
Abstract
Many materials exhibit phase transitions at which both the electronic properties and the crystal structure change. Some authors have argued that the change in electronic order is primary, with the lattice distortion a relatively minor side-effect, and others have argued that the lattice distortions play an essential role in the energetics of the transition. In this paper, we introduce a formalism that resolves this long-standing problem. The methodology works with any electronic structure method that produces solutions of the equation of state determining the electronic order parameter as a function of lattice distortion. We use the formalism to settle the question of the physics of the metal–insulator transitions in the rare-earth perovskite nickelates (RNiO3) and Ruddlesden–Popper calcium ruthenates (Ca2RuO4) in bulk, heterostructure, and epitaxially strained thin film forms, finding that electron-lattice coupling is key to stabilizing the insulating state in both classes of materials.
Similar content being viewed by others


Introduction
The relative importance of electronic and lattice effects in driving phase transitions in quantum materials is the subject of long-standing interest and controversy. One issue of particular focus is the metal to insulator transition (MIT), which in many materials is accompanied by a change in atomic positions, in other words, by a lattice distortion1. Multiple studies have addressed various aspects of the interplay between electronic and lattice contributions to the metal–insulator transition, both theoretically and experimentally. Some works have argued that the lattice plays a crucial role2, while others have emphasize the electronic aspects3; recently, explicit attempts at disentangling the two have appeared4,5,6. Information on the coupling of electronic and structural modes has been used to design and study new materials7,8,9 and to help clarify their interconnected roles. Machine learning approaches are discovering features from comparison of MIT and non-MIT materials10,11,12. Nonetheless, the problem remains an open topic of exploration with a wide variety of theoretical and experimental approaches2,3,4,5,6,7,8,9,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54. Yet, the relative importance of electronic and lattice effects in producing a transition has remained unclear, in part because of the lack of an appropriate theoretical framework and of computational schema for evaluating the needed quantities.
Metal insulator transition phenomena are properly analyzed by constructing an energy landscape in the space of lattice distortions and electronic order parameters as shown in Fig. 1 for NdNiO3. However, construction of the full energy landscape as a function of electronic order parameter and lattice configuration has not been feasible, as current quantum many-body methods do not allow the order parameters to be independently varied. Here we show how to avoid this difficulty by building the needed energy landscapes from solutions of the equation of state (electronic order parameter as a function of lattice configuration). This information is available from modern quantum many-body methods at reasonable computational expense.

Energy landscape for NdNiO3 as a function of electronic disproportionation ΔN and lattice distortion (bond disproportionation) Q as obtained from the energy functional methods introduced in this paper using data from ref. 4.
Our work was inspired by the pioneering work of Fisher and collaborators55, who combined elegant experimental measurements with an insightful free energy analysis to show that the observed second order nematic transition in an iron pnictide material has an intrinsically electronic origin. Our work may be viewed as a generalization of the ideas of Fisher et al. to first order transitions, where a more global view of the energy landscape is required, and as an extension of the insightful equation of state analysis of Peil, Hampel, Ederer, and Georges6 to the computation and interpretation of the full free energy landscape. We make substantial use of concepts put forward in analyses of the metal–insulator transition in epitaxially strained Ca2RuO45 and in nickelate heterostructures4. We apply the methodology developed here to two paradigmatic metal–insulator materials, the perovskite rare earth nickelates, and the Ruddlesden–Popper calcium ruthenates, settling a decades-old controversy by showing that in both cases the energetics associated with the lattice degrees of freedom are essential to the stabilization of the insulating phase.
Results
Framework
Following refs. 4,5,6 we consider a free energy F(ΔN, Q) that depends on an electronic order parameter, ΔN, and a lattice distortion, Q. The precise definitions of ΔN and Q will depend on the specifics of the system. We choose (ΔN, Q) = (0, 0) as the equilibrium configuration of the metallic phase. The insulating state then corresponds to a configuration (ΔN, Q) ≠ (0, 0) and the question of the existence of a purely electronic transition relates to the properties of F as a function of ΔN at Q = 0.
A second order transition corresponds to a linear instability of the (ΔN, Q) = (0, 0) state, in other words to a change in sign of the smallest eigenvalue of the Hessian matrix ∂2F/∂2ΔN, Q evaluated at ΔN = Q = 0. In general, the eigenvector associated with the negative eigenvalue will have components along both ΔN and Q, indicating that the electronic and lattice orders are coupled. A purely electronically driven transition would correspond to a change in sign of ∂2F/∂2ΔN, a lattice-driven transition would correspond to a change in sign of ∂2F/∂2Q, and in a mixed situation, neither single derivative changes sign but the lowest eigenvalue of the Hessian matrix does change sign. In the case of the nematic transition in pnictides, Fisher and co-workers were able55 to experimentally determine ∂2F(Q = 0)/∂ΔN2 and show that it changed sign at a temperature only slightly lower than the observed nematic transition temperature, thereby establishing that a purely electronically driven nematic transition exists in these materials, with a transition temperature that is slightly enhanced by coupling to the lattice.
The transitions of interest in this paper are first order, characterized by the appearance of a new free energy minimum at which ΔN and Q are both different from zero (see e.g., Fig. 1). Study of first order transitions requires global knowledge of the free energy. One may say that the transition is electronically driven if F(ΔN, Q = 0) has an extremum at a ΔN ≠ 0 with ∂2F(ΔN, 0)/∂ΔN2 > 0 and with energy lower than F(0, 0). One may say that the transition is lattice-assisted if F(ΔN, 0) has a local minimum at a ΔN ≠ 0 but that the lattice coupling is required to make this minimum the ground state. Finally, if F(ΔN, Q = 0) has only one minimum, at ΔN = 0 but exhibits a stable minimum with ΔN ≠ 0 at a Q ≠ 0 then we may say that the transition is not electronically driven. The energy functional shown in Fig. 1 shows a transition that is not electronically driven because the global minima are at a Q, ΔN ≠ 0 but along the line Q = 0 the functional has only one minimum, at the origin.
The global knowledge of F required to assess the relative importance of electronic and lattice effects in stabilizing the insulating state has been challenging to obtain. Available quantum many-body methods can, with reasonable computational cost, obtain estimates of the optimal ΔN that minimizes F at fixed atomic positions (Q), but finding the dependence of F on ΔN at fixed Q or on Q at fixed ΔN is difficult, both because it is not straightforward to control ΔN independently of Q and because calculations of energies are very expensive.
However, the metal–insulator transitions of greatest current experimental interest share two features that greatly simplify an analysis. First, the lattice energetics is to good approximation harmonic4,5,6, as shown from calculations4 and by the observation that phonon frequencies change only very slightly (~1%) across the metal–insulator transition. Second, and closely related to the first point, the coupling between the electronic order parameter and lattice modes is linear and is well determined by standard theoretical methods4,5,6, as are the lattice energetics. This means that we may write, schematically,4,5,6:
Requiring stationarity of F with respect to variations in ΔN and Q yields the equations of state4,6
and
Because g and K are known, Eq. 2 can be solved. Substituting the solution back into Eq. 1 produces:
From this point of view, the key question is the magnitude of the lattice stabilization energy \(\frac{1}{8K}{\left(g{{\Delta }}N\right)}^{2}\): is it large enough to create a new extremum at a ΔN ≠ 0? Is it large enough to ensure that the extremum at ΔN = 0 is unstable? Answering this question requires knowledge of Fel.
Peil, Hampel, Ederer, and Georges6 observed that existing many-body methods such as the dynamical mean field method enable computation of electronic properties at fixed lattice configuration Q, in effect solving Eq. 3. Here we take one further step: the ansatz for the electronic energy functional in Eq. 1 means that the computed ΔN(Q) determines dFel(ΔN)/dΔN; the derivative can then be integrated, thereby constructing F, Fel itself. In practice, we perform the integration by fitting the ΔN(Q) results to a polynomial, which is analytically integrated. While this method requires a choice of polynomial form, we empirically find that the uncertainty thereby introduced is small, of the order of a few meV, comparable to the error in performing an explicit energy calculation. This approach is computationally inexpensive, relatively unaffected by noise, and the analytically specified functional forms provide additional insight, in particular enabling a straightforward examination of the global energy landscape.
The method is general: the electronic and lattice order parameters can be of scalar, vector, or tensor character, higher-order couplings can be included and any theoretical method that delivers an equation of state can be used in Eq. 1. The applicability of the model in its current form is, however, restricted to situations in which the orbital basis used to define the electronic order parameter can be clearly defined, and the order parameter energetics can be separated from the energetics of the rest of the material which can then be subsumed into the lattice stiffnesses K. We will discuss the limits of our method’s applicability in more detail in the “Discussion” section, and in the Conclusion mention other materials to which it may be applicable.
We use this approach to analyze the metal–insulator transitions occurring in the rare-earth nickelates and their heterostructures, and in bulk and epitaxially strained Ca2RuO4. We find that in all of these situations the metal–insulator transition is, in the sense defined above, not electronically driven.
It is important to emphasize that the distinction between electronically driven and lattice driven transitions is not the same as the distinction between weak and strong electron correlations. A material may be said to be weakly correlated if density functional band theory (DFT) methods (or their hybrid and +U extensions) adequately describe the behavior and may be said to be strongly correlated if beyond DFT methods are required for the correct description of materials properties. While the weak/strong correlation distinction is not the main focus of this paper we believe the available evidence indicates that beyond DFT methods are needed to properly calculate the electronic term Fel of the materials we study here.
Rare-earth nickelates
The perovskite rare-earth nickelates have the chemical formula RNiO3; when synthesized in bulk form the materials with R = Lu, Y, Sm, Nd, Pr exhibit a first order transition from a high-temperature metal to a low-temperature insulator while LaNiO3 remains metallic down to the lowest measured temperature56. The material properties depend systematically on the choice of rare-earth ion, with LuNiO3 exhibiting the highest metal insulator transition temperature TMIT = 600 K, SmNiO3 having TMIT ≈ 400 K, NdNiO3 having a TMIT ≈ 200 K and PrNiO3 having a TMIT ≈ 130 K17,57,58. The materials may also be grown in heterostructure form4,7,13,17,18,24,48,59,60, with a small number of layers of RNiO3 sandwiched between layers of other materials. The metal–insulator transition temperature in the heterostructures differs from that in the bulk.
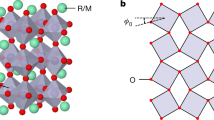
Except for the La-based compound, where the symmetry is slightly different, the high temperature metallic state forms in a Pbnm structure which for present purposes may be regarded as a pseudo-cubic lattice of Ni ions with an O ion at the midpoint of each Ni–Ni bond. The insulating state is characterized by a two-sublattice bond disproportionation, with one Ni sublattice having a short mean Ni–O bond length and the other by a long one, represented qualitatively in Fig. 2. The difference in mean Ni–O bond lengths defines the lattice mode Q appropriate to this transition.

NiO6 octahedra disproportionate electronically and structurally in a three-dimensional checkerboard pattern in RNiO3 metal–insulator transition compounds, with R a rare-earth. In the low-energy model of refs. 4,6, the Ni ions corresponding to the larger octahedra have 1e− \(+\frac{{{\Delta }}N}{2}\) electrons (dark blue) in their eg shell, while those corresponding to the smaller one (light blue) have 1e− \(-\frac{{{\Delta }}N}{2}\). The difference in average bond lengths defines Q for the nickelate materials. Red circles correspond to oxygen atoms, while the green arrow is a measure of the oxygen displacement along the Ni-Ni direction.
In the low T insulating state, the two Ni sublattices differ in electronic configuration. This difference has been characterized in various ways in the literature6,36,44,61,62,63. We follow Peil and collaborators4,6 and define the electronic change ΔN as the difference in the occupation of the eg states obtained from a narrow window Wannier fit of the bands crossing the Fermi level, i.e.,
with NHF (NLF) the total eg occupation on the octahedron with higher (lower) Ni occupation NiO6 octahedra, while Q is defined as the average Ni–O bond length difference between the two, as shown in Fig. 2.
In Fig. 3 we present results for three different bulk-form rare earth nickelates, (LuNiO3, SmNiO3, PrNiO3) as a function of Q, digitized from the calculations of ΔN presented in ref. 6. In this reference, the calculations performed are DFT+DMFT (DFT+Dynamical Mean Field Theory) calculations in the paramagnetic phase. In all compounds, two regimes are found, a low Q regime where ΔN is proportional to Q and a high Q regime where ΔN is large but weakly dependent on Q; the two regimes are separated by a discontinuity.

a Points: Electronic disproportionation as a function of bond disproportionation ΔN(Q) values used for the three RNiO3 materials as obtained from Density Functional Theory + Dynamical Mean Field Theory calculations from ref. 6; Light lines: fits of the points to Eq. 7. Also shown is the Q(ΔN) line obtained from the Q equation of state Eq. 2. Note that the ratio of the lattice stiffness to the linear coupling K/g has the same value for all three materials. b Electronic energy, c total energy Ftot = F of three RNiO3 compounds as a function of electronic order parameter ΔN, minimized over structural order parameter (bond disproportionation) Q. d Total energy as function of lattice distortion Q, minimized over ΔN. Energies per 10 atom unit cell.
To determine the free energy model to which the results should be fit we observe that the two sublattice nature of the insulating state means that the free energy must be invariant under simultaneous inversion Q ↔ −Q, ΔN ↔ −ΔN, so in particular, Fel(ΔN) must be even in ΔN. Motivated by the observed first-order nature of the transition we assume that Fel is a 6th order polynomial in ΔN:
Solving Eq. 2 and eliminating Q from the full free energy then changes the quadratic term \({\chi }_{0}^{-1}\to {\chi }_{0}^{-1}-\frac{{g}^{2}}{4K}\). Note that the shift in the quadratic term involves the parameter g2/K, which varies slightly across the RNiO3 series although as noted in ref. 6 the ratio g/K is almost material-independent. The K for the Lu, Sm, and Pr materials were reported in ref. 6 to be 39.29, 41.45, and 44.47 eV/Å2, while the g are 3.75, 4.02, and 4.24 eV/Å.
Equation 6 implies that the equation of state Eq. 3 takes the explicit form
We fit the points in Fig. 3 to Eq. 7, using the g values presented in ref. 6, obtaining the light solid curves shown in Fig. 3 and the fit parameters given Table 1. It is important to note that the DMFT equation of state shown in Fig. 3 has two branches, with the solution discontinuously changing from one to the other as Q is varied. In our fits we only use the data points shown as symbols in Fig. 3; these points lie outside the region of multistability (i.e., where two ΔN solutions exist within DMFT for the same Q). However, our theoretical energy function correctly reproduces behavior that was not part of the original fit, for example, the locally stable large ΔN at Q = 0 solution for LuNiO36. Including points within the region of multistability does not lead to significant changes to our results. The free energy we obtain here may be thought of as the free energy that would be obtained from the two component Landau theory of ref. 6 if the metal–insulator variable were integrated out.
Panel b of Fig. 3 shows the purely electronic energy Fel corresponding to the fit parameters given in Table 1. We see that for all of the materials the purely electronic theory is characterized by a locally and globally stable metallic minimum at Q = 0. Only for LuNiO3 does the purely electronic theory even exhibit a second, insulating extremum and even in this case it is not energetically favored. Panel c presents the total free energy as a function of ΔN obtained by minimization over the lattice degrees of freedom Q. We see that inclusion of the lattice coupling is necessary to stabilize the ΔN, Q ≠ 0 solution: in other words, it is the electron-lattice coupling that drives the transition.
Table 1 shows that as one moves across the RNiO3 series from R = Lu (most insulating) to R = Pr (least insulating), the linear response electronic susceptibility of the metallic state decreases, consistent with the empirical observation that the Lu material is the strongest insulator and the Pr material is the weakest. We see also that the nonlinear terms in the energy are the same for the Lu and Sm compounds, which are the two strong insulators, but are different in the Pr compound, which is close to the metal–insulator transition. This shows that to understand composition-dependent changes one must consider more than just the variation in linear response susceptibilities. Results to be presented below for superlattices further confirm this point.
The present theory predicts that PrNiO3 is undistorted and metallic, although close to the metal–insulator transition boundary, while the compound is empirically distorted and insulating, although again with a very low transition temperature. We believe this discrepancy arises because the transition in PrNiO3 is from a paramagnetic metal to an antiferromagnetic insulator, in contrast to the Sm and Lu materials where the transition is between two paramagnetic phases. Inclusion of antiferromagnetism in the theory will yield a lower energy for the insulating phase of PrNiO3 at the U, J values used here while a slightly different choice of interaction parameters would also push PrNiO3 to the insulating side of the phase diagram.
We may take the analysis further by substituting the ΔN(Q) obtained from the solution of Eq. 7 into the full free energy expression to obtain the free energy as a function of the lattice coordinate Q shown in the panel d of Fig. 3. This is analogous to the energy that is usually calculated by electronic structure codes, which work at fixed atomic positions. The discontinuities arise from the different branches of the ΔN(Q) curves. The resulting free energy is, to a good approximation, the combination of two parabolic energy curves, one corresponding to the metallic state, and one to the insulating state. We see that in this theory the ΔN = 0, Q = 0 state is stable to lattice distortions for PrNiO3, marginally stable for SmNiO3, and unstable for LuNiO3.
The analysis presented here is based on a fit of the computed ΔN(Q) to a free energy model. We emphasize that this fit is in no way essential to our method; one could simply numerically integrate a suitably dense set of ΔN(Q) data. But for the procedure used here the question of the sensitivity of the results to the choice of free energy arises. To quantify the uncertainties we have refit the PrNiO3 data shown in Fig. 3, now constraining the 4th and 6th order coefficients to have the same values as found in SmNiO3. Results are shown in Fig. 4. We see from panel a that this fit to the data is not quite as good, especially in the small to intermediate Q region. The fitted energy (panel b) is very similar to that shown in Fig. 3, with the value of ΔN at the minimum almost the same in the two cases and the energy of insulating solution in the constrained fit is now slightly lower than the energy of the metallic solution, placing paramagnetic PrNiO3 slightly on the insulating side of the transition. These differences indicate that the systematic uncertainties in the method are not large.

Fits (a) and total energy (b) for PrNiO3 based on Density Functional Theory + Dynamical Mean Field Theory data from ref. 6; green lines correspond to the optimal polynomial fit, red lines to 4th and 6th kept constant to the values extracted for SmNiO3, and only the quadratic term fitted to the data.
Layered structures of NdNiO3/NdAlO3
An important modern direction in quantum materials science is heterostructuring: the ability to synthesize structures in which atomically thin planes of one material alternate with atomically thin planes of another. Previous experimental13 and theoretical4 work analyzed systems of the type shown schematically in Fig. 5 in which a few layers of metal–insulator transition compound NdNiO3 were grown as a thin film sandwiched epitaxially between layers of the wide-gap insulator NdAlO3. The few-layer systems had significantly higher transition temperatures than did bulk NdNiO3. Theoretical DFT+DMFT results are available4 for three systems: bulk NdNiO3 and two heterostructured systems with a 4 unit cell repeat distance: one layer of NdNiO3 followed by three of NdAlO3 (monolayer), and two layers of NdNiO3 followed by two of NdAlO3 (bilayer). The ΔN(Q) results are shown in Fig. 6

a Schematic of superlattice consisting of NdNiO3 layered in between the band insulator NdAlO3; b schematic view of two atomic layers of NdNiO3 sandwiched between NdAlO3 layers as described in ref. 4. Dashed line with red slash: forbidden electronic hopping path, indicating mechanism of dimensional confinement. Zig-zag lines connecting O (red circles) and Al atoms: representation of the increased stiffness to distortions of apical Ni–O bonds as a result of resistance from the Al–O bonds.

a Data showing electronic disproportionation as a function of structural distortion ΔN(Q), from4 (points) and the polynomials (light lines) obtained by fitting the data to the equation of state, Eq. 7. b Electronic and c total energy as functions of electronic order parameter ΔN for bulk NdNiO3 and for superlattices consisting of a bilayer of NdNiO3 alternating with NdAlO3 and a monolayer of NdNiO3 alternating with three layers of NdAlO3 using data from ref. 4. d Total energy as function of octahedral distortion Q after minimizing over electronic disproportionation ΔN. Note change of y-axis scale between panel a and other two panels. Energy normalized to correspond to 10 atom unit cell for the bulk, and per two Ni atoms (one layer) of NdNiO3 in the superlattices.
Two competing effects were found4: the electronic confinement of the electrons in the NdNiO3 by the nearby NdAlO3 layers favors a higher electronic disproportionation ΔN, while the energy cost of imposing a lattice distortion on the adjacent Al-O octahedra is equivalent to an increase in the stiffness K of the NiO6 lattice with a K = 31.6914eV/Å2, for the bulk material, K = 35.6269 eV/Å2 for the bilayer and K = 40.4753 eV/Å2 for the monolayer; all energies per 10 atom unit cell. The values for g are those quoted in ref. 4; we reproduce them here for convenience: g = 3.2212 eV/Å for the bulk, g = 3.3465 eV/Å for the bilayer, and g = 3.3780 eV/Å for the monolayer. Performing an analysis similar to that presented in section “Rare-earth nickelates” leads to the energy curves presented in Fig. 6 and to the fit parameters shown in Table 2. The original data with the fit are presented for convenience in Fig. 6
Turning first to the purely electronic correlation energy shown in the (b) panel of Fig. 6, we observe that neither NdNiO3 nor the heterostructured materials show a purely electronically driven insulating state; however, hints of an inflection point, the precursor of the formation of a higher ΔN extremum, can be seen in bilayer and monolayer curves around ΔN = 1 reflecting the effect of quantum confinement in increasing electron correlation effects. Inclusion of the lattice energy produces a global, insulating minimum in all three materials. The monolayer and bilayer show a significantly larger energy difference between the insulating and the metallic states than does bulk NdNiO3, in agreement with their higher TMIT.
The origin of the differences between monolayer, bilayer, and bulk nickelates is surprising: the modest decrease in the magnitude of g2/K as we pass from bulk to bilayer to monolayer reflects the increase in lattice stiffness, opposing the order, and nearly counterbalances the modest decrease in \({\chi }_{0}^{-1}\) reflecting the increased correlation physics of the confined metallic state. The more important effect, however, is the change in the magnitude of the higher order (ΔN4, ΔN6) terms shown in Table 2. The change can be seen in Fig. 6 from the variation of the critical Q below which the insulating state is not stable. In the calculations presented in the previous subsection the higher order terms were also found to be different in PrNiO3 (proximal to the metal–insulator transition) than in LuNiO3 and SmNiO3 (farther from the transition point).
Metal–insulator transition in bulk Ca2RuO4
Ca2RuO4 forms in a slightly distorted version of the n = 1 Ruddlesden–Popper structure, a layered structure consisting of RuO2 planes separated by pairs of CaO layers. Each Ru is six-fold coordinated by oxygen, forming RuO6 octahedra. The relevant electronic states (actually Ru–O antibonding states) may be thought of as Ru t2g symmetry d-states (dxy, dxz, dyz) with 4 t2g electrons per Ru. Below 340 K the material undergoes a first-order transition from a high temperature metallic to a low temperature insulating phase. No symmetry is broken at the transition: the two phases share the same crystal symmetry but differ in the occupancies of the d orbitals and shape of the RuO6 octahedra, and the relative occupancies of the d-levels. As sketched in Fig. 7, the metallic state is characterized by an approximately equal occupancy of the t2g orbitals, while in the insulating state the xy orbitals are doubly occupied and the xz/yz orbitals each contain a single electron. The appropriate electronic order parameter is the occupancy difference between the xy and xz/yz orbitals
No crystal symmetry protects the orbital occupancies in the high temperature metallic state, which is characterized by a ΔN = NH close to, but not exactly, zero.

a In the metallic phase, all 3 t2g orbitals of Ca2RuO4 are approximately equally occupied; ΔN ≈ 0. b In the insulating phase, the xy orbital is doubly occupied, while the yz and xz are singly occupied; ΔN = 1. c Representation of Q0 structural mode, in which all bond lengths change equally. d Representation of structural mode Q3 in which octahedral volume is preserved and the change in the two in-plane Ru–O bond lengths are equal, and opposite in sign to the change in the apical bond length. The electronic state in b is associated with a structural term of the form −(Q3 − λ0Q0), with λ0 > 0 Legend: a, b: black lines correspond to orbital energy levels, blue circles signify electrons; c, d: purple circle = Ru atom, red circle, O atom; arrows and dotted lines signify bonds and their changes with the two different structural modes.
The change in electron occupancy across the metal–insulator transition occurs simultaneously with a decrease in the Ru–O apical bond length and an increase in the in-plane Ru-O bond length (a flattening of the octahedra). At the transition, there is a change in the lattice constants, and rearrangements of other internal coordinates. The harmonic nature of the lattice Hamiltonian means that most of the lattice modes may be integrated out, leaving an effective theory involving two structural degrees of freedom. The two lattice degrees of freedom are needed, because if the point symmetry of the Ru ion is lower than cubic, then both the unit cell volume change and the relative octahedral distortion couple linearly to the differential level occupancy ΔN.
Following ref. 5 we write the two relevant structural degrees of freedom as a variable Q3 parametrizing a volume preserving change in the c-direction Ru–O bonds relative to the average in-plane Ru–O bonds, and a change Q0 in the octahedral volume and define the lattice variables such that in the high-T metallic state Q3 = Q0 = 0. In terms of changes δx, δy, δz to the three Ru–O bond lengths, defined as:
We now build the free energy. We define the lattice distortion relative to the high-T metallic state as the vector \(\overrightarrow{Q}=({Q}_{3},{Q}_{0})\); the lattice restoring term K is a 2 × 2 matrix with entries determined in previous work5 to be K33 = 17.7, K03 = 7.6, K00 = 46.2 eV/Å2 per formula unit. We write the linear combination of the Q that couples to the electronic disproportionation as \(\overrightarrow{{{{{{{{\mathcal{F}}}}}}}}}\cdot \overrightarrow{Q}\) with \(\overrightarrow{{{{{{{{\mathcal{F}}}}}}}}}={F}_{3}(1,-\lambda )\). Reference 5 finds F3 = 2.8 eV/Å and λ = 0.45.
Here ΔNH is the value of ΔN in the high temperature insulating phase; it is nearly, but not quite, zero.
For the purely electronic energy we chose the 4th order form:
Odd powers occur because no symmetry is broken at the transition.
Figure 8 shows previously published results5 for ΔN as a function of the relevant combination of lattice distortions along with our fit to Eq. 11. As in the previous section we have not used points in the coexistence region for the fit. We find σ0 = −0.009, a = 0.70, b = −1.718, c = 1.28.

a Electronic disproportionation as a function of structural disproportionation ΔN(Q) from5 (heavy black points) and polynomial fit (solid line) used for the equation of state. b Correlated electron free energy, and total free energy as a function of the electronic disproportionation order parameter characterizing the orbital polarization: Fel(ΔN) and Ftotal(ΔN) for bulk Ca2RuO4. c Total energy plotted against orbital disproportionation ΔN for materials grown on four substrates indicated in the legend (NSAT is Nd0.4Sr0.6Al0.7Ta0.3O3) which provide different strains relative to the metallic phase of Ca2RuO4. For compressive strain (NdAlO3), the material is always metallic and the orbital polarization has the opposite sign from the insulating state. d Total free energy versus electronic disproportionation ΔN : Ftotal(ΔN) computed for bulk Ca2RuO4 different values of the linear coupling term F3. e Phase diagram of epitaxially constrained Ca2RuO4 in plane of tensile strain (defined as difference of mean in-plane Ru–O bond length from value in the high temperature structure) in Å and electron-lattice coupling parameter F3. Red dashed line: value of F3 at which metal insulator transition occurs in bulk system. Arrows (red on line) indicate strain imposed by epitaxial growth.
The higher curve (blue) in Fig. 8, panel b, shows the purely electronic free energy resulting from the fit. We see that in the absence of lattice effects the metallic, undistorted state is energetically favored and that there is not even a metastable minimum corresponding to the insulating state, although there is a hint of an inflection point that is a precursor of a higher ΔN extremum. The lower trace (dark orange line) shows \(\bar{F}\), the full free energy after the lattice modes have been integrated out. We see that the inclusion of the lattice energetics qualitatively changes the free energy, strongly favoring large ΔN. Indeed we see that the energy does not have a minimum in the physically allowed range ΔN ≤ 1; rather it is minimized at the boundary ΔN = 1, as ∣ΔN ≤ 1∣ is the largest allowed value. We again conclude that the transition in this compound is driven by the lattice contribution to the energetics.
Epitaxially constrained Ca2RuO4
Ca2RuO4 has also been grown expitaxially on insulating substrates. The epitaxial constraint means that the in-plane lattice parameters of Ca2RuO4 are forced to be equal to the in-plane lattice parameters of the substrate material. A priori, this constraint does not fix the values of the internal coordinates including the the Ru–O in-plane bond length of relevance here. Density functional theory calculations, however5, indicate that in practice the material adapts to the epitaxial constraint by adjusting the in-plane Ru–O bond lengths, rather than by rotating the Ru–O6 octahedra, so that the percentage change in the in-plane Ru–O bond lengths is fixed by the percentage change in the in-plane lattice parameters. The z-direction lattice constants and Ru–O apical bond lengths are still of course free to relax. This means that the theory of the previous section can be simply adapted to the epitaxial case by a change of variables. We write:
and
with δx0 and δy0 fixed by the epitaxial constraint. It is also convenient to define the zero of δz to be the value
that δz would take at ΔN = NH.
We obtain:
with
and
The epitaxial constraint thus has three effects: it increases the net stiffness to lattice distortions by preventing relaxations of the lattice coordinates associated with in-plane bond lengths, making the insulating phase more difficult to obtain. Second, it reduces the electron lattice coupling (Eq. 17) which also makes it more difficult to obtain an insulating phase. Finally, it provides a term linearly proportional to ΔN, which for positive epitaxial strain favors the insulating phase. Panel a of Fig. 9 shows the change in apical Ru–O bond length as a function of epitaxial strain; panel b the occupancy difference ΔN. The first order transition is evident. Note that for an epitaxial strain matching the in-plane lattice constant of the high temperature metallic phase, insufficient lattice energy is available to stabilize the insulating phase. As the tensile strain is increased, the octahedral deformation, which couples linearly to ΔN, increases, and beyond a critical value drives a first order transition.

Equilibrium electronic order parameter characterizing the orbital polarization ΔN (a) and Ni–O apical bond distortion δz (b) versus epitaxial strain defined as the difference in mean in-plane Ru–O bond length relative to the value in the high temperature structure of Ca2RuO4 as imposed through epitaxial strain via in-plane Ni–O bond changes away from the bulk values, δx0, δy0 where δx0 = δy0, computed as described in the main text.
Experiments (refs. 64,65) have studied films grown epitaxially on substrates NdAlO3, LaAlO3, NSAT (Nd0.4Sr0.6Al0.7Ta0.3O3), and NdGaO3 corresponding to changes (relative to the high-T state, and assuming the Ru–O bond length exactly tracks the in-plane strain) in average in-plane Ru–O lattice parameters of −0.04, 0.008, .03, and 0.046 respectively. Figure 8 shows that the theory, with no adjustable parameters, predicts that for the theoretically predicted coupling F3 = 2.8 eV/Å the films grown on NdAlO3 and LaAlO3 should be metallic, while the films grown on NSAT and NdGaO3 should be insulating. In the experiment, the LaAlO3-strained material has a transition at T ≈ 200 K from a high temperature metal to a low temperature weak insulator/bad metal, while the others are metallic and insulating as predicted by the theory. The qualitative agreement is good; the quantitative discrepancies may arise from a more subtle relation between Ru–O bond length and in plane lattice constant than was assumed in ref. 5 or from small systematic errors in the theory. The good correspondence between experiment and theory shows the utility of the methodology introduced here.
Temperature Dependence
The methods presented here provide some insight into the physics of the temperature-driven metal–insulator transition, indicating an important area for future research. We see from Figs. 3 and 6 that for the nickelates the low T energy landscape is characterized by two minima, with energy differences per formula unit ranging from 80 meV (LuNiO3) to 25 meV (NdNiO3). In the nickelates, as the temperature increases, a first order metal–insulator transition occurs. The lattice distortion Q exhibits negligible temperature dependence over the entire insulating phase, indicating that the minima remain robust and the transition is driven by an entropic effect that lifts up the free energy of the large ΔN extremum relative to that of the ΔN = 0 extremum.
To investigate whether the entropic effect arises from the local correlations captured by the dynamical mean field approximation, for bulk NdNiO3 we have extended the calculations shown in Fig. 6 to much higher temperatures by performing calculations with the same methodology and interaction parameters as in4 but changing the electronic temperature. The parameters, as obtained from the fit for certain temperatures can be found in Table 3. Figure 10 shows the results: all of the parameters in the electronic free energy vary systematically with temperature, decreasing in magnitude as the temperature is increased from ~290 to 1000 K; the result is a very weakly first order transition at ≈1080 K (the model actually passes very close to the tricritical point at which the second and fourth order coefficients of the theory simultaneously change sign). This behavior, while theoretically interesting, is inconsistent with the observed strongly first order character and much lower transition temperature behavior, suggesting that either the single site dynamical mean field approximation does not adequately describe the temperature dependence of the electronic contribution to the free energy, or that an entropic effect associated with the lattice degrees of freedom plays an important role. Indeed the neglect of spatial fluctuations generically means that single-site DMFT approximations overestimate transition temperatures66,67. It should also be noted that in this material antiferromagnetism plays an important role in the ordering, and that a direct computation44 of the free energy F(ΔN, Q) using a different DMFT formalism places paramagnetic NdNiO3 on the paramagnetic metal side of the transition and yields a stronger low T temperature dependence, and shows for LuNiO3 a temperature dependence of F(ΔN, Q) in the range T < 500K very similar to that found here for NdNiO3.

a Electronic free energy as function of electronic disproportionation ΔN computed for NdNiO3 at temperatures from 290 to 1160 K. b Total free energy computed for bulk NdNiO3 for the same temperature range. c Total expanded view of total free energy for bulk NdNiO3 at temperatures close to the transition. d Optimal bond disproportionation Q, as obtained by a linear transformation from the electronic disproportionation ΔN, using the lattice stiffness K and electron-lattice coupling term g: Q(T) = \({{\Delta }}N(T)\frac{g}{2K}\). Data for fits provided in Supplementary Note 1.
Ca2RuO4 presents different issues. A substantial temperature dependence is observed68 across the insulating phase. Since the electronic order parameter ΔN in this material is fixed at its largest possible value for a range of couplings, the temperature dependence must imply a temperature dependence of the electron-lattice coupling F3 as proposed for related reasons in5. Within single-site dynamical mean field theory, there is no theoretical justification for a strongly temperature-dependent coupling parameter, but on the assumption that changes in electron-lattice coupling F3 are a proxy for changes in temperature we show in Fig. 8, panel d, the energy computed for different values of F3. We see that a first order transition occurs at an F3 ≈ 1.58 ≈ 0.6 of the F3 we estimated from the low temperature calculation. The phase diagram presented in Fig. 8 panels e, and c, then indicates that films grown on substrates with a tensile epitaxial constraint exhibit significantly higher transition temperatures than found in bulk, roughly consistent with experiment.
Discussion
It is important to critically examine the assumptions that underly our results. The first assumption enabling the construction is that the lattice energy is quadratic in deviations from an equilibrium position at fixed value of the electron order parameter, in other words that the physics controlling the relevant atomic force constants arises from chemical bonds inside the solid that are only weakly affected by the transition from metal to insulator. This and the harmonic nature of the lattice response are found in density functional calculations (see e.g., the supplementary material of ref. 4) and are further supported by the observation that phonon frequencies do not change much across the transitions of interest.
The second assumption is that the coupling between electronic and lattice degrees of freedom is linear, and may be determined a priori. This second assumption has been confirmed by explicit calculations (see, for example, the supplementary information in ref. 4). Further investigation of these assumptions, and identification of compounds in which they break down, is an important topic for future research. One important direction is a detailed comparison of results obtained along the lines indicated here to direct computations of energies and free energies44,62,63,69.
Given these assumptions, a many-body calculation of electronic configuration as a function of lattice distortion may be used to construct free energies, essentially by integrating the equation of state. We accomplish the integration by fitting the equation of state results to a polynomial which is integrated analytically. This procedure is convenient because it avoids ambiguities associated with the coexistence region of the first order transition, but is not necessary.
We applied the methodology to two currently interesting families of compounds, that exhibit first order metal–insulator transitions as the temperature is decreased: the rare earth perovskite nickelates RNiO3, and Ca2RuO4. The two materials were chosen because they exhibit qualitatively different electron-lattice couplings. The transition in the RNiO3 is a symmetry-breaking transition leading to two inequivalent Ni sites and an alternating pattern of long and short Ni–O bond lengths. The transition in Ca2RuO4 is isosymmetric, with no broken crystal symmetries between the two states. In both materials, we find that if the lattice coordinate is set to the value appropriate to the high temperature metallic state, the resulting energy F(ΔN, Q = 0) has only one minimum, at ΔN = 0. It is only after the coupling to the lattice is included that the total functional F(ΔN, Q) acquires a minimum at a ΔN, Q ≠ 0. We, therefore, conclude that lattice effects are essential in driving the metal–insulator transition in both compounds, rather than being merely a minor consequence of a fundamentally electronically driven transition, thereby settling a long-standing controversy. It is important to note however that although the purely electronic theory does not produce a metal–insulator transition, the correlation contribution to the electronic energy is still important. DFT-level calculations would lead to a Fel, which rises so steeply with ΔN that the only extremum would be at ΔN = Q = 0. A beyond-DFT theory is needed to obtain an electronic energy which, although it does not by itself have a minimum at a non-zero ΔN, is “soft” enough to enable the lattice energies to produce the ordered state.
It is important to consider the limitations of our conclusions. First, while the approach introduced here will work with any method that calculates the electronic order parameter as a function of lattice distortion ΔN(Q), in practice the information available in the literature comes from the density functional plus dynamical mean field methodology. While this method successfully produces results that are consistent with many experiments, its precise quantitative accuracy is unknown. The method does require an identification of correlated orbitals, it relies on the assumption that density functional theory gives an adequate account of the energetics of the uncorrelated orbitals, it requires specification of interaction parameters, and its solution of the many body problem involves a stringent locality assumption. Cross checking via other methods (as for example was done for Ca2RuO4 in ref. 70) would be desirable. Further, there are several variants of the DMFT plus DFT methodology, differing in the choice of energy window and the representation of correlated states71; the dependence of our conclusions on the implementation is an interesting question.
The specification of interaction parameters is of particular importance. As shown e.g., in refs. 3,4, as the interaction parameters are increased in magnitude, a transition can be generated in the purely electronic theory. The precise statement made here is that with interaction parameters determined by reproducing physical properties including gaps, and effective masses, and structural distortion, the purely electronic theory does not exhibit a phase transition: electron-lattice coupling plays an essential role in stabilizing the insulating states.
It will also be noted that while the theory presented here reproduces trends and orders of magnitude very well, it has some quantitative deficiencies, for example predicting that PrNiO3 is metallic but close to the phase boundary when in fact it is insulating but close to the phase boundary, with the lowest transition temperature of any bulk member of the RNiO3 nickelate family. This deficiency is likely to be remedied by the inclusion of magnetism in the theory, since in PrNiO3 and NdNiO3, but not in the other compounds studied in this paper, the metal–insulator transition is accompanied by a magnetic transition. Similarly, the phase diagram of epitaxially constrained Ca2RuO4 films is qualitatively correct but exhibits quantitative discrepancies with experiment. We note that in this paper no attempt was made to adjust the parameters from the literature or to fine-tune the fits to obtain better agreement with the experiment. Small changes, reflecting moderate parameter uncertainties and modest systematic errors in the DFT+DMFT approach, would likely cure these discrepancies.
Conclusions
Electronic phase transitions in quantum materials are almost always accompanied by some form of lattice distortion, and in many specific cases, the question of whether the transition is driven by the lattice or by correlated electron energetics has been hotly debated. In theoretical terms, the answer is determined by the properties of a free energy functional F(ΔN, Q) that depends on suitably defined electronic (ΔN) and lattice (Q) modes. For second order transitions, the needed information can often be determined from symmetry arguments along with the values of a small number of parameters determined from the linear response of the symmetry-unbroken state. However, many transitions of current interest, in particular metal–insulator transitions, are first order, so that knowledge of the free energy landscape over a wide range is required. This information has been more difficult to obtain both because of the simple expense of computing an entire energy landscape and because these computations require fixing the electron order parameter at values that do not extremize the energy. In this paper, we proposed a method that constructs the needed free energy from the equation of state (electron order parameter as a function of lattice distortion). The method is computationally feasible and can be applied using any many-body method that obtains the equation of state. We demonstrate the power of the method by using it to resolve the long-standing question of the role of the lattice in the metal–insulator transitions in two representative classes of quantum materials, the perovskite rare earth nickelates, and the Ruddlesden–Popper calcium ruthenates. We find that in both materials the lattice degree of freedom is essential to the metal–insulator transition.
We also observe that while we presented results based on dynamical mean field calculations, the method is generic in nature and can easily be used with other electronic structure methods, including DFT, DFT+U72,73, Gutzwiller74,75, Auxiliary–Boson76,77,78,79—particularly with the advent of new codes that calculate energy and naturally handle symmetry breaking80,81,82,83,84, and quantum Monte Carlo85.
The methods can be applied to quantify the importance of lattice effects in other systems of current interest, including CaFeO386,87 which exhibits charge ordering and lattice disproportionation, perovskite titanates and vanadates, where lattice distortions couple to orbital ordering88,89,90, VO2 where a dimerization instability occurs91, V2O3 where the metal–insulator transition couples to the volume of the material92, and the rare earth manganites where electronic charge, orbital and magnetic ordering are tightly coupled to complex lattice distortions93,94,95, two-dimensional correlated van der Waals dihalides and trihalides MX2, MX3, with M a transition metal ion and X a halogen96,97,98,99, as well as in charge density waves in transition metal dichalcogenides100,101.
The access to the free energy provided by our methods enables additional insights. Issues relating to temperature dependence were discussed in the temperature dependence section “Temperature Dependence”. The energy landscape shown in Fig. 1 makes it clear that only one narrowly defined path connects the metallic and insulating minima; this information may be used to investigate the kinetics of order parameter nucleation if the material is supercooled or superheated across the phase boundary. Further, the differing roles of the electronic and lattice degrees of freedom in defining the energy landscape make possible an analysis of the kinetics of state evolution after excitation. For example, excitation might rapidly heat the electrons (leading to changes in Fel) but only slowly heat the lattice degrees of freedom (which would in any event respond more slowly). Extension of the theory presented here to include the antiferromagnetism that occurs in the rare earth nickelates may help understand recent ultrafast experiments exploring the relation between antiferromagnetism, lattice distortions, and metallicity in NdNiO3102,103.
Methods
The new numerical ΔN(Q) data for NdNiO3 used to build the electronic energies in this paper is obtained by using identical methodology and parameters as in our previous work (ref. 4). We’ve used Quantum Espresso104 for density functional theory calculations, Wannier90105 to build the low-energy Wannier model, and dynamical mean field theory calculations using the TRIQS implementation106, using the continuous time hybridization107 solver. We’ve used the exact same numerical and interaction parameters as in (ref. 4), but with a different electronic temperature which can be obtained by changing the parameter β.
The other DFT+DMFT data used in this work is digitized from previous work, namely from ref. 6 (bulk LuNiO3, SmNiO3, and PrNiO3), from ref. 4 (for bulk NdNiO3 and layered NdNiO3/NdAlO3), and from ref. 5 (Ca2RuO4).
Data obtained either from new calculations or digitizing from previous work, is fit using Matlab and built into our equation of state (3), as appropriate for each material.
Data availability
The data used for this work and scripts used to fit the data can be found in the manuscripts referenced herein, both original and digitized from previous work,4,5,6, can be accessed directly on GitHub at: https://github.com/alexandrub53/EnergyLandscapes.
Code availability
The scripts used to fit the DFT+DMFT data and obtain electronic and total energies throughout the paper can be accessed directly on GitHub at: https://github.com/alexandrub53/EnergyLandscapes.
References
Imada, M., Fujimori, A. & Tokura, Y. Metal–insulator transitions. Rev. Mod. Phys. 70, 1039 (1998).
Mercy, A., Bieder, J., Iniguez, J. & Ghosez, P. Structurally triggered metal–insulator transition in rare-earth nickelates. Nat. Commun. 8, 1677 (2017).
Subedi, A., Peil, O. E. & Georges, A. Low-energy description of the metal–insulator transition in the rare-earth nickelates. Phys. Rev. B - Condens. Matter Mater. Phys. 91, 1 (2015).
Georgescu, A. B., Peil, O. E., Disa, A. S., Georges, A. & Millis, A. J. Disentangling lattice and electronic contributions to the metal–insulator transition from bulk vs. Layer confined RNiO3. Proc. Natl Acad. Sci. USA 116, 14434 (2019).
Han, Q. & Millis, A. Lattice energetics and correlation-driven metal–insulator transitions: The case of Ca2RuO4. Phys. Rev. Lett. 121, 67601 (2018).
Peil, O. E., Hampel, A., Ederer, C. & Georges, A. Mechanism and control parameters of the coupled structural and metal–insulator transition in nickelates. Phys. Rev. B 99, 245127 (2019).
Domínguez, C. et al. Length scales of interfacial coupling between metal and insulator phases in oxides. Nat. Mater. 19, 1182 (2020).
Mundet, B. et al. Near-atomic-scale mapping of electronic phases in rare earth nickelate superlattices. Nano Lett. 21, 2436 (2021).
Liao, Z. et al. Metal–insulator-transition engineering by modulation tilt-control in perovskite nickelates for room temperature optical switching. Proc. Natl Acad. Sci. USA 115, 201807457 (2018).
B. Georgescu, A. et al. Database, features, and machine learning model to identify thermally driven metal–insulator transition compounds. Chem. Mater. 33, 5591 (2021).
Wang, Y, Iyer, A., Chen, W. & Rondinelli, J. M. Featureless adaptive optimization accelerates functional electronic materials design. Appl. Phys. Rev. 7, 041403 (2020).
Fowlie, J., Georgescu, A. B., Mundet, B., del Valle, J. & Tückmantel, P. Machines for materials and materials for machines: Metal–insulator transitions and artificial intelligence. Front. Phys. 9, 1 (2021).
Disa, A. S. et al. Control of hidden ground-state order in NdNiO3 superlattices. Phys. Rev. Mater. 1, 024410 (2017).
Szymanski, N. J., Walters, L. N., Puggioni, D. & Rondinelli, J. M. Design of heteroanionic MoON exhibiting a Peierls metal–insulator transition. Phys. Rev. Lett. 123, 236402 (2019).
Schuelleret, E. C. et al. Structural signatures of the insulator-to-metal transition in BaCo1−xNix S2. Phys. Rev. Mater. 4, 104401 (2020).
Qi, Y. & Rabe, K. M. Electron-lattice coupling contributions to polarization switching in charge-order-induced ferroelectric. Preprint at http://arxiv.org/abs/2103.16466 (2021).
Catalano, S. et al. Rare-earth nickelates RNiO3: Thin films and heterostructures. Rep. Prog. Phys. 81, 046501 (2018).
Ismail-Beigi, S., Walker, F. J., Disa, A. S., Rabe, K. M. & Ahn, C. H. Picoscale materials engineering. Nat. Rev. Mater. 2, 17060 (2017).
Boris, A. V. et al. Dimensionality control of electronic phase transitions in nickel-oxide superlattices. Sci. Rep. 332, 937 (2011).
Kumah, D. P. et al. Tuning the structure of nickelates to achieve two-dimensional electron conduction. Adv. Mater. 26, 1935 (2014).
Middey, S. et al. Disentangled cooperative orderings in artificial rare-earth nickelates. Phys. Rev. Lett. 120, 156801 (2018).
Gray, A. X. et al. Insulating state of ultrathin epitaxial LaNiO3 thin films detected by hard X-ray photoemission. Phys. Rev. B - Condens. Matter Mater. Phys. 84, 1 (2011).
Stemmer, S. & Millis, A. J. Quantum confinement in oxide quantum wells. MRS Bull. 38, 1032 (2013).
Phillips, P. J. et al. Experimental verification of orbital engineering at the atomic scale: Charge transfer and symmetry breaking in nickelate heterostructures. Phys. Rev. B 95, 205131 (2017).
Kotiuga, M. & Rabe, K. M. High-density electron doping of SmNiO3 from first principles. Phys. Rev. Mater. 3, 115002 (2019).
Zhang, J. Y., Kim, H., Mikheev, E., Hauser, A. J. & Stemmer, S. Key role of lattice symmetry in the metal–insulator transition of NdNiO3 films. Sci. Rep. 6, 23652 (2016).
Shamblin, J. et al. Experimental evidence for bipolaron condensation as a mechanism for the metal–insulator transition in rare-earth nickelates. Nat. Commun. 9, 86 (2018).
Meyers, D. et al. Pure electronic metal–insulator transition at the interface of complex oxides. Sci. Rep. 6, 27934 (2016).
Först, M. et al. Multiple supersonic phase fronts launched at a complex-oxide heterointerface. Phys. Rev. Lett. 118, 027401 (2017).
Forst, M. et al. Spatially resolved ultrafast magnetic dynamics initiated at a complex oxide heterointerface. Nat. Mater. 14, 883 (2015a).
Medarde, M., Lacorre, P., Conder, K., Fauth, F. & Furrer, A. Giant 16 O–18 O isotope effect on the metal–insulator transition of rnio3 perovskites (r = rare earth). Phys. Rev. Lett. 80, 2397 (1998).
Hu, W., Catalano, S., Gibert, M., Triscone, J. M. & Cavalleri, A. Broadband terahertz spectroscopy of the insulator–metal transition driven by coherent lattice deformation at the SmNio3/LaAlO 3 interface. Phys. Rev. B 93, 161107(R) (2016).
Caviglia, A. D. et al. Photoinduced melting of magnetic order in the correlated electron insulator NdNiO3. Phys. Rev. B - Condens. Matter Mater. Phys. 88, 220401(R) (2013).
Guzmán-Verri, G. G., Brierley, R. T. & Littlewood, P. B. Cooperative elastic fluctuations provide tuning of the metal–insulator transition. Nature 576, 429 (2019).
Mandalet, B. et al. The driving force for charge ordering in rare earth nickelates. Preprint at https://arxiv.org/abs/1701.06819 (2017).
Park, H., Millis, A. J. & Marianetti, C. A. Influence of quantum confinement and strain on orbital polarization of four-layer LaNiO3 superlattices: A DFT+DMFT study. Phys. Rev. B 93, 1 (2016).
Ruppen, J. et al. Optical spectroscopy and the nature of the insulating state of rare-earth nickelates. Phys. Rev. B - Condens. Matter Mater. Phys. 92, 155145 (2015).
Peil, O. E., Ferrero, M. & Georges, A. Orbital polarization in strained LaNiO3: Structural distortions and correlation effects. Phys. Rev. B - Condens. Matter Mater. Phys. 90, 045128 (2014).
Han, M. J., Wang, X., Marianetti, C. A. & Millis, A. J. Dynamical mean-field theory of nickelate superlattices. Phys. Rev. Lett. 107, 206804 (2011).
Seth, P. et al. Renormalization of effective interactions in a negative charge transfer insulator. Phys. Rev. B 96, 205139 (2017).
Strand, H. U. R. Valence-skipping and negative- U in the d -band from repulsive local Coulomb interaction. Phys. Rev. B - Condens. Matter Mater. Phys. 90, 155108 (2014).
Blanca-Romero, A. & Pentcheva, R. Confinement-induced metal-to-insulator transition in strained LaNiO3/LaAlO3 superlattices. Phys. Rev. B - Condens. Matter Mater. Phys. 84, 195450 (2011).
Janson, O. & Held, K. Finite-temperature phase diagram of (111) nickelate bilayers. Phys. Rev. B 98, 115118 (2018).
Haule, K. & Pascut, G. L. Mott transition and magnetism in rare earth nickelates and its fingerprint on the X-ray scattering. Nat. Sci. Rep. 7, 10375 (2017).
Koohfar, S. et al. Confinement of magnetism in atomically thin La0.7Sr0.3CrO3/La0.7Sr0.3MnO3 heterostructures. npj Quantum Mater. 4, 25 (2019).
Koohfar, S. et al. Effect of strain on magnetic and orbital ordering of LaSrCrO3/LaSrMnO3 heterostructures. Phys. Rev. B 101, 064420 (2020).
Zubko, P., Gariglio, S., Gabay, M., Ghosez, P. & Triscone, J. M. Interface physics in complex oxide heterostructures. Annu. Rev. Condens. Matter Phys. 2, 141 (2011).
Mayet, S. J. et al. Control of octahedral rotations in (LaNiO3)n/(SrMnO3)m superlattices. Phys. Rev. B 83, 153411 (2011).
Lee, D. et al. Isostructural metal–insulator transition in VO2. Science 362, 1037 (2018).
Charnukha, A. et al. Coexisting first and second order electronic phase transitions in a correlated oxide. Nat. Phys. 14, 1056 (2018).
Kartoon, D., Argaman, U. & Makov, G. Driving forces behind the distortion of one-dimensional monatomic chains: Peierls theorem revisited. Phys. Rev. B 98, 165429 (2018).
Argaman, U., Kartoon, D. & Makov, G. Distorted structures in half-filled p-band materials. J. Phys. Condens. Matter 31, 465501 (2019).
Lee, S. et al. Strong orbital polarization in a cobaltate-titanate oxide heterostructure. Phys. Rev. Lett. 123, 117201 (2019).
Nelson, J. N. et al. Interfacial charge transfer and persistent metallicity of ultrathin SrIrO3/SrRuO3 heterostructures. Sci. Adv. 8, eabj0481 (2022).
Chu, J. H., Kue, H., Analytis, J. & Fisher, I. Divergent nematic susceptibility in an iron arsenide superconductor. Science 337, 710 (2012).
Klein, Y. M. et al. RENiO3 Single Crystals (RE = Nd, Sm, Gd, Dy, Y, Ho, Er, Lu) Grown from Molten Salts under 2000 bar of Oxygen Gas Pressure. Cryst. Growth Des. 21, 4230–4241 (2021).
Lacorre, P., Medarde, M., Zacchigna, M., Grioni, M. & Margaritondo, G. Electronic-structure evolution through the metal–insulator transition in (formula presented). Phys. Rev. B - Condens. Matter Mater. Phys. 60, R8426 (1999).
Cheng, J. G., Zhou, J. S., Goodenough, J. B., Alonso, J. A. & Martinez-Lope, M. J. Pressure dependence of metal–insulator transition in perovskites RNiO3 (R = Eu, Y, Lu). Phys. Rev. B - Condens. Matter Mater. Phys. 82, 1 (2010).
Chenet, B. et al. Spatially controlled octahedral rotations and metal–insulator transitions in nickelate superlattices. Nano Lett. 21, 1295 (2021).
Chen, H. et al. Modifying the electronic orbitals of nickelate heterostructures via structural distortions. Phys. Rev. Lett. 110, 1 (2013).
Park, H., Millis, A. J. & Marianetti, C. A. Site-selective Mott transition in rare-earth-element nickelates. Phys. Rev. Lett. 109, 156402 (2012).
Park, H., Millis, A. & Marianetti, C. Total energy calculations using DFT+ DMFT: Computing the pressure phase diagram of the rare earth nickelates. Phys. Rev. B 89, 245133 (2014).
Park, H., Millis, A. J. & Marianetti, C. A. Computing total energies in complex materials using charge self-consistent DFT+DMFT. Phys. Rev. B - Condens. Matter Mater. Phys. 90, 1 (2014).
Nairet, H. et al. Electronic Materials and Applications (The American Ceramic Society, 2017).
Dietl, C. et al. Tailoring the electronic properties of Ca2RuO4 via epitaxial strain. Appl. Phys. Lett. 112, 1 (2018).
Calderón, M. J., Brey, L. & Guinea, F. Surface electronic structure and magnetic properties of doped manganites. Phys. Rev. B - Condens. Matter Mater. Phys. 60, 6698 (1999).
Chattopadhyay, A. & Millis, A. Optical spectral weights and the ferromagnetic transition temperature of colossal-magnetoresistance manganites: Relevance of double exchange to real materials. Phys. Rev. B - Condens. Matter Mater. Phys. 61, 10738 (2000).
Friedt, O. et al. Structural and magnetic aspects of the metal–insulator transition in Ca2−xSrxRuO4. Phys. Rev. B 63, 1744321 (2001).
Haule, K. Exact double counting in combining the dynamical mean field theory and the density functional theory. Phys. Rev. Lett. 115, 196403 (2015).
Hao, H. et al. Metal–insulator and magnetic phase diagram of Ca2RuO4 from auxiliary field quantum Monte Carlo and dynamical mean field theory. Phys. Rev. B 101, 235110 (2020).
Karp, J., Hampel, A. & Millis, A. J. Dependence of DFT+DMFT results on the construction of the correlated orbitals. Phys. Rev. B. 103, 195101 (2021).
Anisimov, V. I., Zaanen, J. & Andersen, O. K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 44, 943 (1991).
Anisimov, V. I., Aryasetiawan, F. & Liechtenstein, A. First-principles calculations of the electronic structure and spectra of strongly correlated systems: the LDA + U method. J. Phys.: Condens. Matter 9, 767 (1997).
Deng, X., Wang, L., Dai, X. & Fang, Z. Local density approximation combined with Gutzwiller method for correlated electron systems: Formalism and applications. Phys. Rev. B - Condens. Matter Mater. Phys. 79, 075114 (2009).
Ho, K. M., Schmalian, J. & Wang, C. Z. Gutzwiller density functional theory for correlated electron systems. Phys. Rev. B - Condens. Matter Mater. Phys. 77, 073101 (2008).
Lau, B. & Millis, A. J. Theory of the magnetic and metal–insulator transitions in RNiO3 bulk and layered structures. Phys. Rev. Lett. 110, 126404 (2013).
de’ Medici, L. & Capone, M. The Iron Pnictide Superconductors (Springer, 2017)
Florens, S. & Georges, A. Slave-rotor mean-field theories of strongly correlated systems and the Mott transition in finite dimensions. Phys. Rev. B 70, 035114 (2004).
Georgescu, A. B. & Ismail-Beigi, S. Generalized slave-particle method for extended Hubbard models. Phys. Rev. B 92, 235117 (2015a).
Georgescu, A. B., Kim, M. & Ismail-Beigi, S. Boson subsidiary solver (BoSS) v1.1. Comput. Phys. Commun. 265, 107991 (2021a).
Georgescu, A. B. & Ismail-Beigi, S. Symmetry breaking in occupation number based slave-particle methods. Phys. Rev. B 96, 165135 (2017).
Georgescu, A. B. & Ismail-Beigi, S. Generalized slave-particle method for extended Hubbard models. Phys. Rev. B - Condens. Matter Mater. Phys. 92, 235117 (2015b).
Maurya, A. K. et al. Mott transition, magnetic and orbital orders in the ground state of the two-band Hubbard model using variational slave-spin mean field formalism. J. Phys.: Condens. Matter 34, 055602 (2022).
Hassan, S. R. & DeMedici, L. Slave spins away from half filling: Cluster mean-field theory of the Hubbard and extended Hubbard models. Phys. Rev. B 81, 035106 (2010).
Jarrell, M. Hubbard model in infinite dimensions: A QMC study. Phys. Rev. Lett. 69, 168 (1992).
Woodward, P., Cox, D. & Moshopoulou, E. Structural studies of charge disproportionation and magnetic order. Phys. Rev. B - Condens. Matter Mater. Phys. 62, 844 (2000).
Rogge, P. C. et al. Electronic structure of negative charge transfer CaFeO3 across the metal–insulator transition. Phys. Rev. Mater. 2, 015002 (2018).
Pavarini, E. et al. Mott transition and suppression of orbital fluctuations in orthorhombic 3d1 perovskites. Phys. Rev. Lett. 92, 176403 (2004).
Beck, S. & Ederer, C. Tailoring interfacial properties in CaVO3 thin films and heterostructures with SrTiO3 and LaAlO3: A DFT+DMFT study. Phys. Rev. Mater. 4, 125002 (2020).
Zhang, X. J., Koch, E. & Pavarini, E. Origin of orbital ordering in YTiO3 and LaTiO3. Phys. Rev. B 102, 035113 (2020).
Tomczak, J. M. & Biermann, S. Effective band structure of correlated materials: The case of VO2. J. Phys. Condens. Matter 19, 365206 (2007).
Leonov, I., Anisimov, V. I. & Vollhardt, D. Metal–insulator transition and lattice instability of paramagnetic V2O3. Phys. Rev. B - Condens. Matter Mater. Phys. 91, 1 (2015).
El Baggari, I. et al. Charge order textures induced by non-linear couplings in a half-doped manganite. Nat. Commun. 12, 1 (2021).
Uhlenbruck, S. et al. Interplay between charge order, magnetism, and structure in La0.875Sr0.125MnO3. Phys. Rev. Lett. 82, 185 (1999).
McLeod, A. S. et al. Multi-messenger nanoprobes of hidden magnetism in a strained manganite. Nat. Mater. 19, 397 (2020).
Georgescu, A. B., Millis, A. J. & Rondinelli, J. M. Trigonal symmetry breaking and its electronic effects in two-dimensional dihalides and trihalides. Preprint at https://arxiv.org/abs/2110.04665 (2021).
Guo, X. et al. Structural monoclinicity and its coupling to layered magnetism in few-layer CrI3. ACS Nano 15, 10444 (2021).
Ju, H. et al. Possible persistence of multiferroic order down to bilayer limit of van der Waals material NiI2. Nano Lett. 21, 5126 (2021).
Wang, D. & Sanyal, B. Systematic study of monolayer to trilayer CrI3: Stacking sequence dependence of electronic structure and magnetism. J. Phys. Chem. C. 125, 18467 (2021).
Santosh, K. C., Zhang, C., Hong, S., Wallace, R. M. & Cho, K. Phase stability of transition metal dichalcogenide by competing ligand field stabilization and charge density wave. 2D Mater. 2, 035019 (2015).
Hovden, R. et al. Atomic lattice disorder in charge-density-wave phases of exfoliated dichalcogenides (1T-TaS2). Proc. Natl Acad. Sci. USA 113, 11420 (2016).
Stoicaet, V. et al. Disentangling electronic and magnetic order in NdNiO3 at ultrafast timescales. Preprint at https://arxiv.org/abs/2004.03694 (2020).
Forst, M. et al. Spatially resolved ultrafast magnetic dynamics initiated at a complex oxide heterointerface. Nat. Mater. 14, 883 (2015).
Giannozzi, P. et al. Quantum ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21, 395502 (2009).
Mostofi, A. A. et al. An updated version of wannier90: A tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 185, 2309 (2014).
Parcollet, O. et al. TRIQS: A toolbox for research on interacting quantum systems. Comput. Phys. Commun. 196, 398 (2015).
Seth, P., Krivenko, I., Ferrero, M. & Parcollet, O. TRIQS/CTHYB: A continuous-time quantum Monte Carlo hybridisation expansion solver for quantum impurity problems. Comput. Phys. Commun. 200, 274 (2016).
Acknowledgements
We are indebted to Antoine Georges and Oleg Peil for inspiration, helpful comments, and advice at various stages of this work. Discussions with Jennifer Fowlie, Claribel Dominguez, Bernat Mundet Bolos, Marta Gibert, and Jean-Marc Triscone stimulated this project. We would also like to acknowledge suggestions to improve this manuscript from Lauren Walters, Sophie Beck, Alexander Hampel, Claribel Dominguez, as well as conversations with James Rondinelli, Kyle Miller, and Danilo Puggioni during the preparation of this manuscript. A.B.G. was partly supported by the Advanced Research Projects Agency-Energy (ARPA-E), U.S. Department of Energy, under Award Number DE-AR0001209. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. The Flatiron Institute is a division of the Simons Foundation.
Author information
Authors and Affiliations
Contributions
A.B.G. and A.J.M. planned the project and edited the final version of the paper, A.B.G. performed the numerical analysis and wrote the first draft of the paper.
Corresponding author
Ethics declarations
Competing interests
All authors declare no competing interests.
Peer review
Peer review information
Communications Physics thanks Olivier Delaire, Robert Green and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Georgescu, A.B., Millis, A.J. Quantifying the role of the lattice in metal–insulator phase transitions. Commun Phys 5, 135 (2022). https://doi.org/10.1038/s42005-022-00909-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-022-00909-z
This article is cited by
-
Through the slopes of a light-induced phase transition
Nature Physics (2024)
-
Picosecond volume expansion drives a later-time insulator–metal transition in a nano-textured Mott insulator
Nature Physics (2024)
-
Uncertainty-aware mixed-variable machine learning for materials design
Scientific Reports (2022)
-
Hole doping in a negative charge transfer insulator
Communications Physics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
