
Abstract
Bicyclic boronates play critical roles in the discovery of functional materials and antibacterial agents, especially against deadly bacterial pathogens. Their practical and convenient preparation is in high demand but with great challenge. Herein, we report an efficient strategy for the preparation of bicyclic boronates through metal-free heteroatom-directed alkenyl sp2-C‒H borylation. This synthetic approach exhibits good functional group compatibility, and the corresponding boronates bearing halides, aryls, acyclic and cyclic frameworks are obtained with high yields (43 examples, up to 95% yield). Furthermore, a gram-scale experiment is conducted, and downstream transformations of the bicyclic boronates are pursued to afford natural products, drug scaffolds, and chiral hemiboronic acid catalysts.
Similar content being viewed by others
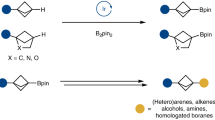
Introduction
Boronic acids1 have been recognized as powerful substances in molecular recognition2,3, chemical biology4,5, materials science6, and catalysis7 in the past decades. Among them, the bicyclic boronates have attracted remarkable attention and research interests for organic and medicinal chemists owing to their unique structural features and properties. Such bicyclic skeletons exist not only in bioactive molecules (for example, taniborbactam8, VNRX-71459, QPX772810, benzazaboroine-211, and naphthoxaborin-112), but also in fluorescent sensors (such as naphthoxaborin-213) and functional materials (for instance, benzazaboroine-114 and BN2VN15) (Fig. 1a)16,17. Particularly, the three compounds under clinical development as β-lactamase inhibitors, including taniborbactam, VNRX-7145, and QPX7728, are regarded as the present hope against deadly bacterial pathogens18, and their scalable synthesis is in urgent need to supply the clinical demands19. Because of the growing importance of bicyclic boronates, substantial efforts have been devoted to their preparations. Recently, boron insertion emerged as a strategy for the synthesis of boron-containing products. In 2016 and 2017, Yorimitsu and coworkers successfully achieved nickel- and manganese-catalyzed boron insertion into the sp2-C − O bond of benzofurans for the construction of bicyclic boronates (Fig. 1b)13,20. Subsequently, they developed transition-metal-free ring-opening borylation of indoles, in which boron atom was inserted into the C2 − N bond using a large excess amount of lithium metal, to furnish 1,2-benzazaborins21. Very recently, Dong, Liu, and coworkers realized the boron insertion into the challenging sp3-C − O bond in alkyl ethers through zinc/nickel tandem catalysis to provide bicyclic boronates (Fig. 1b)22. In addition, the palladium-catalyzed boron-selective biaryl coupling was also an efficient synthetic approach to versatile dibenzoxaborins23. It is worth mentioning that all these methods were based on the catalysis of transition metals (Ni, Zn, Mn, or Pd) or the use of excessive alkali metal (Li), which might limit their applications, particularly in consideration of industrial-scale production and the need to remove trace metals in active pharmaceutical ingredients24.

a Bicyclic boronate framework existing in drug molecules and functional materials. b Transition-metal-catalyzed boron insertion. c Metal-free approach to bicyclic boronates. d Metal-free O-directed alkenyl sp2-C‒H borylation.
Compared with the transition-metal-catalyzed process, the alternative metal-free strategy is usually practical, cost-effective, and environmentally benign. Early studies involving metal-free approach were reported by Dewar25, Paetzold26, and their co-workers, in which unprotected anilines or phenols could be converted to borazaroarenes and boroxaroarenes, respectively (Fig. 1c). However, their transformation suffered from harsh reaction conditions and the narrow substrate range26.
With the recent development of metal-free C‒H borylation of (hetero)arenes using boron reagents, such as BX3 and BH327,28, we became interested in the preparation of cyclic boronates from alkenes via metal-free sp2-C–H borylation29,30. To the best of our knowledge, the related research is quite rare in literature24,31, probably due to the fact that alkenes can undergo cationic polymerization or haloboration under Lewis acidic conditions31. Inspired by the concept of “frustrated Lewis pairs”, we propose that a suitable bulky base, just like a proton sponge, might be compatible with the borylation of alkenes without decreasing the activity of Lewis acid. Herein, our initial strategy is to utilize the oxygen atom in 2-(E)- or (Z)-vinyl anisole 1 as a directing group for sp2-C–H borylation of neighboring olefin (Fig. 1d). Upon treatment of 1 with BBr3, the corresponding borate AE or AZ is produced after the release of methyl bromide. Subsequently, the dienol borate AE or AZ can readily undergo a unique tautomerization through 1,5-sigmatropic rearrangement32, in which 1,5-boron-shift occurred to give homoenone boronate B with a newly formed C‒B bond. Compared with the direct intermolecular electrophilic borylation of terminal alkenes29, our process takes advantage of intramolecular rearrangement of 2-vinylphenoxyborate (AE or AZ) to gain the key ortho-quinone methide intermediate (B)33,34, and such C‒B bond construction can potentially improve the reaction efficiency and exclude the formation of different isomers. Due to the olefin double bond migration during the tautomerization process, both (Z)- and (E)- isomers of substrate 1 form a common homoenone boronate B. Next, under the promotion of a base, the elimination of HBr from intermediate B occurs, and the resulting intermediate C can be quenched by water to produce the desired bicyclic boronate 3. Obviously, the key in our strategy is to select an appropriate base to capture HBr. Because strong base tends to form a tight pair with BBr3 and consequently prevents the cleavage of methyl ether bond, we focused on a range of bulky organic bases to promote this borylation process. As a matter of fact, such a special (Z)-alkenyl hemiboronic acid35 is rarely documented, and its straightforward synthetic access is in great demand36,37. Herein, we report a practical preparation of bicyclic boronates via metal-free heteroatom-directed alkenyl sp2-C‒H borylation.
Results and discussion
Optimization of reaction conditions
Our investigation started with the model reaction of o-isopropenylanisole (1aa), which is a component of the volatile oil of Lippia javanica and shows antimicrobial activities38, and the selected results were summarized in Table 1. Initially, the reaction was carried out with commercially available borylation reagents, such as BBr3, BF3·OEt2, and BCl3 (2.0 equiv, 2a–c) in dichloromethane (DCM) at –60 °C for 0.5 h, however, only BBr3 (2a) produced the desired bicyclic boronate 3aa in 35% yield, albeit with quantitative conversion (entry 1). As we envisioned, an appropriate base may suppress the occurrence of side reactions including cationic polymerization, hydrobromination, and bromoboration of alkene under acidic conditions. Thus some representative aliphatic and heterocyclic aromatic amines, such as N,N-diisopropylethylamine (DIPEA, A1)30, pyridine (A2), and 2,6-lutidine (A3)29, were selected for our reaction (entries 2–4). Both A1 and A2 resulted in low reaction conversion (entries 2 and 3). To our delight, the steric base, 2,6-lutidine A3 led to 100% conversion and afforded 3aa in 38% yield, indicating that Lewis acid BBr3 could be compatible with bulky heterocyclic aromatic base. Several 2,6-disubstituted pyridines, for instance, 2,6-bis(trifluoromethyl)pyridine (A4), 2,6-di-tert-butylpyridine (A5, DTBP), and 2,6-diphenylpyridine (A6), were subsequently tested in our reaction. Among them, 2,6-di-tert-butylpyridine (A5) proved to be the most effective, affording 3aa in almost quantitative yield (entry 6). The use of diphenyl pyridine (A6) considerably reduced the reaction conversion and yield (entry 7), and 2,6-bis(trifluoromethyl)pyridine (A4), which was less basic due to the existence of two -CF3 substitutions, surprisingly improved the yield of 3aa from 38% to 89%. Another less basic base 2,3,5,6-tetramethylpyrazine (A7) was employed, and the desired bicyclic boronate 3aa was achieved with 98% yield, which was quite comparable with the result from A5 (entry 5). Next, the amounts of BBr3 and A5 were investigated. When a proportional excess of A5 (2.0–1.1 equiv) relative to BBr3 (1.5 equiv) was used, the yield of 3aa dramatically decreased and significant amount of uncycylized side product o-isopropenyl phenol 1bc was observed (entries 11–13). Interestingly, maintaining the 1:1 ratio of BBr3 and A5 allowed for further reduction in employing both reagents to 1.1 equiv. without an erosion of yield (entry 12). Lastly, raising or lowering the reaction temperature caused more side-reactions or decreased the reaction conversion, respectively (entries 13 and 14). As a result, the best yield for alkenyl sp2-C–H borylation was achieved when BBr3 (2a, 1.1 equiv) and 2,6-di-tert-butylpyridine (A5, 1.1 equiv) were used in DCM at –60 °C.
Substrate scope of O-directed borylation of terminal alkenes
With the optimal reaction conditions in hand, the scope of substrates was investigated, and the results are summarized in Fig. 2. First, different R1 substituents at alkenyl moiety, such as methyl, ethyl, 2-ethyl methanesulfonate, and phenyl, were evaluated. All reactions proceeded smoothly in moderate to high yields (Fig. 2, 3aa–3ad), and the structure of 3aa was unambiguously confirmed by X-ray crystallographic analysis (CCDC 2143877 (3aa) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.) (Supplementary Note 1 and Data 2). The substrate with R1 = H could also afford the desired product 3ae, albeit with slightly decreased yield of 78% (Fig. 2, 3ae). Next, a variety of substituents R2 at the phenyl ring were investigated, including alkyl (Me-, tBu-, EtCO2CH2-, or CH3(CH2)7NHCH2-), halogen (F-, Cl-, or Br-), trifluoromethyl and ether functionality (MeO- or 4-CN-C6H4O-). All the substitution groups were well tolerated, regardless of their location at C3-, C4-, C5-, or C6-position and their electron-withdrawing or electron-donating properties, in this borylative cyclization, with unambiguous confirmation of 3ah structure by X-ray crystallographic analysis (CCDC 2143875 (3ah) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.) (Fig. 2, 3af–3ax, Supplementary Note 2 and Data 3). It is worth mentioning that methoxyl and N-pivaloyl group, which was reported for directed sp2-C–H borylation of (hetero)arenes24,27,39, did not affect our desired borylation process and only O-directed alkenyl boronic product was obtained in 92% yield (Fig. 2, 3ap). Additionally, free -OH group at phenyl ring was also tolerated although excess base A5 was required to inhibit side reactions, and the corresponding product 3ao was obtained in 56% yield (Fig. 2, 3ao). Naphthalene substrates also worked quite well to afford the desired boronates with moderate yields (Fig. 2, 3ay–3ba). Notably, mono- and di-vinyl substituted chiral 2,2’-dimethoxy-1,1’-binaphthalene substrates 1az and 1ba, also proved to be suitable for this reaction, offering monohemiboronic acid (3az) and bishemiboronic acid (3ba) with excellent yields, respectively (Fig. 2, 3az and 3ba). The borylation of substrate 1az’ bearing free OH group required excess BBr3 for full conversion, and 2’-naphthol product 3az’ was obtained with 75% yield (Fig. 2, 3az’). Two estrone derivatives were also subjected to our reaction conditions. Interestingly, the treatment of methyl ether substate 1bb with excess BBr3 and A5 could deliver the desired boronate 3ba, along with some demethylation side product 1bb’, while the phenol substrate 1bb’ failed to undertake the borylation. At last, various O-linked groups R3, such as H, ethyl (Et), iso-propyl (iPr), t-butyl (tBu), benzyl (Bn), and phenyl (Ph), were evaluated. All substrates, except O-phenyl ether 1bg, proceeded smoothly to give the desired product 3aa with acceptable to moderate yields. Coincidently, the yields for two reactions with substrates 1bc, 1ao, and 1az' were eroded by the existence of free -OH group, probably due to the consumption of A5 by initially released HBr. It is worth mentioning that when the substituent R2 is an electron-withdrawing group such as -CHO (3fa), -CN (3fb), or a substrate with a heteroaromatic ring (3fc), no desired product was detected in our standard conditions.

aReactions were performed with 1 (0.2 mmol), 2a (1.1 equiv, 1.0 M in DCM), A5 (1.1 equiv) in DCM (1.0 mL) under N2 atmosphere, –60 °C. bYield of isolated product. c2a (3.0 equiv, 1.0 M in DCM), A5 (1.1 equiv), –60 °C. d2a (2.0 equiv, 1.0 M in DCM), A5 (2.0 equiv), –60 °C. eA5 (2.0 equiv), –40 °C, 12 h. f2a (10.0 equiv, 1.0 M in DCM), A5 (10.0 equiv), 0 °C, 8 h. g2a (2.0 equiv, 1.0 M in DCM), A5 (2.0 equiv), 0 °C.
Substrate scope of N- and S-directed borylation of terminal alkenes
Upon achieving the above exciting results, we turned our attention to other heteroatoms as a directing group. First, nitrogen atom was evaluated for this borylation reaction. Fortunately, the secondary aromatic amines worked very well as directing group to give desired products with moderate to good yields (Fig. 3, 3bh–3bk). To our surprise, even the NH of heteroarenes, such as indole24,40 and carbazole, could be employed as directing group to promote alkenyl sp2-C‒H borylation with 91% and 72% yields, respectively (Fig. 3, 3bl and 3bm). Notably, the tertiary aromatic amine could also serve as directing group, affording a boronic acid product 3bn in 52% yield (Fig. 3, 3bn). Next, sulfur atom was introduced as directing group for our reaction, and the corresponding alkenyl sp2-C‒H borylation was realized to give (Z)-alkenyl boronic acid product 3bo with 42% yield. In addition, the stereochemistry of 3bh, 3bm, and 3bo was unambiguously established by X-ray crystallographic analysis (CCDC 2143812 (methyl ester of 3bh), 2144121 (3bm), and 2143882 (3bo) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.) (Supplementary Note 3–5 and Data 4–6). It is also worth mentioning that no desired product can be detected when the substrate contains -NH2 (1fd) or -SH (1fe) group under the standard conditions.

aReactions were performed using 1 (0.2 mmol), 2a (2.0 equiv, 1.0 M in DCM), A5 (2.0 equiv) in DCM (1.0 mL) under N2 atmosphere, 0 °C. bYield of isolated product. c2a (1.1 equiv, 1.0 M in DCM), A5 (1.1 equiv) in DCM (1.0 mL), 0 °C.
Substrate scope of O-directed borylation of (E)- and (Z)-alkenes
To further expand the reaction scope, the substrates were extended to internal alkenes. First, all o-(E)-vinylanisole substrates, regardless of different R1 (hydrogen or alkyl group) and R2 (methyl, n-hexyl, phenyl or even forming a ring with R1) substitutions, could successfully afford the corresponding products in moderate to good yields (Fig. 4, 3bp–3bu). Next, the (Z)-alkenyl substrates 1bv and 1bw were subjected to this borylation, as expected, the corresponding bicyclic boronates 3bv and 3bw were successfully obtained with 95% and 81% yields, respectively. When (Z)- and (E)- isomers of trisubstituted alkene 1bx (R1 and R2 = n-butyl) were separately treated with BBr3 and A5, the same cyclization product 3bx was achieved with excellent yield (Fig. 4, 3bx). This result suggested that a mixture of Z/E isomers could lead to one single product. As a matter of fact, the treatment of a (Z)- and (E)-alkenyl mixture 1by (E/Z = 1.06/1.00) under the borylation conditions indeed afforded the desired product 3by in 93% yield, and its structure was further confirmed by X-ray crystallography (CCDC 2196049 (3by) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. crystal data.) (Fig. 4, 3by, Supplementary Note 6 and Data 7).

aReactions were performed using 1 (0.2 mmol), 2a (1.1 equiv, 1.0 M in DCM), A5 (1.1 equiv) in DCM (1.0 mL) under N2 atmosphere, −60 °C. bYield of isolated product. c−20 °C. d0 °C.
Gram-scale experiment and synthetic transformations
To demonstrate the synthetic applicability of this method, a gram-scale experiment of 1aa was carried out and the corresponding product 3aa was isolated with 95% yield. Moreover, several subsequent transformations of this boronic acid 3aa were conducted to illustrate its unique utilities (Fig. 5A). First, the metal-free coupling between boroxine 3aa and tosyl hydrazone delivered the E-alkene 4 in a good yield41, and its stereochemistry was established by X-ray crystallographic analysis (CCDC 2143883 (4) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.) (Supplementary Note 7 and Data 8) . Two Pd-catalyzed reactions of 3aa were also pursued. The Suzuki-Miyaura coupling with 4-bromotoluene led to (Z)-2-(1-phenylprop-1-en-2-yl)phenol 5 with 78% yield, and the carbonylation with carbon monoxide afforded 4-methyl coumarin 7 in 98% yield39. Under the copper-catalyzed conditions, 3-methyl benzofuran 8 was furnished from 3aa through C–O bond formation with 91% yield42. The boronic acid 3aa was also subjected to reduction or oxidation process. Hydrogenation over Pd/C could provide functionalized bicyclic boronate 6 in 90% yield, and the oxidative cleavage of carbon-boron bond with H2O2 resulted in the formation of 3-methyl-2-coumaranone 9 in 92% yield, likely through enol/hemiacetal intermediates15. Similar oxidation of 3bh could generate 1,3-dimethylindole 10 through enol/hemiaminal intermediates in 95% yield (Fig. 5A). Next, a close chiral analog 11 of Hall’s hemiboronic acid catalyst43, in which the terminal phenyl ring was replaced with a cyclohexyl moiety, was designed and synthesized. The application of our protocol could conveniently convert the substrate 1bz to the chiral catalyst 11, which could promote the desymmetrization of 2-phenyl-propan-1,3-diol to provide chiral alcohol (S)-13 in 38% yield and with 85% ee (Fig. 5B). Finally, the utility of our method was demonstrated in a streamlined synthesis of (±)-QPX7728, an ultrabroad-spectrum inhibitor of serine and metallo-β-lactamases (Fig. 5C)10,18. Starting from the commercially available 3-bromo-4-fluoro-2-methoxy benzaldehyde, a four-step sequence, including Wittig reaction (94% yield), boron-insertion (88% yield), Simmons–Smith cyclopropanation (67% yield), and halogen–lithium exchange with dry ice quenching process (75% yield), successfully afforded (±)-QPX7728 in 42% overall yield44. Compared with the previous synthesis19,44, this as an efficient alternative method is allowing for further optimization in the scalable production of QPX7728.

A. Gram-scale experiment and several transformations: (a) p-Anisaldehyde tosylhydrazone (1.3 equiv), K2CO3 (1.3 equiv), dioxane (2.0 mL), 110 °C, 1.5 h; (b) 4-Bromotoluene (1.2 equiv), Pd(PPh3)4 (5 mol %), THF (2.0 mL), K2CO3 (2.0 equiv, 2.0 M aq), 80 °C, 5 h; (c) 10% Pd/C (10 mol %), H2 6 atm, rt, 1.5 h; (d) Pd(OAc)2 (1.0 equiv), CO, DMSO/MeOH (2.0 mL/1.0 mL), rt, 3.0 h; (e) Cu(OAc)2 (0.2 equiv), Ag2CO3 (3.0 equiv), 1,10-phenanthroline (0.22 equiv), EtOH (2.0 mL), H2O (0.1 mL), in air, 80 °C, 22 h; (f) 30% H2O2 (1.0 mL), 3.0 N NaOH (1.0 mL), THF/EtOH (2.0 mL/0.5 mL), rt, 10 min. B. Synthesis and application of chiral hemiboronic acid catalyst: (g) Trityl bromide (1.0 equiv), Et3N (1.0 equiv), DCM (2.0 mL), 60 °C, 4.0 h. C. Practical synthesis of (±)-QPX772.
Mechanistic considerations
To gain further information regarding the reaction mechanism, we carried out 11B NMR experiments (Fig. 6a). Compared with the chemical shift (δ 39.02 ppm) of pure BBr3 in CD2Cl2, the addition of 2,6-di-tert-butylpyridine (A5, 1 equiv) caused a slight high-field shift, giving only one signal (δ 36.40 ppm) in the positive chemical shift region. Interestingly, the addition of 2,6-lutidine (A3, 1 equiv) led to several 11B signals in both positive and negative chemical shift regions, and the major peak might be corresponding to a tight bromoborenium cation complex with 2,6-lutidine45, causing the erosion of reaction yield. As we expected for a less hindered base, pyridine (A2) formed a stable complex with BBr3, and the 11B NMR showed only one signal in the negative chemical shift region (δ ‒7.33 ppm). 1H NMR experiments also showed a similar trend for the coordination of BBr3 with different bases. Upon the addition of 1 equivalent of BBr3, significant downfield shifts were observed for both A3 and A2, while little shifting effect was observed for A5 (Fig. 6b). Therefore, both 11B and 1H NMR spectroscopic characterization strongly supported our initial strategy design, in which sterically bulky base could slightly decrease the Lewis acidity of BBr3 through relatively weak coordination and mainly serve as an effective proton scavenger.

a 11B NMR stacked spectra. b 1H NMR stacked spectra.
Conclusion
In conclusion, we have developed a practical, concise, and convenient synthetic method for the construction of bicyclic boronates through metal-free heteroatom-directed alkenyl sp2-C‒H borylation. The reactions worked efficiently with good to excellent yields and were well compatible with various functional groups. More importantly, the alkene starting materials, in either (Z)- or (E)-form, could produce the desired bicyclic boronates, which significantly expanded the substrate scope of such borylative cyclization. Additionally, a gram-scale reaction for bicyclic boronates and several subsequent synthetic transformations were also presented to elaborate the valuable utilities. In particular, a practical synthesis of the ultrabroad-spectrum β-lactamase inhibitor (±)-QPX7728 was developed.
Method
General procedure for the preparation of product 3aa
A dried Schlenk flask was charged with 2,6-di-tert-butylpyridine (A5, 0.22 mmol, 1.1 equiv) and 0.8 mL of DCM at –60 °C under nitrogen atmosphere. BBr3 (2a, 0.22 mmol, 1.1 equiv; 1.0 M solution in DCM) was subsequently added dropwise while stirring. After the reaction mixture was stirred for 5 min, a solution of 1aa (0.20 mmol, 1.0 equiv) was added dropwise and the resulting mixture was stirred for another 2.0 h at –60 °C. Finally, the reaction was quenched with 2,6-di-tert-butylpyridine (A5, 1.0 equiv), methanol (1.0 mL), and water (0.2 mL). Upon the removal of solvents in vacuo, the residue was purified by flash silica gel (300-400 mesh) chromatography (petroleum ether/ethyl acetate = 5/1) to afford the desired product 3aa (30 mg) in 95% yield as white solid.
Data availability
Detailed experimental procedures and characterization of compounds can be found in the Supplementary Information. The X-ray crystallographic structure reported in this study has been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 2143877 (3aa), 2143875 (3ah), 2143812 (methyl ester of 3bh), 2144121 (3bm), 2143882 (3bo), 2196049 (3by), and 2143883 (4). These data can be obtained free of charge from The CCDC via www.ccdc.cam.ac.uk/data_request/cif. All data are available from the authors upon request. NMR spectra as a separate Supplementary Data 1. All cif files as Supplementary Data 2–8.
References
Hall, D. G. in Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, 1–133 (Wiley‐VCH Verlag GmbH & Co., 2011).
Whyte, G. F., Vilar, R. & Woscholski, R. Molecular recognition with boronic acids-applications in chemical biology. J. Chem. Biol. 6, 161–174 (2013).
Thareja, S., Zhu, M., Ji, X. & Wang, B. Boron-based small molecules in disease detection and treatment (2013–2016). Heterocycl. Commun. 23, 137–153 (2017).
Antonio, J. P. M., Russo, R., Carvalho, C. P., Cal, P. & Gois, P. M. P. Boronic acids as building blocks for the construction of therapeutically useful bioconjugates. Chem. Soc. Rev. 48, 3513–3536 (2019).
Diaz, D. B. & Yudin, A. K. The versatility of boron in biological target engagement. Nat. Chem. 9, 731–742 (2017).
Brooks, W. L. & Sumerlin, B. S. Synthesis and applications of boronic acid-containing polymers: from materials to medicine. Chem. Rev. 116, 1375–1397 (2016).
Hall, D. G. Boronic acid catalysis. Chem. Soc. Rev. 48, 3475–3496 (2019).
Krajnc, A. et al. Bicyclic boronate VNRX-5133 inhibits metallo- and serine-β-lactamases. J. Med. Chem. 62, 8544–8556 (2019).
Trout, R. E. et al. Discovery of VNRX-7145 (VNRX-5236 Etzadroxil): an orally bioavailable beta-lactamase inhibitor for enterobacterales expressing ambler class A, C, and D enzymes. J. Med. Chem. 64, 10155–10166 (2021).
Hecker, S. J. et al. Discovery of cyclic boronic acid QPX7728, an ultrabroad-spectrum inhibitor of serine and metallo-β-lactamases. J. Med. Chem. 63, 7491–7507 (2020).
Rombouts, F. J., Tovar, F., Austin, N., Tresadern, G. & Trabanco, A. A. Benzazaborinines as novel bioisosteric replacements of naphthalene: propranolol as an example. J. Med. Chem. 58, 9287–9295 (2015).
Lu, C. J. et al. Discovery of boron-containing compounds as Aβ aggregation inhibitors and antioxidants for the treatment of Alzheimer’s disease. Med. Chem. Commun. 9, 1862–1870 (2018).
Saito, H., Otsuka, S., Nogi, K. & Yorimitsu, H. Nickel-catalyzed boron insertion into the C2-O bond of benzofurans. J. Am. Chem. Soc. 138, 15315–15318 (2016).
Figueiredo, T. et al. Injectable self-healing hydrogels based on boronate ester formation between hyaluronic acid partners modified with benzoxaborin derivatives and saccharides. Biomacromolecules 21, 230–239 (2020).
van de Wouw, H. L., Lee, J. Y., Awuyah, E. C. & Klausen, R. S. A BN aromatic ring strategy for tunable hydroxy content in polystyrene. Angew. Chem. Int. Ed. 57, 1673–1677 (2018).
Ren, J. et al. Design and synthesis of boron-containing diphenylpyrimidines as potent BTK and JAK3 dual inhibitors. Bioorg. Med. Chem. 28, 115236 (2020).
Plescia, J. & Moitessier, N. Design and discovery of boronic acid drugs. Eur. J. Med. Chem. 195, 112270 (2020).
Lence, E. & González‐Bello, C. Bicyclic boronate β‐lactamase inhibitors: the present hope against deadly bacterial pathogens. Adv. Ther. 4, 2000246–2000267 (2021).
Boyer, S. H. et al. Scalable synthesis of β-Lactamase inhibitor QPX7728 by sequential nickel-catalyzed boron insertion into a benzofuran substrate and enantioselective cyclopropanation of the resulting vinylboronate. Org. Process Res. Dev. 26, 925–935 (2022).
Tsuchiya, S., Saito, H., Nogi, K. & Yorimitsu, H. Manganese-catalyzed ring opening of benzofurans and its application to insertion of heteroatoms into the C2–O bond. Org. Lett. 19, 5557–5560 (2017).
Tsuchiya, S., Saito, H., Nogi, K. & Yorimitsu, H. Aromatic metamorphosis of indoles into 1,2-benzazaborins. Org. Lett. 21, 3855–3860 (2019).
Lyu, H., Kevlishvili, I., Yu, X., Liu, P. & Dong, G. Boron insertion into alkyl ether bonds via zinc/nickel tandem catalysis. Science 372, 175–182 (2021).
Sumida, Y. et al. Boron-selective biaryl coupling approach to versatile dibenzoxaborins and application to concise synthesis of defucogilvocarcin M. Org. Lett. 16, 6240–6243 (2014).
Lv, J. et al. Metal-free directed sp2-C–H borylation. Nature 575, 336–340 (2019).
Dewar, M. J. S. & Dietz, R. New heteroaromatic compounds. Part III. 2,1-Borazaro-naphthalene (1,2-dihydro-1-aza-2-boranaphthalene). J. Chem. Soc. 546, 2728–2730 (1959).
Paetzold, P., Stanescu, C., Stubenrauch, J. R., Bienmüller, M. & Englert, U. 1-Azonia-2-boratanaphthalenes. Z. anorg. allg. Chem. 630, 2632–2640 (2004).
Li, Y. & Wu, X. F. Direct C-H bond borylation of (Hetero)arenes: evolution from noble metal to metal free. Angew. Chem. Int. Ed. 59, 1770–1774 (2020).
Iqbal, S. A., Pahl, J., Yuan, K. & Ingleson, M. J. Intramolecular (directed) electrophilic C-H borylation. Chem. Soc. Rev. 49, 4564–4591 (2020).
Tanaka, S., Saito, Y., Yamamoto, T. & Hattori, T. Electrophilic borylation of terminal alkenes with BBr3/2,6-disubstituted pyridines. Org. Lett. 20, 1828–1831 (2018).
Li, S. et al. Site-fixed hydroboration of terminal and internal alkenes using BX3 /iPr2NEt. Angew. Chem. Int. Ed. 60, 26238–26245 (2021).
Kirschner, S., Yuan, K. & Ingleson, M. J. Haloboration: scope, mechanism and utility. New J. Chem. 45, 14855–14868 (2021).
Van De Water, R. W. & Pettus, T. R. R. o-Quinone methides: intermediates underdeveloped and underutilized in organic synthesis. Tetrahedron 58, 5367–5405 (2002).
Wang, Z. et al. Organocatalytic asymmetric synthesis of 1,1-diarylethanes by transfer hydrogenation. J. Am. Chem. Soc. 137, 383–389 (2015).
Sun, J. & Wang, Z. Recent advances in catalytic asymmetric reactions of o-quinone methides. Synthesis 47, 3629–3644 (2015).
Carreras, J., Caballero, A. & Perez, P. J. Alkenyl boronates: synthesis and applications. Chem. Asian. J. 14, 329–343 (2019).
Bregent, T., Bouillon, J. P. & Poisson, T. Photocatalyzed E→Z contra-thermodynamic isomerization of vinyl boronates with binaphthol. Chemistry 27, 13966–13970 (2021).
Nevesely, T., Wienhold, M., Molloy, J. J. & Gilmour, R. Advances in the E→Z isomerization of alkenes using small molecule photocatalysts. Chem. Rev. 122, 2650–2694 (2022).
Manenzhe, N. J., Potgieter, N. & van Ree, T. Composition and antimicrobial activities of volatile components of Lippia javanica. Phytochemistry 65, 2333–2336 (2004).
Zhou, Q. J., Worm, K. & Dolle, R. E. 10-hydroxy-10,9-boroxarophenanthrenes: versatile synthetic intermediates to 3,4-benzocoumarins and triaryls. J. Org. Chem. 69, 5147–5149 (2004).
Wen, J. & Shi, Z. From C4 to C7: innovative strategies for site-selective functionalization of indole C-H bonds. Acc. Chem. Res. 54, 1723–1736 (2021).
Barluenga, J., Tomas-Gamasa, M., Aznar, F. & Valdes, C. Metal-free carbon-carbon bond-forming reductive coupling between boronic acids and tosylhydrazones. Nat. Chem. 1, 494–499 (2009).
Sumida, Y. et al. Synthesis of dibenzofurans by Cu-catalyzed deborylative ring contraction of dibenzoxaborins. Org. Lett. 22, 6687–6691 (2020).
Estrada, C. D., Ang, H. T., Vetter, K. M., Ponich, A. A. & Hall, D. G. Enantioselective desymmetrization of 2-Aryl-1,3-propanediols by direct O-Alkylation with a rationally designed chiral hemiboronic acid catalyst that mitigates substrate conformational poisoning. J. Am. Chem. Soc. 143, 4162–4167 (2021).
Durka, K., Luliński, S., Dąbrowski, M. & Serwatowski, J. Is carbon dioxide able to activate halogen/lithium exchange? Eur. J. Org. Chem. 2014, 4562–4570 (2014).
Mansaray, H. B. et al. Modelling fundamental arene-borane contacts: spontaneous formation of a dibromoborenium cation driven by interaction between a borane Lewis acid and an arene pi system. Chem. Commun. 47, 12295–12297 (2011).
Acknowledgements
Financial support was generously provided by the National Key R&D Program of China (2022YFF1202600), the National Natural Science Foundation of China (22071155, 21871184, and 81903423), the Science and Technology Commission of Shanghai Municipality (21ZR1460700, 20XD1403600, 19YF1449300, and 20400750300), the Shanghai Municipal Education Commission (2019-01-07-00-10-E00072), and the Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (No: ZYYCXTD-202004).
Author information
Authors and Affiliations
Contributions
P.-Y.P., G.-S.Z. and X.-L.L. performed the synthetic experiments and analysed the experimental data; M.-L.G. and D.G. performed the antibacterial experiments; J.-W.Z. performed the X-ray crystallographic analysis; G.-Q.L., Q.-H.L. and P.T. directed the research and prepared the manuscript. All the authors discussed the results and commented on the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
P.T., Q.-H. L., P.-Y.P., M.-L.G., G.-S.Z. and G.-Q.L. are inventors on CN patent application no. 2022104886919 filed by the Shanghai University of Traditional Chinese Medicine covering the preparation of bicyclic boronates and their applications in antibacterial agents. Other authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, PY., Zhang, GS., Gong, ML. et al. A practical preparation of bicyclic boronates via metal-free heteroatom-directed alkenyl sp2-C‒H borylation. Commun Chem 6, 176 (2023). https://doi.org/10.1038/s42004-023-00976-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-023-00976-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
