
Abstract
The study of ultrafast dynamics is a new tool to understand and control the properties of correlated oxides. By enhancing some properties and realizing new dynamically excited phrases, this tool has opened new routes for technological applications. LaCoO3 is one paradigmatic example where the strong electron, spin, and lattice coupling induced by electronic correlations results in a low-temperature spin transition and a high-temperature semiconductor-to-metal transition that is still not completely understood. Here, we monitor ultrafast metallization in LaCoO3 using time-resolved soft x-ray reflectivity experiments. While the process is entangled at the Co L3 edge, the time information of the different channels is decrypted at different resonant energies of the O K edge. Metallization is shown to occur via transient electronic, spin, and lattice separation. Our results agree with the thermodynamical model and demonstrate the potential of femtosecond soft x-ray experiments at the O K edge to understand correlated oxides.
Similar content being viewed by others


Introduction
The phase diagram of LaCoO3 (LCO) as a function of temperature with its low-temperature (~100 K) spin transition (ST) and a high-temperature (300–600 K) broad semiconductor-to-metal transition (SMT) has been derived over the years through detailed analysis of magnetic susceptibility, resistivity, atomic structure, phonon spectrum, and heat capacity experiments1,2,3,4,5,6,7,8,9,10,11,12. The complex behavior of the system with temperature is thought to be the consequence of the strong interplay between charge, spin, and lattice degrees of freedom. The change in spin required to explain the magnetic susceptibility anomalies13 is accompanied by an increase of the ionic radius8, which, in turn, induces an anomalous lattice expansion responsible for the SMT12. Although the spin state has been considered the driving mechanism responsible for all changes in the system with temperature6,12,13,14,15, the ultimate understanding of the system has remained elusive due to the lack of consensus regarding the strength of electronic correlations. As a result, a longstanding debate has split the community around two models. The first, derived empirically by Goodenough13 from structural and transport measurements, proposed a view based on an ionic picture of the Co d6 atom in an octahedral oxygen environment, where the t2g and eg orbitals are split by the crystal field Δ. The ST and the SMT were interpreted as a gradual temperature transition from a low spin (LS) \(\left[ {t_{2g}^6,S = 0} \right]\) to a high spin (HS) \(\left[ {t_{2g}^4 e_g^2,S = 2} \right]\) and from an HS to an intermediate spin state (IS) \(\left[ {t_{2g}^5 e_g^1,S = 1} \right]\) state, respectively. The increase of the ionic radius from rLS = 0.545 Å to rHS = 0.610 Å8 would explain the anomalous lattice expansion below room temperature (ST) and the increase of the eg–O2p overlap inducing metallization above room temperature (SMT). Theory refinements to the ionic picture included the trigonal distortion of the R\(\bar 3\)c structure and the spin–orbit coupling. The resulting energy diagram has the first two states around the Fermi energy (EF) separated from each other by 11 meV (corresponds to 100 K), the ground state being non-magnetic, while the first excited state has a moment of m = 2.32 μB12.
The second model emerged theoretically in an attempt to include electronic correlations and Co–O hybridization within the density function theory (DFT) with large on-site Coulomb interaction, U, DFT+U formalism. Using U = 7.8 eV, the IS state appears at lower energy than the HS state, thus explaining the ST as a LS–IS one14. According to this model, the system would also exhibit orbital ordering to prevent the metallic ground state as a consequence of the large U value, as shown by local spin density approximation (LSDA)+U calculations16. A semiconducting ground state with a gap close to the experimental one requires lower U values, U ~ 3 eV or U ~ 6 eV16. Following this route, combined cluster calculations with U = 5.5 eV and experiments seem to have balanced the discussion about the ST in favor of the LS–HS crossover with a gradual increase of the HS population with temperature15. This result has been supported by advanced DFT including correlations within the dynamical mean-field theory (DMFT), DFT+DMFT17,18,19. Furthermore, dynamical charge fluctuations favor short-range order at room temperature in a configuration where the two cobalt (Co) atoms in the unit cell connected by an oxygen (O) atom are in a different spin state, Co(LS)–O–Co(HS)19. This configuration was shown to be favored by the charge imbalance that spontaneously appears in the system when the constraint of equal charge between the two Co atoms in a unit cell is relaxed in calculations with moderate U ~ 3 eV20. The SMT is explained naturally as a transition from the room temperature Co(LS)–O–Co(HS) state to the HS metallic state at high temperature. (Recent resonant inelastic x-ray experiments and theoretical calculations have proposed a new scenario in which mobile IS excitons propagating in a LS background contribute to the low-temperature physics of the system, see refs. 21,22.) As an attempt to verify the role of charge and spin fluctuations in the properties of LCO, soft x-ray time-resolved spectroscopic studies with picosecond resolution across the SMT were performed23. The high-temperature phase transition was chosen due to its technological relevance in numerous environmentally friendly energy production domains24,25,26,27,28,29,30,31. Laser excitation with 800 nm photons allowed the realization of a transient metallic state with Co charge disproportionation and a higher spin state than the thermally stabilized one. This observation was deduced by comparing the evolution with temperature and time delay of soft x-ray reflectivity measurements at the O K and Co L edges23.
In this contribution, the SMT in LCO is addressed by means of time-resolved optical pump—soft x-ray probe experiments with femtosecond resolution and DFT+DMFT calculations of the optical reflectivity. Calculations of the measured time-resolved soft x-ray spectra including electronic correlations and lattice dynamics are still not theoretically available. They would require a multi-orbital Schwinger–Keldysh-formalism for interacting electron systems, while only model single-band attempts exists32. There are attempts to calculate realistic dynamical core–hole screening in the x-ray absorption spectra, but only in non-correlated DFT approach33. Since LCO is definitely correlated system, we did a best possible theoretical DFT+DMFT analysis trying to understand the gap collapse, the charge disproportionation, enhancement of the spin and lattice deformations upon laser excitation. The combined study has allowed the understanding of the properties of the laser-induced transient metallic state disentangling the interplay between the electronic, spin, and lattice degrees of freedom upon metallization. The 800 nm optical laser excitation depopulates electrons from the pseudo-t2g orbitals into the pseudo-eg ones (see Methods). As a consequence, charge transfer between the pseudo-eg and the O-2p orbitals is enhanced and the HS state population increases. The larger ionic radius of the HS state induces a lattice distortion with a deformation of the bond angles and a change of interatomic distances, explaining the metallic character of LCO at high temperature. The emerging picture presents the transient metallization as a four-step process in a way similar to the ionic model proposed by Goodenough6 to explain the evolution of LCO with temperature. Thus, our studies become the first experimental demonstration of Goodenough metallization model6,13. Insight into the electronic properties of the laser-excited transient metallic state and the optical excitation mechanism have been derived from DFT+DMFT calculations of the optical conducitivity.
Results
Femtosecond time-resolved reflectivity
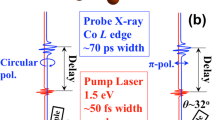
Optical pump—soft x-ray probe reflectivity experiments with femtosecond time resolution were performed at room temperature using the FemtoSpeX-scattering experimental station at the storage ring-based fs-slicing source FemtoSpeX of the BESSY II synchrotron at Helmholtz Zemtrum Berlin (HZB). The two beamlines, the PGM beamline (E/ΔE > 5000) and the ZPM beamline (E/ΔE ~ 500) of the FemtoSpeX facility, have been used to record laser-induced changes at the O K and the Co L3 edges with 50 ps and 120 fs time resolution, respectively. Despite the reduced energy resolution of the ZPM beamline, all spectral features seen at the PGM beamline can clearly be assigned to the ZPM spectra (see Supplementary Fig. 1). The dynamics of LCO was characterized by measuring time-delay scans at the photon energies resonant with the relevant spectroscopic signatures of the SMT transition12,15,23. At the O K edge, we have focused on the resonant energies probing oxygen hybridized with different Co orbitals, namely the pseudo-t2g orbitals, the pseudo-eg orbitals, and the Co-4sp. From the measured spectra, the sum O–Co-pseudo-eg + O–Co-pseudo-t2g, and difference O–Co-pseudo−eg–O–Co-pseudo-t2g, were calculated to understand the underlying physics in the system. The spectra are depicted in Fig. 1: O–Co-pseudo-t2g (Fig. 1a), O–Co-pseudo-eg (Fig. 1b), sum spectra (Fig. 1c), O-Co-4sp (Fig. 1d), and difference spectra (Fig. 1e). At the Co L3 edge, both the white line (WL) and its high-energy shoulder (HSS) ascribed to the HS state population change (Co-HS) have been investigated (Fig. 1f). The data show intensity variations taking place at different time delays, depending on the different resonant energies. The effect can be clearly seen when plotting all the measured data together (see Supplementary Fig. 2). This observation provides clear evidence that the relaxation of the excited states occurs at different time scales for different resonant energies.

Delay scans measured at room temperature at different resonant energies of the O K edge. a O–Co-pseudo-t2g. b O–Co-pseudo-eg. c O–Co-pseudo-eg+O–Co-pseudo-t2g sum spectrum showing compensation of population transfer below ~0.5 ps. At larger time scales, the difference signal increases due to the change in population of the O–Co-pseudo-eg orbitals, a consequence of the charge transfer enhancement. d Delay scan at the O–Co-4sp resonant energy. e O–Co-pseudo-eg–O–Co-pseudo-t2g difference spectrum together with the delay scan at the Co L3 white line (WL). The agreement between the two spectra is quite remarkable. f Delay scan at the Co L3 WL and its high spin (HS) shoulder (HSS). The experimental data are displayed as markers, the fit function as solid lines, and the different components as filled curves
Time constants
To determine the different time scales observed at different resonant energies, a fitting to all the delay scans in Fig. 1 was done. In the first step, a modified three-temperature model (3TM), previously applied to isostructural manganites34, was used. However, the χ2 value of the fits could be improved by a factor of three by allowing the exponential relaxation of a given component to be delayed by the time constant of the previous one. This cascade decay implies that each relaxation channel becomes the source of the next (see Methods). A fit to all the delay scans is possible with five time constants. For some delay scans, only two components were necessary. The time constants were obtained from a self-consistent fit of all delay scans. The fifth time constant [τ5] was used to account for the slow relaxation of the system observed in the picosecond experiments23. The time constant values obtained from the self-consistent fit are τ1 = 0.17 ps, τ2 = 0.52 ps, τ3 = 0.91 ps, τ4 = 1.5 ps, and τ5 = 45 ps. The fit laser pulse duration was 0.125 ps, which matches the 0.13 ps laser pulse duration measured experimentally. The final fit curves are displayed in Fig. 1a–f as solid lines, whereas the individual components are displayed as filled curves. The characteristic time scales contributing to each spectra are indicated in the inset labels.
The delay scan at the O K edge in the O–Co-pseudo-t2g region (Fig. 1a) can be fitted with the time constants τ1 = 0.17 ps, τ2 = 0.52 ps, and τ4 = 1.5 ps. The first two components have the dominating intensity corresponding to the sub-picosecond intensity peak. This first increase of the reflectivity corresponds to the initial depopulation of the O–Co-pseudo-t2g orbitals upon laser excitation. It is followed by a decay, which stabilizes around τ4 = 1.5 ps. The best fit of the time delay scan at the O–Co-pseudo-eg resonant energy (Fig. 1b) was obtained with the ultrafast components τ1 = 0.17 ps, τ3 = 0.91 ps, and τ4 = 1.50 ps and the slow relaxation channel, τ5 = 45 ps. The shortest time delay fits the first drop of the reflectivity and corresponds to the population of the O–Co-pseudo-eg orbitals. It is the counterpart of the depopulation of the O–Co-pseudo-t2g orbitals observed in Fig. 1a. After a very weak recovery, a second broad minimum at around τ4 = 1.50 ps can be observed. It is much more pronounced than in the delay scan measured in the O–Co-pseudo-t2g region. The fact that the initial reflectivity value does not recover after 3 ps is due to the contribution of the slow relaxation channel, τ5 = 45 ps, obtained from picosecond studies23. The reflectivity changes at the shortest time scale, τ1, indicate a perfect redistribution of the population between the O–Co-pseudo-t2g and O–Co-pseudo-eg orbitals, as required to increase the HS of the system. At larger time scales, the reflectivity changes at the two delay scans no longer compensate. The need for different time constants and intensities to fit each scan indicates that other channels besides electron redistribution contribute to the changes in reflectivity at these resonant energies. Indeed, the [O–Co-pseudo-eg+O–Co-pseudo-t2g] sum spectra (Fig. 1c) shows that the intensity cancels out for time delays below ~0.5 ps. From larger time delays, the intensity continues decreasing until it reaches a minimum around the time delay constant, τ4 = 1.50 ps, followed by a slow recovery. A good fit of the sum spectra can be done with only τ4 = 1.50 ps and τ5 = 45 ps components. The same time constants are required to fit the delay scan measured at the O–Co–4sp resonant energy (Fig. 1d). As in the sum delay spectrum, the ultrafast excitation below 0.20 ps is hardly visible. The fact that changes at the O–Co–4sp resonant energy are related to modifications in bond length and angles12,23 indicates that the time delay τ4 is related to structural lattice modifications.
In Fig. 1e, the [O–Co-pseudo-eg—O–Co-pseudo-t2g] difference delay scan is displayed. The intensity changes as a function of time delay reproduce quite well those measured at the Co L3 WL and HSS resonant energies (Fig. 1f). Indeed, the same time constants τ1 = 0.17 ps, τ3 = 0.91 ps, τ4 = 1.5 ps, and τ5 = 45 ps with the same intensities are able to fit all delay scans. The first drop at short time delays [τ1 = 0.17 ps] can be related to bleaching resulting from ultrafast carrier excitation35. Afterwards, a second broad minimum with almost the same intensity reduction is observed around the time delay τ3 = 0.91 ps. The intensity of this time constant is much larger than that observed at the O–Co-pseudo-eg, indicating a relation with the increase of the HS population in the system. From there on, the reflectivity recovers slowly to \(\frac{1}{3}\) of the initial drop after 2.5 ps. To fit this part of the reflectivity, components with time delays τ4 = 1.5 ps and τ5 = 45 ps are required. The evolution at short time delays is in agreement with recent results obtained for ferromagnetic LCO thin films36. Moreover, the saturation-like behavior after three picoseconds reproduces the results measured with picosecond resolution23. The decrease of intensity with time delay can be explained by the increase of the HS population upon laser excitation that is enhanced by charge disproportionation as deduced previously23.
The similarity of the [O–Co-pseudo-eg − O–Co-pseudo-t2g] difference delay scan with the delay scans measured at the Co L3 edge and that of the [O–Co-pseudo-eg + O–Co-pseudo-t2g] difference delay scan with the delays scan at the O–Co–4sp resonant energy can be understood by assuming that the scans at the O–Co-pseudo-t2g and at the O–Co-pseudo-eg can be decomposed in five contributions: orbital electron transfer [OT], change transfer enhancement [CE], spin stabilization [SS], lattice modification [LM], and slow relaxation [SR]. Assuming O–Co-pseudo-t2g ∝ [OT + CE − SS + LM] and O–Co-pseudo-eg ∝ [−OT + CE + SS + LM + SR], we obtain [O–Co-pseudo-eg + O–Co-pseudo-t2g] ∝ [CE + LM + SR] and [O–Co-pseudo-eg − O–Co-pseudo-t2g] ∝ [−OT − SS + SR]. The proposed decomposition agrees with the five time constants required to fit the different delay scans and allows the following assignment: τ1 corresponds to the orbital electron transfer contribution, OT; τ2 to the charge transfer enhancement, CE; τ3 is related to the spin stabilization, SS; and τ4 to the lattice modification, LM. The last constant indicates the slow relaxation, SR. All together, the presence of different time scales points toward an ultrafast metallization mechanism in which the charge, spin, and lattice degrees of freedom evolve with distinct time scales. For the charge channel, the analysis is able to distinguish between the redistribution of electrons between different orbitals and the change in hybridization.
Metallization model
The understanding of the mechanism leading to an ultrafast time separation of the charge, spin, and lattice is sketched in Fig. 2. It can be formulated in four steps, indicated by panels I–IV. For each panel the top figures, Fig. 2a, c, e, g, are a representation of the two CoO6 octahedra per unit cell and their evolution upon laser excitation. Figure 2b, d, f, h display the changes in the DOS obtained from the DFT+DMFT calculations.

Four-step model describing the time evolution of LaCoO3 after laser excitation: (I) Carrier excitation: The femtosecond laser pulse (λ = 800 nm, hν = 1.55 eV) excites the sample, producing carriers and instantaneous gap collapse. (II) Charge transfer enhancement: After carrier relaxation, the charge transfer is enhanced by the larger hybridization of the O–Co-pseudo-eg with the O-2p orbitals. (III) Spin stabilization: The spin configuration in the system is stabilized, which also increases the Co radius. (IV) Lattice modification: The lattice relaxes to accommodate the changes of the spin state and the ionic radius. The change of bonds and angles results in a transient metallic state. In (I)-(a) and (II)-(c), the lattice structure and value of the bond angle at room temperature has been used. In (III)-(e), the value 16X implies that the bond angle has changed to an unknown value. In (IV)-(g), the bond angle and lattice structure at 850 K is provided to indicate the expected bond angle in the excited state. The lattice structures, rhomboedral R\(\bar 3\)c, were plotted using VESTA53. The Co atoms are in the center of an O octahedra (indicated by the shaded area around the central Co atom) and have a different color to indicate the change in spin state and ionic radius at each time step. The density of states (DOS) in (I)-(b) corresponds to the ground state calculations as in Karolak et al.20. In (II)-(d)–(III)-(f), the ground state DOS has been convoluted with a Fermi function at 2000 K to represent the electronic excitation of the system due to the laser pulse. In (IV)-(h), the DOS in a high spin (HS) state with significant charge imbalance is displayed (see text for details)
Carrier excitation
The first step, panel I—Fig. 2a, b, takes place within τ1 = 0.17 ps. The sample, initially at room temperature in a semiconducting state, is excited by the femtosecond pulse, creating hot carriers (electrons and holes) inducing a gap collapse, as it has been recently observed in other oxides, like V2O337. Since the dd transitions are not dipolar-allowed, electrons with larger O-2p character around the Co–O bonds are primarily excited38. In the case of LCO, the ultrafast carrier excitation starts by the OT between O–Co-pseudo-t2g and O–Co-pseudo-eg orbitals because the reflectivity at these two resonance energies has a maximum and minimum, respectively. However, their difference spectrum [O–Co-pseudo-eg − O–Co-pseudo-t2g, Fig. 1e] as well as the spectra at the Co L−edge [Fig. 1f] show a large drop of intensity indicative of bleaching. The hot carriers have much higher energy than the crystal field and the exchange coupling energy resulting in an ill-defined out-of-equilibrium spin state.
Charge transfer enhancement
This process, panel II—Fig. 2c, d, takes places after carrier thermalization. It corresponds to the increased change transfer in the system. Its characteristic time scale is τ2 = 0.52 ps and can be deduced from the fact that the reflectivity in the [O–Co-pseudo-eg + O–Co-pseudo-t2g] sum spectrum (Fig. 1c) does not cancel out for time delays larger than τ2. This effect can be understood by the larger hybridization of the O–Co-pseudo-eg orbitals pointing towards the O-2p orbitals. As a consequence, the charge transfer with oxygen improves upon population of the O–Co-pseudo-eg orbitals. This effect is seen by the small changes observed in the O–Co-pseudo-t2g region (Fig. 1a) above 1 ps and the large decrease of the reflectivity observed at the O–Co-pseudo-eg resonant energy (Fig. 1b) in this area. More subtle effects, like the redistribution of population between different O–Co-pseudo-eg orbitals, cannot be addressed due to the low-energy resolution of the ZPM monochromator.
Spin stabilization
The third step, panel III—Fig. 2e, f, corresponds to the stabilization of the spin system and is represented by the time scale τ3 = 0.91 ps. The process starts after the breaking of the population balance around a time delay of 0.52 ps and stabilizes at around a time delay of 0.91 ps. In the new spin configuration, both Co-excited atoms will be in a charge-disproportionated HS state, as determined from the comparison to the thermally excited state23. To get insight into the nature of the disproportionated HS state, DFT+DMFT calculations of the optical conductivity have been performed following the same approach as in Karolak et al.20 and benchmarked with the experimental results referenced therein. The equilibrium state of the system at room temperature is a 50% incoherent mixture of LS and HS states located on both sides of the oxygen atom—Co(LS)–O–Co(HS)—with no long-range order19,20. This configuration is favored by the charge imbalance that appears in the system when the constraint of equal charge between the two Co atoms in a unit cell is relaxed20. It also explains the semiconducting paramagnetic state of the system and the absence of orbital ordering at room temperature. The charge imbalance induces strong modifications of the optical conductivity in the region below 2 eV (Fig. 3a). At the laser excitation energy used in the experiments, 1.55 eV (800 nm), the optical conductivities of the LS and HS states are very similar to each other. This contrasts with the calculations obtained for pure LS and HS states that result in a weaker excitation of the LS state compared to the excitation for the HS state. The different conductivities can be understood by taking into account the contribution of d5, S = 1/2 states to the LS state, and of d7, S = 3/2 states to the HS configuration. Both contributions result in a large modification of the density of states (DOS) and a reduction of the gap compared to the scenario of LS and HS in a pure d6 electronic configuration (Fig. 3b, d). In the pure LS state, the large gap, slightly larger than the laser photon energy, explains the very low intraband transition probability. In the case of the HS state, the gap is much smaller and therefore the transition is more efficient and the conductivity much higher. In the case of the Co(LS)–O–Co(HS) configuration, the small equilibrium charge disproportionation20 makes possible both LS and HS intraband transitions. Furthermore, interband transitions between the two atoms will also be allowed. The ones from occupied HS states to unoccupied LS states will have higher probably for two reasons: the smaller energy gap between these states, on the one hand, and the Pauli exclusion principle, on the other. The latter will make all transitions from occupied HS O–Co-pseudo-t2g to empty LS O–Co-pseudo-eg possible, whereas only Pauli-satisfying transitions will be available from the occupied LS O–Co-pseudo-eg states to the partially empty HS O–Co-pseudo-eg orbitals.

a Optical conductivities for LaCoO3 in the electronic configurations. (b–d). b and c DOS of the pure low spin (LS) d6 and high spin (HS) d6 configurations, respectively. d Density of states (DOS) of the LS and HS atoms for the charge-imbalanced electronic configuration at room temperature. In this latter case, the data are atom-resolved in order to be able to compare them with the results obtained for the LS (b) and HS (c) states. The length of the arrows in (a–d) indicates the energy of the optical pump (1.55 eV, 800 nm). It is seen that, in the mixed configuration at room temperature, both the LS and HS are similarly excited. In contrast, in the pure d6 configuration, the HS atoms will be primarily excited
As a consequence, the laser-excited state is expected to have a transient metallic state in which the charge disproportionation is enhanced by the laser excitation. Furthermore, the spin state of the excited fraction of the system will have the two atoms in a HS state. This fact, together with the increase of charge disproportionation, explains the differences in the thermally induced metallization of the system23. This conclusion is expected to hold even assuming any of the other initial state models considered for the system12. An alternative explanation to the spin change could be done in terms of laser-induced correlation enhancement39. In this case, the increase of U could result in an excited metallic IS state similar to that proposed initially by LSDA+U calculations14.
Lattice modification
The last step of the process, panel IV—Fig. 2g, h, takes places at around τ4 = 1.50 ps. It corresponds to the stabilization of the metallic state through the structural lattice relaxation. This occurs due to the change in hybridization and the increase of the ionic radius of Co, a consequence of the higher HS content and disproportionation of the system. In Fig. 2—IV, atoms 1 and 2 have d occupancies of 5.83 and 6.17, respectively, and are both in a HS state. This state will be long living, therefore explaining the slow relaxation constant of 45 ps. The ionic radius of the LS and HS states in a pure d6 configuration are rLS = 0.545 Å and rHS = 0.610 Å. Increasing the HS percentage will result in a lattice change consequence of the CoO6 distortion related to the higher ionic radius. This effect will be enhanced, taking into account that the increase of the charge disproportionation implies an increase of the d5 (Co4+) and d7 (Co2+) population. The ionic radius of the HS Co4+ and Co2+ states are \(r_{{\mathrm {HS}},4^ + }\) = 0.530 Å and \(r_{{\mathrm {HS}},2^ + }\) = 0.745 Å, respectively40. As a consequence, an increase of the ionic radius will result in a lattice deformation, both bond angles and lengths, therefore explaining the dynamics observed at the Co-4sp region. In Fig. 2–IV, the bond angle of the thermally activated phase at 850 K is provided to indicate the expected increase in bond angle required to stabilize metallization. The change in both length and angle is expected to improve the hybridization with the O 2p orbitals. This will further enhance the charge transfer and will contribute to the stabilization of the metallic excited state of the system. A similar interpretation can be done assuming that enhanced correlations stabilize the IS state spin configuration since the radius is larger and some other lattice deformations, like Jahn–Teller distortions, can be stabilized14.
The slow relaxation constant required to fit the data, τ5 = 45 ps, is related to laser-induced excitation and how the laser-induced transient metallic state system goes back to the equilibrium state. Therefore, it does not need to be considered as part of the ultrafast metallization process.
Relation to static phonon experiments
The time scales extracted from the reflectivity data are coherent with recent modifications observed in some phonon modes with inelastic X-ray scattering (IXS)41. More concretely, the Eg, A2g phonon modes at 24 meV corresponding to La vibration have shown to soften upon increasing temperature. This phonon energy corresponds to the fastest time scale of 0.17 ps observed in the experiments reported here. The softening of the phonon mode is coherent with the depopulation of the O–Co-pseudo-t2g orbitals pointing towards the La atoms and also with the increase of the ionic radius of the HS state. The second phonon mode that has shown modifications upon increasing temperature across the semiconductor to metal transition is the Eg mode. This mode, corresponding to a rotational mode of O atoms (or a vibration of the CoO6 octahedra), hardens upon increasing the temperature. This hardening is coherent with the increase of the oscillation amplitude of the oxygen atoms perpendicular to the Co–O recently reported and also with the increase on the ionic radius of the Co atoms as the HS spin population of the system increases41. The energy of this mode, 5 meV, corresponds to a time scale of 0.827 ps. This time scale matches the 0.91 ps time scale ascribed to the increase of the HS state and to the change of ~50% in the population unbalance, a consequence of the enhanced hybridization.
The proposed ultrafast metallization model of LCO can be understood by taking into account that the structure is naturally strained by the large size of the La3+ ions, which forces a rhombohedral lattice, and by the large variations of the Co ion size with spin configuration. In this respect, the results prove experimentally the thermodynamical metallization model of the system in which the change in the O–Co-pseudo-t2g and O–Co-pseudo-eg population with temperature results in an increase of the HS state. The larger radius of the latter, in turn, induces the structural changes with temperature. The ultrafast metallization reported here follows the same path, with the difference that the laser excitation enhances the charge disproportionation already present under equilibrium conditions. It is interesting to note that, while the process is entangled at the Co L3 edge, the time information of the different channels involved is decrypted at different resonant energies of the O K edge.
Discussion
In summary, combined optical pump—soft x-ray reflectivity experiments with femtosecond resolution and theoretical DFT+DMFT calculations have been used to address the ultrafast metallization of LCO upon laser excitation. The latter shows intensity variations at different time delays for different resonant energies that allow extracting the relevant time constants with less uncertainty on the fitting procedure. Metallization stabilizes within less than 3 ps in a four-step process in which the increase of ionic radius accompanying the HS state enhancement after carrier thermalization modifies bond lengths in the CoO6 octahedra and increases the Co–O–Co angle, as happens upon thermal excitation of the system. The experiments support the theoretical model within DFT+DMFT with moderate U that favors correlation-induced charge and spin fluctuations. Our findings are the first experimental demonstration of the adiabatic thermal transition model in LCO widely accepted in the literature. Furthermore, the results indicate that a comprehensive analysis of the ultrafast dynamics of perovskites can be obtaining by performing time-resolved experiments with soft x-rays at the O K only. Most of the limits imposed by femtosliced experiments at synchrotrons—reduced time delay range and energy resolution—are overcome at x-ray free-electron lasers.
Methods
d orbitals in a rhombohedral environment
The atomic structure of LCO is rhombohedral (R\(\bar c\)3). This means that the description of the d orbitals in terms of the octahedral t2g, eg basis is not rigorous. Instead linear combinations of them are required. They will be referred to in the following as pseudo-t2g and pseudo-eg.
Soft x-ray time-resolved experiments
Time-resolved soft x-ray reflectivity studies at room temperature were performed at the femtoslicing beamline of the BESSY II synchrotron radiation source of the Hemholtz-Zentrum Berlin (HZB)42,43,44. Experiments were carried out in (001) terminated, 4 × 4 mm2 single crystal cuts from a well-characterized large crystal45. A (θ, 2θ) = (5°, 10°) geometrical configuration was used for the experiments. The pump pulses were produced by a Ti:Sapphire laser at λ = 800 nm (1.55 eV), 6 kHz maximum repetition rate, 50 fs pulse duration, and 2 mJ maximum pulse energy. The x-ray pulse duration of about 100 fs in the femtoslicing operation mode ensures a global time resolution of 130 fs42,43,44. The probe soft x-ray femtosliced photons were produced using Bragg Fresnel reflection zone plate monochromators (ZPMs). One of the ZPMs was optimized to operate in the energy range of the O K edge, 2.21–2.36 nm (560.9–525.3 eV). The second one covered the Co L edge energy range, 1.54–1.60 nm (774.87–805.06 eV).
The penetration length of the excitation laser with λ = 800 nm is ~359 nm, as obtained from Nomerovannaya et al.46. This value is larger than the penetration depth of the soft x-rays at the O K edge (<340 nm) and the Co L edge (<298 nm) obtained from the Center for X-ray Optics (http://www.cxro.lbl.gov/). The values indicate that the sampled area was fully excited by the pump pulse and therefore that an interpretation of the data based in the ultrasonic propagation of the metallic phase in a system where both semiconducting and metallic phases coexist seems unlikely47.
Data fitting
The fit was done using a Levenberg–Marquardt algorithm to search for the minimum value of χ2 defined as: \({\sum} \left( {\left( {y - y_i} \right){\mathrm{/}}\sigma _i} \right)^2\). We initially model the delay scans using a series of exponential decays following a model proposed for isostructural manganites34. However, a fit with three times better χ2 values could be obtained using a series of exponential decays in which the pulse duration and time constant of component i were the values from component i − 11. In total, a good fit could be obtained with a maximum of four components. In some cases, only two were necessary. The time constants were obtained from a self-consistent fit of all delay scans. This was achieved by first fitting all delay scans with free parameters and then optimizing the values so that, overall, the best fit for all delay scans taken together was obtained. The laser pulse duration was estimated to be 0.125 ps, which matches the typically 0.13 ps measured laser pulse duration. The first relaxation constant, τ1 = 0.17 ps, was clearly obtained from the Co L3 and O–Co-pseudo-t2g, while τ5 = 45.00 ps was fixed from ps data23. τ2 = 0.52 ps was obtained from the fit of the O–Co-pseudo-eg–O–Co-pseudo-t2g different spectrum. The two τ3 values, 0.91 and 1.5 ps, were required to provide the best fit at the Co L3 and O–Co-pseudo-eg, respectively.
Theoretical calculations
We performed DFT+DMFT calculations of the static optical reflectivity following the recipe outlined in our previous work20. We set up an effective multiorbital Hubbard model for the two-atomic unit cell constructed via DFT, which has the following Hamiltonian:
where \(\hat H^0\) is the DFT Hamiltonian and \(\hat c_{\alpha i\sigma }^\dagger\) is the creation operator of an electron on site α in Wannier state i and spin σ. Since we focus only on the Co 3d states, the double counting, \(\propto \mu _\alpha ^{{\mathrm {DC}}}\), amounts to a shift of the chemical potential for the Co 3d shell and is determined self consistently. To obtain the full Coulomb interaction tensor, we employ the parametrization via Slater integrals48 connected to the average direct and exchange couplings U and J: F0 = U, J = 1/14(F2 + F4), and F4 = 0.625F2. We use U = 3 eV and different Hund’s rule couplings J ranging from 0.5 to 1.0 eV20. We treat the two Co sites (α) in the unit cell independently using the inhomogeneous or real space DMFT49.
The impurity models are subsequently solved using exact diagonalization (ED, Lanczos method50). Here, it is necessary to approximate the continuous hybridization function Δ(iωn) with a finite cluster containing nS sites and its corresponding hybridization Δcl(iωn)
The optimal set of bath parameters εk and Vmk is found by fitting the full lattice hybridization function. We have employed a five-site cluster, which may appear to be a very crude approximation. However, comparison with continuous time quantum Monte Carlo calculations20 shows that the spectra of both methods agree very well. The reason is that the states of the Co ions are dominated very strongly by a few local atomic states that we include exactly in the ED and not so much by itinerant states that we approximate. Since ED allows direct evaluation of the self-energy matrix \(\underline {\underline \sum } \left( \omega \right)\) on the real energy axis, we can quickly calculate the spectral matrices \(\underline {\underline A } \left( {{\bf{k}},\omega } \right)\), as needed for the evaluation of optical properties (see below).
The calculation of the optical conductivity in DFT+DMFT requires the evaluation of the following expression:
where V is the unit cell volume, f denotes the Fermi function, \(\underline {\underline v } ^\alpha \left( {\bf{k}} \right)\) are the matrix elements of the momentum operator, and \(\underline {\underline A } \left( {{\bf{k}},\omega } \right)\) is the momentum-resolved spectral matrix51,52. For the quick calculation of the momentum-resolved spectra on any k grid, we have used ED, as explained above. The remaining problem of the calculation of the momentum matrix elements is tackled using the improved Peierls approximation as implemented in the “woptic” package51. This methodology was shown in P. Wissgott et al.51 to work well for the low-frequency part of the optical conductivity, i.e. for the case of transition metal oxides dominated by d − d transitions. The package uses an adaptive k sampling procedure, which calculates the necessary quantities on iteratively improved meshes in the Brillouin zone.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
References
Heikes, R. R., Miller, R. C. & Mazelsky, R. Magnetic and electrical anomalies in LaCoO3. Physica 30, 1600 (1964).
Blasse, G. Magnetic properties of mixed metal oxides containing trivalent cobalt. J. Appl. Phys. 36, 879 (1965).
Menyuk, N., Dwight, K. & Raccah, P. M. Low temperature crystallographic and magnetic study of LaCoO3. J. Phys. Chem. Sol. 28, 549 (1967).
Jonker, G. H. Magnetic and semiconducting properties of Perovskites containing manganese and cobalt. J. Appl. Phys. 37, 1424 (1966).
Naiman, C. S., Gilmore, R., DiBartolo, B., Linz, A. & Santoro, R. Interpretation of the magnetic properties of LaCoO3. J. Appl. Phys. 36, 1044 (1965).
Raccah, P. M. & Goodenough, J. B. First-order localized-electron collective-electron transition in LaCoO3. Phys. Rev. 155, 932 (1967).
Bhide, V. G., Rajoria, D. S., Rama Rao, G. & Rao, C. N. R. Mössbauer studies of the high-spin-low-spin equilibria and the localized-collective electron transition in LaCoO3. Phys. Rev. B 6, 1021 (1972).
Radaelli, P. G. & Cheong, S.-W. Structural phenomena associated with the spin-state transition in LaCoO3. Phys. Rev. B 66, 094408 (2002).
Yamaguchi, S., Okimoto, Y. & Tokura, Y. Local lattice distortion during the spin-state transition in LaCoO3. Phys. Rev. B 55, R8666 (1997).
Kobayashi, Y. et al. Inelastic neutron scattering study of phonons and magnetic excitations in LaCoO3. Phys. Rev. B 72, 174405 (2005).
Stølen, S., Grønvold, F., Brinks, H., Atake, T. & Mori, H. Energetics of the spin transition in LaCoO3. Phys. Rev. B 55, 14103 (1997).
Ivanova, N. B., Ovchinnikov, S. G., Korshunov, M. M., Eremin, I. M. & Kazak, N. V. The features of spin, charge and orbital orderings in the cobaltites. Phys. Usp. 179, 837 (2009).
Goodenough, J. B. An interpretation of the magnetic properties of the perovskite-type mixed crystals La1−x Srx CoO3−λ. J. Phys. Chem. Sol. 6, 287 (1958).
Korotin, M. A. et al. Intermediate-spin state and properties of LaCoO3. Phys. Rev. B 54, 5309 (1996).
Haverkort, M. W. et al. Spin state transition in LaCoO3 studied using soft X-ray absorption spectroscopy and magnetic circular dichroism. Phys. Rev. Lett. 97, 176405 (2006).
Yang, Z., Huang, Z., Ye, L. & Xie, X. Influence of parameters U and J in the LSDA + U method on electronic structure of the perovskite LaMO3 (M=C, Mn, Fe, Co, Ni). Phys. Rev. B 60, 15674 (1999).
Kuneš, J. & Křápek, V. Disproportionation and metallization at low-spin to high-spin transition in multiorbital Mott systems. Phys. Rev. Lett. 106, 256401 (2011).
Zhang, G., Gorelov, E., Koch, E. & Pavarini, E. Importance of exchange anisotropy and superexchange for the spin-state transitions in RcoO3 (R=rare earth) cobaltates. Phys. Rev. B 86, 184413 (2012).
Křápek, V. et al. Spin state transition and covalent bonding in LaCoO3. Phys. Rev. B 86, 195104 (2012).
Karolak, M., Izquierdo, M., Molodtsov, S. L. & Lichtenstein, A. I. Correlation-driven charge and spin fluctuations in LaCoO3. Phys. Rev. Lett. 115, 046401 (2015).
Wang, R.-P. et al. Excitonic dispersion of the intermediate-spin state in LaCoO3 revealed by resonant inelastic X-ray scattering. Phys. Rev. B 98, 035149 (2018).
Sotnikov, A. & Kuneš, J. Field-induced exciton condensation in LaCoO3. Sci. Rep. 6, 30510 (2016).
Izquierdo, M. et al. Laser-induced charge-disproportionated metallic state in LaCoO3. Phys. Rev. B 90, 235128 (2014).
Taskin, A. A., Lavrov, A. N. & Ando, Y. Origin of the large thermoelectric power in oxygen-variable RbaCo2O5+x (R=Gd,Nd). Phys. Rev. B 73, 121101 (2006).
Büchler, O., Serra, J., Meulenberg, W., Sebold, D. & Buchkremer, H. Preparation and properties of thin La1−xSrxCo1−yFeyO3−d perovskitic membranes supported on tailored ceramic substrates. Solid State Ion. 178, 91 (2007).
Li, N., Borseave, A., Deloume, J.-P. & Gaillard, F. Catalytic combustion of toluene over a Sr and Fe substituted LaCoO3 perovskite. Solid State Ion. 179, 1396 (2008).
Lee, Y. L., Kleis, J., Rossmeisl, J. & Morgan, D. Ab initio energetics of LaBO3 (001) (B=Mn, Fe, Co, and Ni) for solid oxide fuel cell cathodes. Phys. Rev. B 80, 224101 (2009).
Kim, C. H., Qi, G., Dahlberg, K. & Li, W. Strontium-doped perovskites rival platinum catalysts for treating NOx in simulated diesel exhaust. Science 327, 1624 (2010).
Alvarez-Galván, M. C. et al. Surface reactivity of LaCoO3 and Ru/LaCoO3 towards CO, CO2 and C3H8: effect of H2 and O2 pretreatments. Appl. Catal. B 102, 291 (2011).
Kubicek, M. et al. Tensile lattice strain accelerates oxygen surface exchange and diffusion in La1−xSrxCoO3−δ. Thin Films, ACS Nano 7, 3276 (2013).
Vereshchagin, S. N. et al. Methane oxidation over A-site ordered and disordered Sr0.8Gd0.2CoO3−δ perovskites. Chem. Commun. 50, 6112 (2014).
Aoki, H. et al. Nonequilibrium dynamical mean-field theory and its applications. Rev. Mod. Phys. 86, 779 (2014).
Wessely, O. et al. Ab Initio Theory of Dynamical Core-Hole Screening in Graphite from X-Ray Absorption Spectra. Phys. Rev. Lett. 94, 167401 (2005).
Bielecki, J. et al. Two-component heat diffusion observed in LaMnO3 and La0.7Ca0.3MnO3. Phys. Rev. B 81, 064434 (2010).
Raiser, D. et al. Evolution of hot polaron states with a nanosecond lifetime in a manganite perovskite. Adv. Energy Mater. 7, 1602174 (2017).
Bielecki, J., Rata, A. D. & Borjesson, L. Femtosecond optical reflectivity measurements of lattice-mediated spin repulsions in photoexcited LaCoO3 thin films. Phys. Rev. B 89, 035129 (2014).
Lantz, G. et al. Ultrafast evolution and transient phases of a prototype out-of-equilibrium Mott–Hubbard material. Nat. Commun. 8, 13917 (2017).
Yamaguchi, S., Okimoto, Y., Ishibashi, K. & Tokura, Y. Magneto-optical Kerr effects in perovskite-type transition-metal oxides: La1−xSrxMnO3 and La1−xSrxCoO3. Phys. Rev. B 58, 6862 (1998).
Ido, K., Ohgoe, T. & Imada, M. Correlation-induced superconductivity dynamically stabilized and enhanced by laser irradiation. Sci. Adv. 3, e1700718 (2017).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A32, 751 (1976).
Sikolenko, V. V. et al. Correlated oxygen displacements and phonon mode changes in LaCoO3 single crystal. Physica B 536, 597 (2018).
Brzhezinskaya, M. et al. A novel monochromator for experiments with ultrashort X-ray pulses. J. Synchrotron Radiat. 20, 522 (2013).
Abo-Bakr, M., Feikes, J., Holldack, K., Wüstefeld, G. & Hübers, H. W. Steady-state far-infrared coherent synchrotron radiation detected at BESSY II. Phys. Rev. Lett. 88, 254801 (2002).
Stamm, C. et al. Femtosecond modification of electron localization and transfer of angular momentum in nickel. Nat. Mater. 6, 740 (2007).
Prabhakaran, D., Boothroyd, A., Wondre, F. & Prior, T. Bulk single crystal growth and magnetic studies of La1−xSrxCoO3+δ. J. Cryst. Growth 275, e827–e832 (2005).
Nomerovannaya, L. V. et al. The influence of the Co3+ spin state on the optical properties of LaCoO3 and HoCoO3. J. Phys.: Cond. Matter 16, 5129 (2004).
Okimoto, Y. et al. Ultrasonic propagation of a metallic domain in Pr0.5Ca0.5CoO3 undergoing a photoinduced insulator-metal transition. Phys. Rev. Lett. 103, 027402 (2009).
Slater, J. C. Quantum Theory of Atomic Structure, Vol. 1 (McGraw-Hill, New York, 1960).
Potthoff, M. & Nolting, W. Surface metal-insulator transition in the Hubbard model. Phys. Rev. B 59, 2549 (1999).
Capone, M., de Medici, L. & Georges, A. Solving the dynamical mean-field theory at very low temperatures using the Lanczos exact diagonalization. Phys. Rev. B 76, 245116 (2007).
Wissgott, P., Kuneš, J., Toschi, A. & Held, K. Dipole matrix element approach versus Peierls approximation for optical conductivity. Phys. Rev. B 85, 205133 (2012).
Tomczak, J. M. & Biermann, S. Optical properties of correlated materials: generalized Peierls approach and its application to VO2. Phys. Rev. B 80, 085117 (2009).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272 (2011).
Acknowledgements
We thank Karsten Holldack, Niko Pontius, and Christian Schüessler-Langeheine, for experimental support at the FemtoSpeX facility during the experiments. We acknowledge Alexander Fölisch for supporting the realization of the experiments at the FEMTOSPEX and Kurt Ament for editing the manuscript. M.I. acknowledges BMBF Grant no. 05K12GU2 for financial support.
Author information
Authors and Affiliations
Contributions
M.I. and S.L.M. conceived the experiment. D.P. and A.T.B. prepared and characterized the samples. M.I. conducted the experiment with support from members of the FemtoSpeX group. M.I. analyzed the data and wrote the manuscript with contributions from S.L.M., M.K., and A.S. M.K. and A.L. performed the theoretical calculations. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Izquierdo, M., Karolak, M., Prabhakaran, D. et al. Monitoring ultrafast metallization in LaCoO3 with femtosecond soft x-ray spectroscopy. Commun Phys 2, 8 (2019). https://doi.org/10.1038/s42005-019-0109-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-019-0109-9
This article is cited by
-
Photo-induced antiferromagnetic-ferromagnetic and spin-state transition in a double-perovskite cobalt oxide thin film
Communications Physics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
