section increasing by order of magnitude when scaling between the adjunct resonances87. With recent advancements in ytterbium lasers operating at 1030 nm88,89,90, alongside novel detection schemes91, the second near-infrared window of the tissue has become an increasingly fitting candidate for label-free nonlinear imaging92,93.
When conducting in vivo deep-tissue nonlinear microscopy, it is crucial to evaluate various photodamage mechanisms collectively since they operate in unison at differing intensities upon the interaction of femtosecond pulses with dense tissue. Our study of the long-range focusing identifies two different cavitation regimes. The damage is confined and well-controlled at low repetition rates due to plasma-based ablation and sudden local temperature rise. At high repetition rates, the damage becomes collateral due to plasma-mediated photochemistry and is not influenced by the thermal accumulation of consecutive laser pulses. Our findings agree well with the results of a theoretical model developed by Liang et al.68. Fluorescence labels are employed to visualize photodamage occurring in the tissue and study their influence on the photodamage. While the mKate2 fluorphore has a two-photon absorption pathway when irradiated with femtosecond pulses at 1030 nm, GFP does not have a strong nonlinear absorption path. The values shown in Fig. 2f demonstrate a negligible influence of the ROS activated by two-photon absorption in mKate2 on photodamage at a low repetition rate of 127 kHz. Furthermore, our study reveals that at peak intensities exceeding the CNS damage threshold, no discernible damage is evident at the epidermal level. This behavior can be associated with the four times lower peak intensity on the skin than the spinal cord. Moreover, we demonstrate that it is possible to extend the dwell time; therefore, photon flux, extensively and noninvasively for irradiation at 1030 nm, as long as the peak intensity is below the low-plasma density threshold. In this regime, the associated photon flux to the peak intensity damage threshold can be exceeded by increasing the dwell time without observation of the damage in the sample. This behavior is not observed for irradiation at the harmonics at 515 nm and 343 nm, emphasizing the lower scaling of the free electron density versus peak intensity at shorter irradiation wavelengths68.
These findings significantly contribute to advancements in innovative microscopy techniques, like multimodal microscopy94 or femtosecond fieldoscopy, in which high spatial resolution, label-free images with attosecond temporal resolution can be captured91,95,96,97. We not only lay down guidelines for emerging nonlinear imaging techniques but also suggest a strategy for inducing highly precise, localized injuries, which is of interest to various research fields such as neuroregeneration52,98.
Methods
Irradiation and live imaging
An Yb:KGW amplifier (CARBIDE from Light Conversion) was used to irradiate zebrafish. 20 μJ (on the microscope: from 3.75 pJ up to 14.2 nJ) of the output of the laser was frequency doubled in a 1.5 mm thick BBO crystal with 33.7∘ type I phase matching angle. A second BBO crystal was used for frequency tripling of the laser via cascaded second-order effects by sum frequency generation between 1030 nm and 515 nm pulses99. The crystal was 1.5 mm thick with a phase-matching angle of 62.8∘, type II. A high reflective mirror (Eksma, 042-515) was used as a beam splitter to separate the fundamental from the second-harmonic signal. Afterward, the second harmonic beam was separated from the third harmonic beam by a harmonic separator (Eksma, 0423535pht). The samples were irradiated at 1030 nm, 515 nm and 343 nm. A reflective objective with a numerical aperture 0.5, magnification of 40, and a focal length of 5 mm (Thorlabs) was used to focus the beam on the zebrafish spinal cord. The beam size of the three beams was scaled to 5.1 mm to fill the 5.1 mm aperture of a reflective objective. The size of the beams at the focus was calculated using Abbe’s resolution limit, resulting in beam sizes of 0.2 μm, 0.3 μm, and 0.6 μm for respective irradiation wavelengths. The irradiation peak intensity was calculated based on these values. As the Rayleigh length of the focused beam is shorter than the transversal length of the samples, a CCD camera is used to ensure the light is focused on the spinal cord. To calculate the peak intensity of the laser pulses on the skin of the zebrafish, it was assumed that the distance from the spinal cord to the skin of 3 dpf zebrafish is 100 μm. This information was used to calculate the beam size and the corresponding peak intensity of the irradiation beams on the skin. Live examination and imaging before and after irradiation were done using the following commercial microscope setups: 1) Plan-Apochromat 10x/0.45 M27 objective or Plan-Apochromat 20x/0.8 objective on a Zeiss LSM 980 confocal microscope. The detector ranges used were 490-588 nm for EGFP, 597-695 nm for mKate2 and mCherry, and 641-695 nm for Alexa fluor 647. We used a main beam splitter (MBS) 488/594 for the detection of EGFP, mKate2, and mCherry, and an MBS 488/561/639 for the detection of Alexa fluor 647. 2) Plan-Apochromat 1.0x or 2.0x objective on a Leica M205 FCA stereo microscope equipped with emission filters ET525/50 nm (GFP) and ET630/75 nm (mKate2, mCherry), and a Leica DMC6200 C color camera. For repetitive imaging, larvae were released from agarose after imaging, transferred to embryo medium at 28.5 ∘C, and re-mounted the following day. The emission peak for mKate2 is at 633 nm, for mCherry at 610 nm, for EGFP at 507 nm, and for Alexa fluor at 671 nm.
Zebrafish husbandry and transgenic lines
All zebrafish lines were maintained and reared in a research fish facility (Tecniplast, Italy) under a 14/10 h light/dark cycle according to FELASA recommendations100,101. All experimental procedures were done on larval zebrafish aged up to 120 hours post-fertilization (hpf), derived from voluntarily mating adult zebrafish (permit: I/39/FNC of the Amt für Veterinärwesen und gesundheitlichen Verbraucherschutz Stadt Erlangen). Procedures on zebrafish larvae up to 120 hpf is not regulated as animal experiments by the European Commission Directive 2010/63/EU. For all irradiation experiments, we used 72 hpf zebrafish of either sex. For experimental analyses, we used the following available zebrafish lines: wild-type AB, BAC(pdgfrb:Gal4ff)ncv24102, 5xUAS:EGFPzf82103, mpeg1:mCherrygl23104, elavl3:rasmKate2mps152, her4.3:GFP-Fmps9105, krtt1c19e:EGFPsq1744106, and elavl3:GFP-Fmps10105. The combination of BAC(pdgfrb:Gal4ff) and 5xUAS:EGFP transgenic lines were abbreviated as pdgfrb:GFP. Embryos were treated with 0.00375 % 1-phenyl-2-thiourea (Sigma-Aldrich #P7629) beginning at 24 hpf to prevent pigmentation.
Zebrafish mounting
Zebrafish larvae were anesthetized in E3 medium containing 0.02% Ethyl 3-aminobenzoate methanesulfonate (MS-222; PharmaQ #Tricaine PharmaQ) and mounted in a lateral position in 1% low melting point agarose (Ultra-PureTM Low Melting Point, Invitrogen Cat#16520) between two microscope cover glasses. Larvae were covered with 0.01% MS-222-containing E3 medium to keep preparations from drying out.
Whole-mount TUNEL labelling
Terminally anesthetized larvae were fixed in 4% PFA (Thermo Fisher Scientific Cat#28908) in PBS overnight at 4∘C. After removing the head and tail using micro scissors, larvae were permeabilized by subsequent incubation in acetone and Proteinase K (Invitrogen Cat#25530-049) as described elsewhere107. Samples were re-fixed in 4% PFA in PBS and Click-iT TUNEL Alexa Fluor 647 Imaging Assay (Thermo Fisher Cat#C10247) was performed according to the manufacturer’s protocol to label apoptotic cells. Briefly, samples were equilibrated in TdT reaction buffer for 30 min at room temperature, followed by incubation in TdT reaction cocktail overnight at room temperature. Samples were washed in PBTx and mounted in 75% glycerol in PBS. Imaging was done using a Plan-Apochromat 20x/0.8 objective on a Zeiss LSM 980 confocal microscope.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Talone, B. et al. Phototoxicity induced in living hela cells by focused femtosecond laser pulses: a data-driven approach. Biomed. Opt. Express 12, 7886–7905 (2021).
Zhang, X., Dorlhiac, G., Landry, M. P. & Streets, A. Phototoxic effects of nonlinear optical microscopy on cell cycle, oxidative states, and gene expression. Sci. Rep. 12, 18796 (2022).
Wagner, M. et al. Light dose is a limiting factor to maintain cell viability in fluorescence microscopy and single molecule detection. Int. J. Mol. Sci. 11, 956–966 (2010).
Stelzer, E. H. Light-sheet fluorescence microscopy for quantitative biology. Nat. methods 12, 23–26 (2015).
Zhen, S., van Iersel, M. W. & Bugbee, B. Photosynthesis in sun and shade: the surprising importance of far-red photons. N. Phytologist 236, 538–546 (2022).
König, K., Becker, T., Fischer, P., Riemann, I. & Halbhuber, K.-J. Pulse-length dependence of cellular response to intense near-infrared laser pulses in multiphoton microscopes. Opt. Lett. 24, 113–115 (1999).
Koester, H. J., Baur, D., Uhl, R. & Hell, S. W. Ca2+ fluorescence imaging with pico- and femtosecond two-photon excitation: Signal and photodamage. Biophys. J. 77, 2226–2236 (1999).
König, K. Laser tweezers and multiphoton microscopes in life sciences. Histochem. Cell Biol. 114, 79–92 (2000).
Denk, W., Strickler, J. H. & Webb, W. W. Two-photon laser scanning fluorescence microscopy. Science 248, 73–76 (1990).
Kerr, J. N. & Denk, W. Imaging in vivo: watching the brain in action. Nat. Rev. Neurosci. 9, 195–205 (2008).
Horton, N. G. et al. In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nat. photonics 7, 205–209 (2013).
Andresen, V. et al. Infrared multiphoton microscopy: subcellular-resolved deep tissue imaging. Curr. Opin. Biotechnol. 20, 54–62 (2009).
Velasco, M. G. M. et al. 3d super-resolution deep-tissue imaging in living mice. Optica 8, 442–450 (2021).
Calovi, S., Soria, F. N. & Tønnesen, J. Super-resolution sted microscopy in live brain tissue. Neurobiol. Dis. 156, 105420 (2021).
Helmchen, F. & Denk, W. Deep tissue two-photon microscopy. Nat. methods 2, 932–940 (2005).
Weigelin, B., Bakker, G.-J. & Friedl, P. Third harmonic generation microscopy of cells and tissue organization. J. Cell Sci. 129, 245–255 (2016).
Squier, J. A., Müller, M., Brakenhoff, G. & Wilson, K. R. Third harmonic generation microscopy. Opt. express 3, 315–324 (1998).
Witte, S. et al. Label-free live brain imaging and targeted patching with third-harmonic generation microscopy. Proc. Natl Acad. Sci. 108, 5970–5975 (2011).
Debarre, D., Olivier, N., Supatto, W. & Beaurepaire, E. Mitigating phototoxicity during multiphoton microscopy of live drosophila embryos in the 1.0–1.2 μ m wavelength range. PLoS One 9, e104250 (2014).
Ji, M. et al. Rapid, label-free detection of brain tumors with stimulated raman scattering microscopy. Sci. Transl. Med. 5, 201ra119 (2013).
Müller, M., Squier, J., Wilson, K. & Brakenhoff, G. 3d microscopy of transparent objects using third-harmonic generation. J. Microsc. 191, 266–274 (1998).
Barad, Y., Eisenberg, H., Horowitz, M. & Silberberg, Y. Nonlinear scanning laser microscopy by third harmonic generation. Appl. Phys. Lett. 70, 922–924 (1997).
Cheng, J.-X. & Xie, X. S. Coherent anti-stokes raman scattering microscopy: Instrumentation, theory, and applications. J. Phys. Chem. B 108, 827–840 (2004).
Tosheva, K. L., Yuan, Y., Pereira, P. M., Culley, S. & Henriques, R. Between life and death: strategies to reduce phototoxicity in super-resolution microscopy. J. Phys. D: Appl. Phys. 53, 163001 (2020).
Tirlapur, U. K., König, K., Peuckert, C., Krieg, R. & Halbhuber, K.-J. Femtosecond near-infrared laser pulses elicit generation of reactive oxygen species in mammalian cells leading to apoptosis-like death. Exp. cell Res. 263, 88–97 (2001).
Khan, I., Tang, E. & Arany, P. Molecular pathway of near-infrared laser phototoxicity involves atf-4 orchestrated er stress. Sci. Rep. 5, 1–14 (2015).
Dillenburg, C. S., Almeida, L. O., Martins, M. D., Squarize, C. H. & Castilho, R. M. Laser phototherapy triggers the production of reactive oxygen species in oral epithelial cells without inducing dna damage. J. Biomed. Opt. 19, 048002–048002 (2014).
Olivié, G. et al. Wavelength dependence of femtosecond laser ablation threshold of corneal stroma. Opt. express 16, 4121–4129 (2008).
Sarpe-Tudoran, C., Assion, A., Wollenhaupt, M., Winter, M. & Baumert, T. Plasma dynamics of water breakdown at a water surface induced by femtosecond laser pulses. Appl. Phys. Lett. 88, 261109 (2006).
Astafiev, A. et al. Femtosecond laser microsurgery of mouse oocytes: Formation and dynamics of cavitation bubbles under the action of sharply focused laser radiation on various oocyte zones. Russian J. Phys. Chem. B 17, 148–158 (2023).
Vogel, A., Noack, J., Huettmann, G. & Paltauf, G. Femtosecond-laser-produced low-density plasmas in transparent biological media: a tool for the creation of chemical, thermal, and thermomechanical effects below the optical breakdown threshold. In Commercial and Biomedical Applications of Ultrafast and Free-Electron Lasers, vol.4633, 23–37 (SPIE, 2002).
Linz, N., Freidank, S., Liang, X.-X. & Vogel, A. Wavelength dependence of femtosecond laser-induced breakdown in water and implications for laser surgery. Phys. Rev. B 94, 024113 (2016).
Hemmer, E., Benayas, A., Légaré, F. & Vetrone, F. Exploiting the biological windows: current perspectives on fluorescent bioprobes emitting above 1000 nm. Nanoscale Horiz. 1, 168–184 (2016).
Xu, W., Wang, D. & Tang, B. Z. Nir-ii aiegens: a win–win integration towards bioapplications. Angew. Chem. Int. Ed. 60, 7476–7487 (2021).
Hong, G., Antaris, A. L. & Dai, H. Near-infrared fluorophores for biomedical imaging. Nat. Biomed. Eng. 1, 0010 (2017).
Smith, A. M., Mancini, M. C. & Nie, S. Second window for in vivo imaging. Nat. Nanotechnol. 4, 710–711 (2009).
Weissleder, R. A clearer vision for in vivo imaging. Nat. Biotechnol. 19, 316–317 (2001).
Laissue, P. P., Alghamdi, R. A., Tomancak, P., Reynaud, E. G. & Shroff, H. Assessing phototoxicity in live fluorescence imaging. Nat. methods 14, 657–661 (2017).
Wäldchen, S., Lehmann, J., Klein, T., Van De Linde, S. & Sauer, M. Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 5, 1–12 (2015).
Olivier, N. et al. Cell lineage reconstruction of early zebrafish embryos using label-free nonlinear microscopy. Science 329, 967–971 (2010).
Ferrer Ortas, J. et al. Label-free imaging of red blood cells and oxygenation with color third-order sum-frequency generation microscopy. Light.: Sci. Appl. 12, 29 (2023).
Sun, C.-K. et al. Higher harmonic generation microscopy for developmental biology. J. Struct. Biol. 147, 19–30 (2004).
Oron, D. et al. Depth-resolved structural imaging by third-harmonic generation microscopy. J. Struct. Biol. 147, 3–11 (2004).
Wu, W., Liu, Q., Brandt, C. & Tang, S. Dual-wavelength multimodal multiphoton microscope with sma-based depth scanning. Biomed. Opt. Express 13, 2754–2771 (2022).
You, S. et al. Intravital imaging by simultaneous label-free autofluorescence-multiharmonic microscopy. Nat. Commun. 9, 2125 (2018).
Clark, M. G., Gonzalez, G. A. & Zhang, C. Pulse-picking multimodal nonlinear optical microscopy. Anal. Chem. 94, 15405–15414 (2022).
Matsui, T. et al. Label-free multiphoton excitation imaging as a promising diagnostic tool for breast cancer. Cancer Sci. 113, 2916 (2022).
Chen, I.-H., Chu, S.-W., Sun, C.-K., Cheng, P.-C. & Lin, B.-L. Wavelength dependent damage in biological multi-photon confocal microscopy: a micro-spectroscopic comparison between femtosecond ti: sapphire and cr: forsterite laser sources. Optical Quantum Electron. 34, 1251–1266 (2002).
Magidson, V. & Khodjakov, A. Chapter 23 - circumventing photodamage in live-cell microscopy. In Sluder, G. & Wolf, D. E. (eds.) Digital Microscopy, vol. 114 of Methods in Cell Biology, 545–560 (Academic Press, 2013). https://www.sciencedirect.com/science/article/pii/B9780124077614000233.
Fu, Y., Wang, H., Shi, R. & Cheng, J.-X. Characterization of photodamage in coherent anti-stokes raman scattering microscopy. Opt. Express 14, 3942–3951 (2006).
Maioli, V. et al. Fast in vivo multiphoton light-sheet microscopy with optimal pulse frequency. Biomed. Opt. Express 11, 6012–6026 (2020).
Tsata, V. et al. A switch in pdgfrb+ cell-derived ecm composition prevents inhibitory scarring and promotes axon regeneration in the zebrafish spinal cord. Developmental cell 56, 509–524 (2021).
Tsarouchas, T.et al. Dynamic control of proinflammatory cytokines il-1beta and tnf-alpha by macrophages in zebrafish spinal cord regeneration. nat commun. 2018; 9 (1): 4670. Tech. Rep., Epub 2018/11/09. https://doi.org/10.1038/s41467-018-07036-wPMID:30405119.
Mohanty, S., Rapp, A., Monajembashi, S., Gupta, P. & Greulich, K. Comet assay measurements of dna damage in cells by laser microbeams and trapping beams with wavelengths spanning a range of 308 nm to 1064 nm. Radiat. Res. 157, 378–385 (2002).
He, H., Chan, K. T., Kong, S. K. & Lee, R. K. Y. Mechanism of oxidative stress generation in cells by localized near-infrared femtosecond laser excitation. Appl. Phys. Lett. 95, 233702 (2009).
Rapp, A. & Greulich, K. O. Why soft uv-a damages dna: An optical micromanipulation study. In Optical Trapping and Optical Micromanipulation X, vol. 8810, 162–169 (SPIE, 2013).
Lambert, T. J. Fpbase: a community-editable fluorescent protein database. Nat. methods 16, 277–278 (2019).
Drobizhev, M., Makarov, N. S., Tillo, S. E., Hughes, T. E. & Rebane, A. Two-photon absorption properties of fluorescent proteins. Nat. methods 8, 393–399 (2011).
Wehner, D. et al. Wnt signaling controls pro-regenerative collagen xii in functional spinal cord regeneration in zebrafish. Nat. Commun. 8, 126 (2017).
Venugopalan, V., Guerra III, A., Nahen, K. & Vogel, A. Role of laser-induced plasma formation in pulsed cellular microsurgery and micromanipulation. Phys. Rev. Lett. 88, 078103 (2002).
Hammer, D. X. et al. Experimental investigation of ultrashort pulse laser-induced breakdown thresholds in aqueous media. IEEE J. Quantum Electron. 32, 670–678 (1996).
Noack, J. & Vogel, A. Laser-induced plasma formation in water at nanosecond to femtosecond time scales: calculation of thresholds, absorption coefficients, and energy density. IEEE J. Quantum Electron. 35, 1156–1167 (1999).
Docchio, F., Sacchi, C. A. & Marshall, J. Experimental investigation of optical breakdown thresholds in ocular media under single pulse irradiation with different pulse durations. Lasers Ophthalmol. 1, 83–93 (1986).
Sacchi, C. Laser-induced electric breakdown in water. Josa b 8, 337–345 (1991).
Williams, F., Varma, S. & Hillenius, S. Liquid water as a lone-pair amorphous semiconductor. J. Chem. Phys. 64, 1549–1554 (1976).
Wilkes, Z. et al. Direct measurements of the nonlinear index of refraction of water at 815 and 407 nm using single-shot supercontinuum spectral interferometry. Appl. Phys. Lett. 94, 211102 (2009).
Vogel, A., Nahen, K., Theisen, D. & Noack, J. Plasma formation in water by picosecond and nanosecond nd:yag laser pulses. i. optical breakdown at threshold and superthreshold irradiance. IEEE J. Sel. Top. Quantum Electron. 2, 847–860 (1996).
Liang, X.-X., Zhang, Z. & Vogel, A. Multi-rate-equation modeling of the energy spectrum of laser-induced conduction band electrons in water. Opt. express 27, 4672–4693 (2019).
Vogel, A., Noack, J., Hüttman, G. & Paltauf, G. Mechanisms of femtosecond laser nanosurgery of cells and tissues. Appl. Phys. B 81, 1015–1047 (2005).
Liang, X.-X. & Vogel, A. Probing neuronal functions with precise and targeted laser ablation in the living cortex: comment. Optica 9, 868–871 (2022).
Simanovskii, D. et al. Cellular tolerance to pulsed hyperthermia. Phys. Rev. E 74, 011915 (2006).
Mao, S. et al. Dynamics of femtosecond laser interactions with dielectrics. Appl. Phys. A 79, 1695–1709 (2004).
Schmalz, M. et al. Dissection of dna damage and repair pathways in live cells by femtosecond laser microirradiation and free-electron modeling. Proc. Natl Acad. Sci. 120, e2220132120 (2023).
Boudaíffa, B., Cloutier, P., Hunting, D., Huels, M. A. & Sanche, L. Resonant formation of dna strand breaks by low-energy (3 to 20 ev) electrons. Science 287, 1658–1660 (2000).
Nikogosyan, D. N., Oraevsky, A. A. & Rupasov, V. I. Two-photon ionization and dissociation of liquid water by powerful laser uv radiation. Chem. Phys. 77, 131–143 (1983).
Huels, M. A., Boudaïffa, B., Cloutier, P., Hunting, D. & Sanche, L. Single, double, and multiple double strand breaks induced in dna by 3 - 100 ev electrons. J. Am. Chem. Soc. 125, 4467–4477 (2003).
Vogel, A., Linz, N., Freidank, S. & Paltauf, G. Femtosecond-laser-induced nanocavitation in water: implications for optical breakdown threshold and cell surgery. Phys. Rev. Lett. 100, 038102 (2008).
Vogel, A., Busch, S. & Parlitz, U. Shock wave emission and cavitation bubble generation by picosecond and nanosecond optical breakdown in water. J. Acoustical Soc. Am. 100, 148–165 (1996).
Vogel, A. et al. Energy balance of optical breakdown in water at nanosecond to femtosecond time scales. Appl. Phys. B: Lasers & Opt. 68, 271–280 (1999).
Meesungnoen, J., Jay-Gerin, J.-P., Filali-Mouhim, A. & Mankhetkorn, S. Low-energy electron penetration range in liquid water. Radiat. Res. 158, 657–660 (2002).
Kai, T., Yokoya, A., Ukai, M., Fujii, K. & Watanabe, R. Dynamic behavior of secondary electrons in liquid water at the earliest stage upon irradiation: implications for dna damage localization mechanism. J. Phys. Chem. A 120, 8228–8233 (2016).
van Stroe-Biezen, S., Everaerts, F., Janssen, L. & Tacken, R. Diffusion coefficients of oxygen, hydrogen peroxide and glucose in a hydrogel. Analytica Chim. Acta 273, 553–560 (1993).
Vogel, A. et al. Influence of optical aberrations on laser-induced plasma formation in water and their consequences for intraocular photodisruption. Appl. Opt. 38, 3636–3643 (1999).
Hutson, M. S. & Ma, X. Plasma and cavitation dynamics during pulsed laser microsurgery in vivo. Phys. Rev. Lett. 99, 158104 (2007).
Hammer, D. X. et al. Shielding properties of laser-induced breakdown in water for pulse durations from 5 ns to 125 fs. Appl. Opt. 36, 5630–5640 (1997).
Arnold, C., Heisterkamp, A., Ertmer, W. & Lubatschowski, H. Streak formation as side effect of optical breakdown during processing the bulk of transparent kerr media with ultra-short laser pulses. Appl. Phys. B 80, 247–253 (2005).
Curcio, J. A. & Petty, C. C. The near infrared absorption spectrum of liquid water. J. Opt. Soc. Am. 41, 302–304 (1951).
Fattahi, H. et al. Third-generation femtosecond technology. Optica 1, 45–63 (2014).
Wang, H., Alismail, A., Barbiero, G., Wendl, M. & Fattahi, H. Cross-polarized, multi-octave supercontinuum generation. Opt. Lett. 42, 2595–2598 (2017).
Buberl, T., Alismail, A., Wang, H., Karpowicz, N. & Fattahi, H. Self-compressed, spectral broadening of a yb:yag thin-disk amplifier. Opt. Express 24, 10286–10294 (2016).
Herbst, A. et al. Recent advances in petahertz electric field sampling. J. Phys. B: Atomic, Mol. Opt. Phys. 55, 172001 (2022).
Kenry, D. Y. & Liu, B. Recent advances of optical imaging in the second near-infrared window. Adv. Mater. 30, 1802394 (2018).
Golovynskyi, S. et al. Optical windows for head tissues in near-infrared and short-wave infrared regions: Approaching transcranial light applications. J. Biophotonics 11, e201800141 (2018).
Guesmi, K. et al. Dual-color deep-tissue three-photon microscopy with a multiband infrared laser. Light.: Sci. Appl. 7, 12 (2018).
Srivastava, A. et al. Near-petahertz fieldoscopy of liquid. arXiv preprint arXiv:2310.20512 (2023).
Fattahi, H. Method and apparatus for creating a microscopic sample image of a molecular vibrational response of a sample (2023). US Patent App. 18/024,120.
Scheffter, K.et al. Compressed sensing of field-resolved molecular fingerprints beyond the nyquist frequency. Ultrafast Sci. 4, 0062 (2024).
Matrone, G. et al. Laser-targeted ablation of the zebrafish embryonic ventricle: a novel model of cardiac injury and repair. Int. J. Cardiol. 168, 3913–3919 (2013).
Homann, C., Schriever, C., Baum, P. & Riedle, E. Octave wide tunable uv-pumped nopa: pulses down to 20 fs at 0.5 mhz repetition rate. Opt. express 16, 5746–5756 (2008).
Brand, M., Granato, M. & Nüsslein-Volhard, C. Keeping and raising zebrafish. In Zebrafish: A Practical Approach (eds Nüsslein-Volhard, C. & Dahm, R.) (Oxford University Press, 2002).
Aleström, P. et al. Zebrafish: Housing and husbandry recommendations. Lab. Anim. 54, 213–224 (2020).
Ando, K. et al. Clarification of mural cell coverage of vascular endothelial cells by live imaging of zebrafish. Development 143, 1328–1339 (2016).
Asakawa, K. et al. Genetic dissection of neural circuits by tol2 transposon-mediated gal4 gene and enhancer trapping in zebrafish. Proc. Natl Acad. Sci. 105, 1255–1260 (2008).
Ellett, F., Pase, L., Hayman, J. W., Andrianopoulos, A. & Lieschke, G. J. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood, J. Am. Soc. Hematol. 117, e49–e56 (2011).
Kolb, J. et al. Small leucine-rich proteoglycans inhibit cns regeneration by modifying the structural and mechanical properties of the lesion environment. Nat. Commun. 14, 6814 (2023).
Lee, R. T., Asharani, P. & Carney, T. J. Basal keratinocytes contribute to all strata of the adult zebrafish epidermis. PloS one 9, e84858 (2014).
John, N., Kolb, J. & Wehner, D. Mechanical spinal cord transection in larval zebrafish and subsequent whole-mount histological processing. STAR Protoc. 3, 101093 (2022).
Acknowledgements
We thank Casandra Cecilia Carrillo Mendez and Olga Stelmakh for excellent fish care. H.F. acknowledges financial support from the Max Planck Society. D.W. acknowledges financial support from the Deutsche Forschungsgemeinschaft (project number 460333672 - CRC1540 Exploring Brain Mechanics (subproject B05)). S.J. acknowledges the Erlangen Graduate School scholarship in Advanced Optical Technologies by the Bavarian State Ministry for Science and Art.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
H.F. and D.W. designed the study. S.J. and A.H. performed the measurements. N.J., J.K., and D.W. prepared the samples and performed imaging. S.J., A.H., K.S., D.W., and H.F. performed the data analysis. H.F., D.W., and S.J. wrote the manuscript. All authors reviewed and contributed to the manuscript text.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Communications Physics thanks Nirmal Mazmuder, Pierre Mahou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jun, S., Herbst, A., Scheffter, K. et al. Nonlinear dynamics of femtosecond laser interaction with the central nervous system in zebrafish. Commun Phys 7, 161 (2024). https://doi.org/10.1038/s42005-024-01653-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-024-01653-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
