
Abstract
Over seventy years ago, Niels Bohr described how the charge state of an atomic ion moving through a solid changes dynamically as a result of electron capture and loss processes, eventually resulting in an equilibrium charge state. Although obvious, this process has so far eluded direct experimental observation. By peeling a solid, such as graphite, layer by layer, and studying the transmission of highly charged ions through single-, bi- and trilayer graphene, we can now observe dynamical changes in ion charge states with monolayer precision. In addition we present a first-principles approach based on the virtual photon model for interparticle energy transfer to corroborate our findings. Our model that uses a Gaussian shaped dynamic polarisability rather than a spatial delta function is a major step in providing a self-consistent description for interparticle de-excitation processes at the limit of small separations.
Similar content being viewed by others

Introduction
The simple idea of observing a charged particle while interacting with matter has captivated scientists for decades. At the beginning of the 20th century, Ernest Rutherford was the first to experimentally realise the scattering of α and β particles by matter1 and thereby paved the way for a new field of research: ion–solid interaction. The findings of these early experiments laid the foundation of today’s atomic physics in solids and studying the interaction of ions with matter has been a hot topic ever since. Not only have a number of different industrial applications developed2,3, but also essential methods for medical treatment of certain types of tumours4,5. Besides that, knowledge of ion-solid interaction is fundamental for many analysis techniques6,7 and materials studies, e.g. in plasma–wall interaction8,9 and astrophysical science10,11.
While factors like kinematics, energy deposition, material response, and the emission of secondary particles have already been studied extensively12,13,14,15,16,17,18,19, the charge state of the ion has been mostly disregarded. For ions with moderate energies, Niels Bohr considered counteracting electron capture and loss processes upon entering a sample and deduced a relation for an equilibrium charge state qeq the projectile accommodates20. The value qeq ~ v/v0 × Z1/3 (and \({q}_{{{{{\rm{eq}}}}}} \sim Z\left(1-{{{{{{{{\rm{e}}}}}}}}}^{-\frac{v}{{v}_{0}}{Z}^{-2/3}}\right)\) for higher v, respectively) is velocity dependent and refers to the Bohr velocity v0 = 2.19 × 106 m s−1, i.e. for slow projectiles v < v0 ions are neutralised when travelling through a solid and for v > v0 they are stripped. Measured equilibrium charge state distributions for ions transmitted through foils in various energy ranges may be found in literature21,22,23 and for high ion velocities, the code ETACHA provides a good description24. However, the issue of how and how fast this equilibrium charge state is reached is not yet completely resolved. This is due to the lack of possibilities of measuring ion charge states within a solid. In addition, the effect of the ion passing the surface on the way out of material was discussed as a possible source for obscuring the measured charge state with respect to the actual value in the material25.
While material damage and intentional modification are driven by the ion’s energy deposition, this energy deposition per monolayer (or per unit path length) is determined by the ion’s charge state. In fact, the electronic energy loss scales as qn, n ≈ 220,26 and the nuclear energy transfer is also affected27,28. Previous attempts to observe charge states in a solid relied on transmission through thin foils or performing backscattering spectroscopy29,30. A conclusive answer could not be given using either method because the experiment selected certain trajectories including close collisions (backscattering) or surface effects, and contamination could not be neglected25,31,32,33. Another approach was the detection of emitted X-rays from a Be target with high resolution34,35. This approach fails for slow ions which are stopped in a fraction of the radiative lifetime.
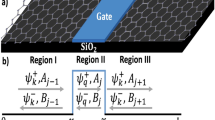
Two-dimensional (2D) materials offer the unique opportunity to disassemble a solid, e.g. graphite, layer by layer in order to solve this 100-year-old puzzle (Fig. 1). We perform transmission experiments of slow (v < v0) highly charged ions (HCIs) with single-layer graphene36 (SLG), bilayer graphene (BLG) and trilayer graphene (TLG) and study the neutralisation behaviour taking two different approaches: variation of the projectiles’ velocity for constant graphene layer numbers as well as variation of the number of graphene layers for constant velocities. In general, ions have not yet reached their equilibrium charge state after transmission through a single layer of graphene37. We derive charge state dependent characteristic velocities vn necessary for neutralisation in one layer of graphene, i.e. incident charge decay by a factor of 1/e. In either experimental approach, we find the resulting vn to be in excellent agreement with one another. This already indicates that surface effects have only a minor influence at the velocities probed.

While for a bulk material information on the ion’s charge state is unattainable after the ion enters the material, ions can be detected after transmission for atomically thin material layers. In this work, we peel graphite layer by layer and thus study the interaction of highly charged Xe ions and single-layer (SLG), bilayer (BLG) and trilayer graphene (TLG) to gain an understanding of the charge exchange dynamics of ions inside a solid. Even before reaching the material, approaching ions resonantly capture electrons from the material and a hollow atom is formed38,56,57. De-excitation of such hollow atoms in close proximity to the material layer further dominates the interaction processes. Exemplary charge states are (approximately) taken from Supplementary Information Fig. S1 and were found in experiments using 116 keV Xe30+ as a projectile. Graphene layers were rendered using the software VESTA82.
While theory has already been well-advanced in describing models for charge transfer from solids to approaching (highly charged) ions for several decades38, the subsequent processes leading to full neutralisation and de-excitation have not been understood in their entirety. In recent experiments with HCIs and two-dimensional (2D) materials37,39,40, new insights in participating de-excitation mechanisms have been found, whereupon interatomic Coulombic decay (ICD)41,42,43,44,45,46,47,48,49,50 was proposed to be the dominant mechanism in HCI neutralisation and de-excitation51. The present study is accompanied by first-principles calculations applying the virtual photon model for ICD41 on the neutralisation mechanisms of HCIs. In our present study HCIs serve as an experimental toolkit, because transmitted ions are not fully neutralised but still keep a small charge enabling easy experimental access. Our conclusions, however, do not depend on the fact that the ions are highly charged and are therefore largely universal.
Results
Ion velocity variation for a constant number of material layers
We find a clear difference between the exit charge states of HCIs transmitted through one, two or three layers of graphene (Supplementary Note 1). In Fig. 2a, the mean number of captured electrons ne for Xe30+ and various incident projectile velocities v is presented. We observe that ne is strongly dependent on the velocity of the impinging projectile.

In a, the number of captured electrons is given in dependence of the inverse projectile velocity. Exponential fits are added according to Eq. (1). To take into account a longer interaction zone due to the increasing number of material layers nL in comparison to single-layer graphene (SLG, circles), bilayer graphene (BLG, triangles) and trilayer graphene (TLG, squares) data points are shifted to v/2 and v/3 in b, respectively. The resulting universal charge exchange behaviour is presented in b for Xe30+ (red) as well as for Xe20+ (blue) and Xe40+ (orange). Error bars were calculated taking into account the influence of limitations in the ion beam setup on the evaluation of mean exit charge states and the mean distribution of exit charge states in spectra obtained using an electrostatic analyser (ESTAT), respectively.
As suggested by Bohr and Lindhard52 and applied by Brandt53, Hattass54 and Gruber37 our experimental data can be fitted by the simple exponential expression
The number of captured electrons ne is calculated as the difference of incident qin and mean exit projectile charge state \({\overline{q}}_{{{{{\rm{out}}}}}}\). Exponential decay is assumed for the interaction time, with τn being the neutralisation time constant. The interaction time t can further be expressed as
where the interaction length d is given as the product of the number of graphene layers nL and the interaction distance around each material layer dn, which we assume to be the same for each graphene layer. In Eq. (2) we have assumed that dn is smaller than the interlayer spacing of the material. A justification will follow in the discussion of the paper (Section “Discussion”). For a constant number of graphene layers, i.e., d = const. the exponent in Eq. (1) simplifies to \(-\frac{1}{v}/\frac{1}{{v}_{n,v}}\). In the case of SLG, the remaining fitting parameter vn,v then describes a charge state dependent characteristic velocity necessary for neutralisation (charge decay to qin/e) within one material layer. The second index v is used to indicate that the experimentally determined neutralisation velocity vn,v stems from a variation of projectile velocity v.
To see whether ne for ion transmission through BLG and TLG scales linearly with the increasing material thickness and thus the time spent interacting with the material, we re-scaled the x-axis for BLG and TLG in Fig. 2b by v/2 and v/3, respectively, in order to take into account the number of material layers. This combines measurements with all three samples to show a universal behaviour. Such behaviour is also found for other charge states, e.g. data for Xeq+, q = {20, 40}, have been added in Fig. 2b.
Variation of the number of material layers for constant ion velocities
As a second approach directly resulting from Eq. (2) we can also alter the number of graphene layers nL for constant ion velocity v in order to vary the interaction time of projectile and material. The SLG, BLG and TLG structures of commercially acquired samples were confirmed using scanning transmission electron microscopy (STEM). Atomic resolution images of appropriate SLG, BLG and TLG samples from Graphenea are shown in Fig. 3 together with their fast Fourier transforms as insets.

Intensities in atomic resolution high-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) images, as well as their corresponding fast Fourier transforms (inset) clearly confirm single-layer (SLG, panel a), bilayer (BLG, panel b) and trilayer (TLG, panel c) nature of Graphenea samples. Importantly, average HAADF intensity linearly scales with increasing graphene layer number. The relative layer orientation of individual layers in BLG and TLG is random (evidenced by the Moire patterns in the HAADF images and the multiple sets of six-fold spots in the fast Fourier transform). This is due to multiple sequential single-layer transfers to build up the BLG and TLG rather than multilayer growth.
The dependence of the number of captured electrons ne on the number of penetrated material layers nL is shown for various incident projectile energies and charge states in Supplementary Note 2. According to Eqs. (1) and (2) we find a characteristic neutralisation length \({n}_{{L}_{n}}\), i.e. the number of material layers necessary for neutralisation. \({n}_{{L}_{n}}\) is naturally dependent on the ion velocity v and thus is presented as a function of the projectile velocity v in Fig. 4 for various incident charge states.

The neutralisation length \({n}_{{L}_{n}}\) in number of graphene layers is extracted from fits for different projectile velocities and charge states (Xe20+: blue circles, Xe27+: pink stars, Xe30+: red squares and Xe35+: yellow triangles) as explained in Supplementary Information Fig. S2. We find that the number of layers needed for highly charged ion neutralisation depends linearly on the projectile energy. The error stems from the difference in fitting parameters considering the extreme cases of errors for the data presented in Supplementary Information Fig. S2.
We find that the number of layers \({n}_{{L}_{n}}\) necessary for neutralisation increases linearly with the projectile velocity v. As the slope of the linear relation in Fig. 4 is of dimension time per distance it represents an inverse velocity \(1/{v}_{n,{n}_{L}}\). Thus, for each charge state, we again find a characteristic neutralisation velocity \({v}_{n,{n}_{L}}\) necessary for neutralisation within one graphene layer. Here, the second part of the index signifies that this neutralisation velocity \({v}_{n,{n}_{L}}\) was derived from studying the interaction of HCIs with samples of different numbers of material layers nL at constant velocities v.
Discussion
Neutralisation dynamics of HCIs
Figure 5 shows neutralisation velocities found in the velocity (vn,v) and layer (\({v}_{n,{n}_{L}}\)) analysis. In either way, we found neutralisation velocities that are charge state-dependent and decrease linearly with increasing incident ion charge state. Our resulting data correspond well within the given uncertainties and thus we conclude that vn,v = \({v}_{n,{n}_{L}}\) = vn. The maximum ion velocity vn for which the ion neutralises during transmission of a single layer of graphene can then be described using Eq. (3) depending only on its charge state q. Coloured areas in Fig. 5 above and below this linear relation, respectively, indicate charge state/velocity combinations that will lead on average to neutral (blue) or still charged (orange) particles after transmission through SLG.

Comparison of the characteristic neutralisation velocities vn calculated from two approaches: variation of projectile velocity for one layer of material (Fig. 2, vn,v, green squares) and variation of the number of layers for specific energies (Fig. 4, \({v}_{n,{n}_{L}}\), red circles). The uncertainty stems from the difference in fitting parameters considering the extreme cases of errors for the data presented in Figs. 2 and 4, respectively. A linear fit incorporating all data points is added in blue and given in Eq. (3). Coloured areas above and below this line, respectively, indicate charge state/velocity combinations that will lead to neutral (blue) or still charged (orange) particles after transmission through a single layer of graphene.
In terms of interaction time, this means that for a higher incident charge state a longer interaction time is needed for neutralisation, i.e. the ion needs to be travelling more slowly through a single layer of graphene, or at a given velocity the material needs to be thicker.
More generally we find that velocity and thickness variation can be directly linked via the simple relation in Eq. (2), namely the interaction time. Hence, the charge exchange depends only on the time the ion spends in close proximity to the material layer: we find the same number of captured electrons ne for an ion with velocity v interacting with a single layer of graphene and an ion with 2v transmitting through BLG (Fig. 6). The interaction time scales linearly with the number of material layers (see Fig. 2 and Eq. (2)). The stopping and charge exchange of atomic particles penetrating through a material is usually described using corresponding cross-sections leaving only a velocity dependence20.

We find that the exit charge state of the ion depends only on the interaction time spent in close proximity to the material layer, i.e., the charge exchange is the same for an ion with velocity v transmitting through a single layer of graphene and an ion with velocity 2v transmitting through bilayer graphene. Graphene layers were rendered using the software VESTA82. In the zoom-out, the interlayer spacing between two graphene layers and an effective interatomic Coulombic decay (ICD) range are marked.
Recently, we used the critical distance for resonant charge transfer stemming from the classical over the barrier (COB) model38 as effective interaction length dn ~ 9 Å and an upper estimate for the interaction time of HCIs with a single layer of graphene given the incident projectile velocity v37. This distance of 9 Å is significantly larger than the interlayer spacing of multiple graphene layers and a graphite sample (~3.34 Å 55). The linear scaling of the interaction time with a number of graphene layers indicates, however, that the interaction length determining the neutralisation process dn needs to be even smaller than the graphene interlayer spacing. If this were not the case, a non-linear behaviour would be expected. This justifies the assumption made in Eq. (2), where we replaced the total interaction length with dn times the number of material layers.
When ions reach a distance to a surface smaller than or equal to the critical distance of the COB model38 the formation of a hollow atom characterised by empty inner and occupied outer (high n Rydberg) states is initiated (Fig. 1)38,56,57. Following our conclusion above we suggest that the interaction time of HCIs and materials is thus not primarily determined by the hollow atom formation but rather by the de-excitation of the same. We recently identified interatomic Coulombic decay (ICD) as a dominant process in the de-excitation of hollow atoms40,51. In general, ICD is described by a rate Γ exhibiting a strong dependence on interatomic distances R, i.e. Γ ~ 1/R6 at large interatomic separations44,58. For small R an even stronger dependence is expected59. Thus, the de-excitation of a hollow atom happens primarily in a limited area around the graphene layer with diminishing interatomic separation R of the hollow atom and material atoms.
Ab-initio model description of HCI de-excitation
In order to model the local de-excitation dynamics dominating the final charge state after passing one material layer in a truly atomistic picture, we developed an approach to treat ICD at small interatomic separations (see “Methods” section). Our theoretical considerations are based on the virtual photon model of ICD41. The resulting ICD rate Γ for five different incidents Xe charge states is given in Fig. 7a in dependence of the interatomic separation R. For the figure legend we used n = qin, which is a good approximation for the principal quantum number n into which resonant charge transfer occurs initially37,38,60. One can observe that Γ is indeed enhanced substantially at R smaller than the interlayer spacing of graphene layers/graphite.

In a, Γ is shown in dependence of the interatomic separation R. Γ(R) increases significantly at R smaller than the interlayer spacing of graphene (dashed grey line) and ultimately reaches a plateau value at the limit of small separations R. The ICD rate Γ is shown for five incident Xe charge states (Xe28+: blue, Xe29+: orange, Xe30+: red, Xe31+: green, and Xe32+: violet) assuming n ≈ qin. Plateau values are added as dashed lines in the corresponding colours and separately given for all possible incident Xe charge states in b both in eV and s−1.
One particularly noteworthy outcome of this model is the plateau value at a limit of small separations. Since Γ is in general unknown at R < 2 Å and diverging in theoretical models ~1/R6 41, its value was recently extrapolated as an ad hoc assumption based on various data found in the literature to small interatomic distances also leading to a plateau value, which is independent of the incident charge state28. By contrast, in the virtual photon model applied here plateau values for each incident charge state come out naturally and match in the order of magnitude with the rate Γ found in ref. 28. The ICD rate plateau value Γ(R → 0) decreases with decreasing incident charge state and amounts to insignificant values for low charge states compared to well-known Auger rates, e.g. for q = 1 → Γ(R → 0) ~ 102 s−1 (10−13 eV) (Fig. 7b)61. This implies that ICD becomes substantial only in the de-excitation and neutralisation of HCIs whereas for lowly charged ions as important in ion-induced Auger electron spectroscopy62 and low energy ion scattering (LEIS), which uses protons or He, common intraatomic Auger processes with rates of 1014–1015 s−1 (0.1–1 eV) dominate. The vanishing rate of ICD for lower charged ions is well in agreement with the observation of intraatomic Auger de-excitation for low-charged ions (i.e. the absence of ICD) in methods like LEIS32.
For an estimate on the effective ICD range (Fig. 6), we used the separation R at which Γ(R) has decreased to a fraction of 1/e of its plateau value. This effective interaction distance for ICD was (numerically) found to be dn ≈ 1.69 Å independent of the incident charge state.
The value of the reciprocal of the ICD rate Γ(R) corresponds to the lifetime of the de-excitation process at a given interatomic separation R. Assuming that the ion needs to spend at least this ICD lifetime within a region of ±dn around a carbon atom (marked as dashed pink line in the zoom-out in Fig. 6) we can identify a maximum ion velocity for neutralisation within one material layer. For incident ion charge states 28–32 with the corresponding Γ(R) shown in Fig. 7, we thereby find neutralisation velocities vn in the range of 0.8–2.1 nm fs−1 which agrees well with our experimentally found neutralisation velocities presented in Fig. 5. Note, that this consideration is only a rough estimation for central collisions and is not fully comparable to our experiment.
Implementation of the ab initio model in experiment simulations
In the experiment, we observe ions scattered within a detector angle of ±0.5∘ measured from the forwarding scattering direction. Thus, there is only a limited range of impact parameters that we access in our ion beam spectrometer. Wilhelm and Grande recently presented a code based on a time-dependent potential (TDPot) to directly simulate the measured interaction of HCIs and atomically thin materials28. In TDPot we define a simulation cell based on the discussed sample explicitly taking into account material geometry, i.e. possible impact parameters. The ICD rate from our presented model, however, includes only weak dependences on material properties in terms of atom radii, the photon absorption cross-section of carbon, and the Xe energy level splitting. In fact, experimentally found ICD rates from various materials also do not differ strongly28. Recent publications have successfully applied this model to carbon nanomembranes and molybdenum disulfide, respectively, using extrapolated ICD rates from experimental data40,63. A short description of the code and underlying model can be found in Supplementary Note 3; for further information, the reader is referred to the original publication28.
When introducing the ICD rate Γ (see Eq. (19) and Fig. 7) in the code TDPot we simulated the mean number of captured electrons ne for q = {28, 30, 32} and derive neutralisation velocities vn as shown for experimental data in Fig. 2. The model allows the determination of the charge exchange for individual trajectories (not only central collisions) with Γ(R) and the material structure as the only necessary input. For each charge state, we again set n = qin, as an approximation for the principal quantum number n into which resonant charge transfer occurs initially37,38,60. Simulated data points can be found in Supplementary Note 4. We again find an agreement in order of magnitude of resulting vn compared to experimental results. For example, with incident ion charge states 28–32 we find vn between 0.6 and 1.7 nm fs−1. These values are smaller than our simple estimation based on the ICD lifetime, which is reasonable since small impact parameters with high ICD rates (and large scattering angles) are not accessible in our experiment28. However, using Γ(R) from Eq. (19), we currently cannot reproduce the exact values found in the experiment. This might follow from the fact that dynamical screening (reducing the effective incoming charge) is not included in our quasi-static first-principles calculation in the dipole approximation. Please note, that TDPot specialises in the description of neutralisation dynamics of slow HCIs and reaches its limits when it comes to smaller incident charge states (where common Auger processes dominate, see Fig. 7b) and higher velocities (when electron loss cannot be neglected anymore). For further discussion and simulations covering these limits, the reader is referred to Supplementary Note 5.
Neutralisation time constants
By taking 2 × dn = 3.38 Å (from our simple estimate above) as a constant trajectory-independent estimate for the interaction distance of each material layer and going back to Eq. (2) we can also find a new estimation of the characteristic neutralisation times of HCIs. As an example, for Xe20+ to Xe40+ we find neutralisation time constants ranging from τn ≈ 0.8 fs (qin = 20) to τn ≈ 2 fs (qin = 40). These values indicate that the neutralisation process is ~40% faster than discussed by Gruber et al.37 which even strengthens the conclusion of the same work that graphene responds to a strong and localised electric field (introduced for example by an HCI) in an ultra-fast way. Being aware of the time scales of interaction processes of charged particles and material can thus help to approach material properties under extreme conditions as shown in the case of local current densities in graphene in ref. 37. Aside from material properties, material compositions are often studied using charged particles64. In ion-beam spectroscopy techniques like Rutherford backscattering spectrometry and low or medium energy ion scattering, material composition and depth profiling are obtained via consideration of collision kinematics and ion stopping. Since the electronic energy loss depends on q it is highly beneficial to understand charge exchange processes inside a material to enhance depth profiling resolution. In addition to material studies, ion neutralisation in graphene is also part of a recently presented new setup of a 229Th nuclear clock65.
While we observed the neutralisation time dependence using the particular model system of highly charged Xe on graphene, our theoretical predictions are independent of the particular choice of the ion-target combination and therefore universal, whereas they are dominant for slow HCIs and become less significant for lower charge states and/or higher velocities. Our combined experimental and theoretical effort finally bridges the gap between atomic physics of isolated atoms and molecules in the gas phase and understanding interparticle energy transfer in a solid and during a heavy particle scattering event.
Methods
Ion beam spectroscopy
The ion beam spectrometer at TU Wien is equipped with a Dresden EBIS-A electron beam ion source66 producing Xe1+ to Xe44+ ions with kinetic energies of 1–400 keV. A Wien filter is used to select specific charge states for irradiation67. Our experiments are performed in transmission geometry: projectiles are transmitted through thin materials and detected afterwards angle-resolved on a position-sensitive RoentDek delay line microchannel plate (MCP) detector68. Determination of exit charge states can be done by analysing the deflection of particles after transmission of the sample in a pair of deflection plates. In combination with the MCP signal, an electron detector close to the target position69 allows us to measure the time of flight (TOF) of projectiles, recorded in coincidence with exit charge states in a list mode. For more details on the spectrometer please see70.
Samples of SLG, BLG and TLG studied in this work were commercially acquired from Graphenea71. Freestanding sample areas are found on holes with 2 μm diameter on a 10–20 nm thick Quantifoil support both placed on Au TEM grids. The materials are grown by chemical vapour deposition and transferred (multiple times) onto the TEM grid applying a polymer-based transfer technique. We heat samples in situ using a Lasertack laser diode (6 W, 445 nm)72 and Ohmic heating (400 °C) to remove residuals from production and transfer processes as well as contaminants adventitiously adsorbed while sample handling in ambient conditions according to the procedure described in73,74. Filtering exit charge states for their TOFs discriminates signals from both the Quantifoil support and still contaminated areas. To prevent re-adsorption of contaminants during measurements at a base pressure of 5 × 10−9 mbar, we keep samples at 180 °C.
Electron microscopy
STEM was done in a Nion UltraSTEM 100 at 60 kV. Images were taken via a high-angle annular dark-field detector.
Theoretical model
The theory underpinning the experiment is based on the virtual photon model of ICD41, which was later generalised to arbitrary environments in58. The main result was the following expression for the ICD rate Γ
where γD is the spontaneous decay rate of the donor transition with frequency ωD, σA(E) is the photoionisation cross-section of the acceptor at energy ED = ℏωD, and \({\mathsf{G}}({{{{{{{\bf{r}}}}}}}},{{{{{{{\bf{r}}}}}}}}^{\prime} ,\omega )\) is the dyadic Green’s tensor describing the propagation of electromagnetic excitations of frequency ω from point \({{{{{{{\bf{r}}}}}}}}^{\prime}\) to r.
In its original41 and generalised58 forms, the virtual photon model applies only when donor and acceptor are sufficiently far apart that there is no appreciable wave function overlap, but this is not the situation found in the experiment described here. In principle, one should use a full ab initio electronic structure calculation, but for highly charged ions in asymmetric environments, this is computationally infeasible. In order to remedy this, we adopt a technique from the theory of the van der Waals forces (recently applied to Auger decay75), whereby the dynamic polarisability α(ω) of donor and acceptor are taken to be ‘smeared out’ over a finite region (see, e.g.76), instead of being taken as a spatial delta function as is implicitly done in the virtual photon model. In particular, we take
where a is a characteristic length, roughly corresponding to the spatial extent of the atomic wave function. As discussed in detail in ref. 77, this leads to a modified Green’s tensor which, upon substitution into Eq. (4) leads to
with
where R is the interatomic separation, and the parameters aD and aA describe the spatial extents of the Xe donor and C acceptor, respectively. At large separations R (or, equivalently, small radii aD and aA), equations (6) and (7) together reproduce the well-known point-like rate
At small separations, the rate becomes
in contrast to the point-like rate, which diverges at small separations.
We now need to choose some reasonable parameters for the various radii, the energy level splitting of the Xe donor, and the cross-section of the C acceptor. The Xe donor is in a high Rydberg state of principal quantum number n, so we take aD = a0n2/Z ≈ 9 Å and energy level splittings of
where m and k are the principal quantum numbers of the two states at hand, and ERyd is the Rydberg energy. For the carbon acceptor, we use the van der Waals radius of 1.7Å, and the tabulated photoionisation cross-sections found in refs. 78,79,80.
The process of decay from a multiply-occupied high Rydberg state can be complex, consisting of a cascade decay through different paths. In order to take this into account, we use the following rate equation for the population Pk of level k
where γk→m is the decay rate connecting level k to level m. The expected (virtual) photon population Fk for a particular frequency k after a given cascade can then be found from
The decay rates we use are those for hydrogen-like atoms, scaled by the appropriate factor Z4 for nuclear charge Z. We, therefore, use the following as a weighting factor in the expression for the ICD rate
where \({\gamma }_{k\to m}^{{{{{{{{\rm{H}}}}}}}}}\) are the hydrogen-like decay rates81, so that the decay rate from a particular level n is
To a good approximation, the values of the weighting g(k, m) calculated from the from level n turn out to be equal at the 0.1% level to the those that would be found by taking the transition n → n − 1, so that we may simplify by taking
where C(n) is a fitted function of n found from the numerical solution of the rate equations
The relevant hydrogen-like rates are well-approximated by
with A = 4.86 × 10−6 eV. As shown in Supplementary Note 6, the photoionisation cross-section of carbon is well-approximated (in the relevant range of energies) by
where σ0 = 0.115 Å2. Combining Eqs. (14), (15) and (18), we get
Using the various parameters discussed in this section, at the limit of small separations R this can be simplified to
As shown in Fig. 7, this agrees with the full position-dependence found by evaluating Eq. (19).
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Rutherford, E. LXXIX. The scattering of α and β particles by matter and the structure of the atom. Lond. Edinb. Dublin Philos. Mag. J. Sci. 21, 669–688 (1911).
Chason, E. et al. Ion beams in silicon processing and characterization. J. Appl. Phys. 81, 6513–6561 (1997).
Stevie, F. A. et al. Applications of focused ion beams in microelectronics production, design and development. Surf. Interface Anal. 23, 61–68 (1995).
Chu, W. T., Ludewigt, B. A. & Renner, T. R. Instrumentation for treatment of cancer using proton and light-ion beams. Rev. Sci. Instrum. 64, 2055–2122 (1993).
Schulz-Ertner, D. & Tsujii, H. Particle radiation therapy using proton and heavier ion beams. J. Clin. Oncol. 25, 953–964 (2007).
Perrière, J. Rutherford backscattering spectrometry. Vacuum 37, 429–432 (1987).
Arnold Bik, W. M. & Habraken, F. H. P. M. Elastic recoil detection. Rep. Prog. Phys. 56, 859–902 (1993).
Schmid, K. et al. Interaction of nitrogen plasmas with tungsten. Nucl. Fusion 50, 025006 (2010).
Stadlmayr, R. et al. Erosion of iron-tungsten model films by deuterium ion irradiation: a benchmark for TRI3DYN. Phys. Scr. T171, 014021 (2020).
Szabo, P. S. et al. Solar wind sputtering of wollastonite as a lunar analogue material—comparisons between experiments and simulations. Icarus 314, 98–105 (2018).
Nénon, Q. et al. Phobos surface sputtering as inferred from MAVEN ion observations. J. Geophys. Res. Planets 124, 3385–3401 (2019).
Honig, R. E. Sputtering of surfaces by positive ion beams of low energy. J. Appl. Phys. 29, 549–555 (1958).
Facsko, S. et al. Formation of ordered nanoscale semiconductor dots by ion sputtering. Science 285, 1551–1553 (1999).
Facsko, S. et al. Ion-induced formation of regular nanostructures on amorphous GaSb surfaces. Appl. Phys. Lett. 80, 130–132 (2002).
Aumayr, F. & Winter, H. Potential electron emission from metal and insulator surfaces. in Slow Heavy-Particle Induced Electron Emission from Solid Surfaces, 79–112 (Springer Berlin Heidelberg, Berlin, Heidelberg, 2007).
Winter, H. Kinetic electron emission for grazing scattering of atoms and ions from surfaces. in Slow Heavy-Particle Induced Electron Emission from Solid Surfaces, 113–151 (Springer Berlin Heidelberg, Berlin, Heidelberg, 2007).
Gomes, D. R., Turkin, A. A., Vainchtein, D. I. & De Hosson, J. T. On the mechanism of ion-induced bending of nanostructures. Appl. Surf. Sci. 446, 151–159 (2018).
De Zwart, S. T. et al. Sputtering of silicon by multiply charged ions. Surf. Sci. 177, L939–L946 (1986).
Lohmann, S. & Primetzhofer, D. Disparate energy scaling of trajectory-dependent electronic excitations for slow protons and He ions. Phys. Rev. Lett. 124, 096601 (2020).
Bohr, N. The Penetration of Atomic Particles Through Matter (Munksgaard, Copenhagen, 1948).
Armstrong, J. C., Mullendore, J. V., Harris, W. R. & Marion, J. B. Equilibrium charge-state fractions of 0.2 to 6.5 MeV helium ions in carbon. Proc. Phys. Soc. 86, 1283–1295 (1965).
Shima, K., Ishihara, T., Miyoshi, T. & Mikumo, T. Equilibrium charge-state distributions of 35–146-MeV Cu ions behind carbon foils. Phys. Rev. A 28, 2162–2168 (1983).
Betz, H. Charge states and charge-changing cross sections of fast heavy ions penetrating through gaseous and solid media. Rev. Mod. Phys. 44, 465–539 (1972).
Rozet, J. P., Stéphan, C. & Vernhet, D. ETACHA: a program for calculating charge states at GANIL energies. Nucl. Instrum. Methods Phys. Res. B 107, 67–70 (1996).
Primetzhofer, D., Spitz, M., Taglauer, E. & Bauer, P. Resonant charge transfer in low-energy ion scattering: information depth in the reionization regime. Surf. Sci. 605, 1913–1917 (2011).
Schiwietz, G. & Grande, P. L. Stopping of protons-improved accuracy of the uca model. Nucl. Instrum. Methods Phys. Res. Sect. B 273, 1–5 (2012).
Biersack, J. P. The effect of high charge states on the stopping and ranges of ions in solids. Nucl. Instrum. Methods Phys. Res. B 80-81, 12–15 (1993).
Wilhelm, R. A. & Grande, P. L. Unraveling energy loss processes of low energy heavy ions in 2D materials. Commun. Phys. 2, 89 (2019).
Jamecsny, S. & Carstanjen, H. D. Depth dependent charge state distributions of heavy MeV ions in RBS and ERD experiments. Nucl. Instrum. Methods Phys. Res. B 125, 128–132 (1997).
Niehus, H., Heiland, W. & Taglauer, E. Low-energy ion scattering at surfaces. Surf. Sci. Rep. 17, 213–303 (1993).
Goldberg, E. C., Monreal, R., Flores, F., Brongersma, H. H. & Bauer, P. New model for ion neutralization at surfaces. Surf. Sci. 440, L875–L880 (1999).
Wang, N. P. et al. Low-energy ion neutralization at surfaces: resonant and Auger processes. Phys. Rev. A 64, 012901 (2001).
Draxler, M., Gruber, R., Brongersma, H. H. & Bauer, P. Velocity scaling of ion neutralization in low energy ion scattering. Phys. Rev. Lett. 89, 263201 (2002).
Zhao, Y. et al. X-ray spectroscopy of hollow argon atoms formed on a beryllium surface. Nucl. Instrum. Methods Phys. Res., B 245, 72–75 (2006).
Zhao, Y. et al. X-ray emission of hollow atoms formed by highly charged argon and xenon ions below a beryllium surface. Nucl. Instrum. Methods Phys. Res. B 258, 121–124 (2007).
Novoselov, K. S. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Gruber, E. et al. Ultrafast electronic response of graphene to a strong and localized electric field. Nat. Commun. 7, 13948 (2016).
Burgdörfer, J., Lerner, P. & Meyer, F. W. Above-surface neutralization of highly charged ions: the classical over-the-barrier model. Phys. Rev. A 44, 5674–5685 (1991).
Schwestka, J. et al. Charge-exchange-driven low-energy electron splash induced by heavy ion impact on condensed matter. J. Phys. Chem. Lett. 2019, 4805–4811 (2019).
Creutzburg, S. et al. Vanishing influence of the band gap on the charge exchange of slow highly charged ions in freestanding single-layer MoS2. Phys. Rev. B 102, 045408 (2020).
Averbukh, V., Müller, I. B. & Cederbaum, L. S. Mechanism of interatomic coulombic decay in clusters. Phys. Rev. Lett. 93, 263002 (2004).
Jahnke, T. et al. Experimental observation of interatomic Coulombic decay in neon dimers. Phys. Rev. Lett. 93, 163401 (2004).
Marburger, S., Kugeler, O., Hergenhahn, U. & Möller, T. Experimental evidence for interatomic Coulombic decay in Ne clusters. Phys. Rev. Lett. 90, 203401 (2003).
Cederbaum, L. S., Zobeley, J. & Tarantelli, F. Giant intermolecular decay and fragmentation of clusters. Phys. Rev. Lett. 79, 4778–4781 (1997).
Bande, A., Gokhberg, K. & Cederbaum, L. S. Dynamics of interatomic Coulombic decay in quantum dots. J. Chem. Phys. 135, 144112 (2011).
Trinter, F. et al. Evolution of interatomic Coulombic decay in the time domain. Phys. Rev. Lett. 111, 093401 (2013).
Trinter, F. et al. Resonant Auger decay driving intermolecular Coulombic decay in molecular dimers. Nature 505, 664–666 (2014).
Haller, A., Peláez, D. & Bande, A. Inter-Coulombic decay in laterally arranged quantum dots controlled by polarized lasers. J. Phys. Chem. C. 123, 14754–14765 (2019).
Kim, H.-K. et al. Ion-impact-induced interatomic Coulombic decay in neon and argon dimers. Phys. Rev. A 88, 042707 (2013).
Iskandar, W. et al. Interatomic Coulombic decay as a new source of low energy electrons in slow ion-dimer collisions. Phys. Rev. Lett. 114, 033201 (2015).
Wilhelm, R. A. et al. Interatomic Coulombic decay: the mechanism for rapid deexcitation of hollow atoms. Phys. Rev. Lett. 119, 103401 (2017).
Bohr, N. & Lindhard, J. Electron Capture and Loss by Heavy Ions Penetrating Through Matter (I kommission hos Munksgaard, 1954).
Brandt, W., Laubert, R., Mourino, M. & Schwarzschild, A. Dynamic screening of projectile charges in solids measured by target x-ray emission. Phys. Rev. Lett. 30, 358–361 (1973).
Hattass, M. et al. Charge equilibration time of slow, highly charged ions in solids. Phys. Rev. Lett. 82, 4795–4798 (1999).
Guo, Y., Guo, W. & Chen, C. Tuning field-induced energy gap of bilayer graphene via interlayer spacing. Appl. Phys. Lett. 92, 243101 (2008).
Winter, H. & Aumayr, F. Hollow atoms. J. Phys. B 32, R39–R65 (1999).
Arnau, A. et al. Interaction of slow multicharged ions with solid surfaces. Surf. Sci. Rep. 27, 113–239 (1997).
Hemmerich, J. L., Bennett, R. & Buhmann, S. Y. The influence of retardation and dielectric environments on interatomic Coulombic decay. Nat. Commun. 9, 2934 (2018).
Averbukh, V. & Cederbaum, L. S. Interatomic electronic decay in endohedral fullerenes. Phys. Rev. Lett. 96, 053401 (2006).
Tőkési, K., Wirtz, L., Lemell, C. & Burgdörfer, J. Hollow-ion formation in microcapillaries. Phys. Rev. A 64, 042902 (2001).
Goebl, D., Monreal, R. C., Valdés, D., Primetzhofer, D. & Bauer, P. Calculation of Auger-neutralization probabilities for He+-ions in LEIS. Nucl. Instrum. Methods Phys. Res. B 269, 1296–1299 (2011).
Valeri, S. Auger electron emission by ion impact on solid surfaces. Surf. Sci. Rep. 17, 85–150 (1993).
Wilhelm, R. A. On the highly charged ion transmission spectroscopy applied to 2d materials. in Journal of Physics: Conference Series 6, 062010 (IOP Publishing, 2020).
Nastasi, M., Mayer, J. W. & Wang, Y. Ion Beam Analysis: Fundamentals and Applications (CRC Press, 2014).
Seiferle, B. et al. Energy of the 229Th nuclear clock transition. Nature 573, 243–246 (2019).
Zschornack, G. et al. Compact electron beam ion sources/traps: review and prospects (invited). Rev. Sci. Instrum. 79, 02A703 (2008).
Schmidt, M., Peng, H., Zschornack, G. & Sykora, S. A compact electron beam ion source with integrated Wien filter providing mass and charge state separated beams of highly charged ions. Rev. Sci. Instrum. 80, 063301 (2009).
RoentDek Handels GmbH. CoboldPC Software. http://www.roentdek.com/ (2020).
Lemell, C., Stöckl, J., Winter, H. & Aumayr, F. A versatile electron detector for studies on ion-surface scattering. Rev. Sci. Instrum. 70, 1653–1657 (1999).
Schwestka, J. et al. A versatile ion beam spectrometer for studies of ion interaction with 2D materials. Rev. Sci. Instrum. 89, 085101 (2018).
Graphenea. Graphene on TEM grids. https://www.graphenea.com (2020).
Lasertack GmbH. Lasertack—New Laser Generation. NUBM44. https://www.lasertack.com/6w-445nm-laserdiodenmodul-nubm44 (2020).
Tripathi, M. et al. Cleaning graphene: comparing heat treatments in air and in vacuum. Phys. Status Solidi RRL 11, 1700124 (2017).
Niggas, A. et al. The role of contaminations in ion beam spectroscopy with freestanding 2D materials: a study on thermal treatment. J. Chem. Phys. 153, 014702 (2020).
Franz, J., Bennett, R. & Buhmann, S. Y. Auger decay in dispersing and absorbing environments. Phys. Rev. A 104, 013103 (2021).
Mahanty, J. & Ninham, B. W. Dispersion contributions to surface energy. J. Chem. Phys. 59, 6157–6162 (1973).
Thiyam, P. et al. Intermolecular Casimir-Polder forces in water and near surfaces. Phys. Rev. E 90, 032122 (2014).
Henry, R. J. W. Photoionization cross-sections for atoms and ions of carbon, nitrogen, oxygen, and neon. Astrophys. J. 161, 1153–1155 (1970).
Moore, C. Ionization Potentials and Ionization Limits Derived from the Analyses of Optical Spectra (Office of Standard Reference Data, National Bureau of Standards, 1970).
Barfield, W. D. & Huebner, W. F. On the calculation of scattering cross sections from absorption cross sections. J. Quant. Spectrosc. Radiat. Transf. 16, 27–34 (1976).
Kramida, A., Ralchenko, Y., Reader, J. & NIST ASD Team. NIST Atomic Spectra Database (version 5.8). https://physics.nist.gov/asd [November 11, 2020] (2020).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
The authors acknowledge funding from the Austrian Science Fund (FWF): Y 1174-N36, I 4914-N. A.N., B.C.B and R.A.W. acknowledge funding from TU Wien’s competitive Innovative Projects programme. J.S. appreciates funding from the Doctoral College TU-D.
Author information
Authors and Affiliations
Contributions
A.N., J.S., R.A.W. and F.A. designed the research, A.N, S.C., J.S. and B.W. performed ion beam spectroscopy measurements, T.G., D.E. and B.C.B. conducted electron microscopy analysis of used materials. P.L.G. and R.A.W. developed the simulation code used which was applied by A.N. J.P.M. provided radiative absorption cross-sections for comparison. R.B. developed the first-principles model presented. A.N. and R.B. wrote the paper. All authors commented.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Physics thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Niggas, A., Creutzburg, S., Schwestka, J. et al. Peeling graphite layer by layer reveals the charge exchange dynamics of ions inside a solid. Commun Phys 4, 180 (2021). https://doi.org/10.1038/s42005-021-00686-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-021-00686-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
