
Abstract
Although the electrochemiluminescence (ECL) of metal nanoclusters has been reported, revealing the correlation between structure and ECL at an atomic level is highly challenging. Here, we reported the impact of the metal core of Au20(SAdm)12(CHT)4 (Au20-AC for short; SAdm = 1-adamantanethiolate; CHT= cyclohexanethiol) and its isomer Au20(TBBT)16 (TBBT = 4-tert-butylthiophenol) on their solution-state and solid-state electrochemiluminescence. In self-annihilation ECL experiments, Au20-AC showed a strong cathodic ECL but a weak anodic ECL, while the ECL signal of Au20(TBBT)16 was weak and barely detectable. Density functional theory (DFT) calculations showed that the Au7 kernel of [Au20-AC]- is metastable, weakening its anodic ECL. Au20-AC in solution-state displayed an intense co-reactant ECL in the near-infrared region, which is 7 times higher than that of standard Ru(bpy)32+. The strongest solid-state ECL emissions of Au20-AC and Au20(TBBT)16 were at 860 and 770 nm, respectively — 15 nm red-shifted for Au20-AC and 20 nm blue-shifted for Au20(TBBT)16, compared to their corresponding solid-state photoluminescence (PL) emissions. This work shows that ECL is significantly affected by the subtle differences of the metal core, and offers a potential basis for sensing and immunoassay platforms based on atomically precise emissive metal nanoclusters.
Similar content being viewed by others
Introduction
Electrogenerated chemiluminescence entails the generation of electronically excited species at electrode surfaces which emit light upon their relaxation to a lower-level state. ECL is a highly sensitive and selective analytical technique, with a low detection limit in absence of background light1,2,3,4,5,6,7,8,9. Metal nanoclusters have been demonstrated to be excellent ECL luminophores due to their high intensity and efficiency10,11,12,13,14,15,16,17,18. Determined structure and fascinating properties of metal nanoclusters provide important insights for structure-property correlations and important guidance for designing functional nanomaterials19,20,21,22. Although progress of ECL of metal nanocluster has been made, the influence factors of ECL of metal nanoclusters have been unrevealed. Structural isomers of metal nanocluster are ideal models for understanding the effect of different structural moieties on ECL properties23,24,25,26. Thus far, only once case of correlation between structure and ECL property of metal nanocluster has been reported27, yet whether the different influencing factors affect the behavior of ECLs in solution is still unknown. Recently, solid-state ECL (SSECL) has been developed after the aggregation-induced ECL (AIECL) proposed28,29,30,31,32,33. Aggregation-induced emission (AIE) materials provide the possibility for AIECL. In metal nanocluster, Xie et al. revealed gold nanoclusters with long Au-SR motifs could generate strong emissions due to the aggregation of motifs on the metal cores32, which is desirable for the ECL of metal nanocluster in solid state.
Herein, we report the preparation and structure of Au20(SAdm)12(CHT)4 (abbrev. Au20-AC below, SAdm = 1-adamantanethiol, CHT = cyclohexanethiol), whose structure is similar to that of Au20-Iso1, as predicted by Pei and co-workers34; Au20-AC contain one Au7 core, two Au2(SR)3, one Au3(SR)4 and one long Au6(SR)6 motif. We explored self-annihilation and coreactant ECLs of Au20-AC and its isomer Au20(TBBT)16 (TBBT = 4-tert-butylthiophenol, reported by Jin et al.35) in solution and solid states. Electrochemistry, ECL, PL and density functional theory (DFT) simulation have been employed to establish a correlation between structure and ECL performance in different states. It is observed that Au20-AC displayed an intense cathodic ECL and a weak anodic ECL, while Au20(TBBT)16 showed weak cathodic and anodic ECLs in self-annihilation pathway. DFT calculations indicate that the anion radical is destabilized due to metal core distortion for Au20-AC, leading to weak anodic ECL. ECL spectrum of Au20-AC in solution state is centered at 830 nm in the presence of TPrA. The oxidative reduction SSECL of both Au20 clusters in phosphate buffer solution (PBS, pH = 7.5) was observed in the near-infrared region. Overall, our correlation of the structure of these Au metal nanocluster isomers with their ECL performance constitutes a possible approach towards the design of intense ECL emitters and the development of associated detection platforms6,36,37,38.
Results and discussion
Structure and characterization of Au20-AC
Au20-AC was formed by ligand etching Au18(CHT)14 nanoclusters with HSAdm39 and purified by TLC. As shown in Supplementary Fig. 1, three bands corresponding to Au20-AC, Au16 and Au21 nanoclusters from top to bottom, were observed. The UV-vis absorption spectra of these three nanoclusters are shown in Supplementary Fig. 2. Rhombic crystals of Au20-AC could be obtained within 2~3 days using a mixture of methanol and CH2Cl2.
The structure of Au20-AC was determined by single-crystal X-ray diffraction (Supplementary Data 1), which revealed that its crystals adopt the monoclinic space group C2/c. Full details are presented in Fig. 1 and Supplementary Table 1. Structurally, Au20-AC is almost identical to the predicted Au20-Iso1, and contains a Au7 kernel comprised of two fused tetrahedra (Fig. 1a) arranged linearly through a common Au atom, different to the twisted tetrahedra found in Au20(TBBT)16. The average Au−Au bond length in the Au7 kernel of Au20-AC (2.730 Å) is slightly (0.40%) longer than that in Au20(TBBT)16 (2.719 Å). Au2(SR)3, Au3(SR)4 and circle-like Au6(SR)6 motifs were observed to cap the core tetrahedra (Fig. 1d, e, b). Interestingly, the mixed thiol ligands HSAdm and CHT comprise the Au3(SR)4 and Au6(SR)6 motifs, forming Au3(SAdm)2(CHT)2 and Au6(SAdm)4(CHT)2 motifs. The energy of Au20-Iso1 was predicted to be comparable to or even lower than that of Au20(TBBT)1634. The use of mixed ligands instead of the more usual single thiol ligand might facilitate the experimental preparation of Au20-Iso1. Regarding the Au20-AC superstructure, the cyclohexane units present in the Au6(SAdm)4(CHT)2 motifs adopt a chair configuration and are arranged in a head-to-head pattern due to the non-covalent interactions of C…H and H…H (Fig. 1g), contributing to the high stability of Au20-AC in the aggregated state.

a The di-tetrahedral Au7 kernel. b The circular Au6(SAdm)4(CHT)2 motif. c The structure of kernel and motifs in Au20(SAdm)12(CHT)4. d The dimeric Au2(SAdm)3 motif. e The trimeric Au3(SAdm)2(CHT)2 motif. f The total structure of Au20(SAdm)12(CHT)4, all H atoms are omitted for clarity. g The arrangement of cyclohexane rings in a head-to-head pattern in neighboring CHT of Au20-AC. (Color labels: green, yellow = Au; red = S; grey, blue = C; white = H).
The composition of Au20-AC was further confirmed by electrospray ionization mass spectrometry, X-ray photoelectron spectroscopy and thermogravimetric analysis. Cesium acetate (CsOAc) was added, to form adducts. As shown in Supplementary Fig. 3, the electrospray ionization mass spectra depicts a series of peaks, each separated by 52 Da and corresponding to a series of different Au20-AC moieties. [Au20(C10H15S)16-n(C6H11S)n + Cs]+ (n = 4~9) containing HSAdm and CHT ligands in different combinations, reflecting their dynamic equilibrium in the solution state. However, the crystal state Au20-AC was determined by single crystal diffraction to contain 12 HSAdm and 4 CHT ligands. In mass spectrometry, the isotopic peaks of [Au20(SAdm)12(CHT)4 + Cs]+ are consistent with the simulated spectra (Supplementary Fig. 3). The composition of Au20-AC was also confirmed by X-ray photoelectron spectrum (Supplementary Fig. 4) and thermogravimetric analysis (Supplementary Fig. 5).
Photoluminescence and voltammetry analysis of Au20 isomers
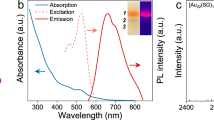
Au20-AC and Au20(TBBT)16 in solid state were intensely photoluminescent. Figure 2a shows the PL spectra of solid Au20-AC (red line) and Au20(TBBT)16 (blue line); the peaks are at 845 and 790 nm, respectively. A solution of Au20-AC was non-fluorescent at the same excitation wavelength, reflecting its strong AIE behaviour (Supplementary Fig. 6). In the solution state, van der Waals forces drive the rotation of molecules until their surface motifs establish stable interactions with neighbouring nanoclusters; the free rotation of the ligands and any dissociation/association of the thiol ligands results in the loss of non-radiative energy40. Accordingly, Au20-AC was barely luminescent in solution. However, the ligands of aggregated and crystallized nanoclusters are fixed. This restriction of intramolecular rotation (RIR) and suppression of any disaggregation and association processes inhibits the non-radiative decay of excited states, resulting in intense AIE in the solid state. The ligands of Au20-AC are non-aromatic and thus unable to π–π stack, but their long-range interaction (between unit cells) may still affect their PL40. Aggregated Au20-AC showed a longer PL lifetime (2.19 μs) than aggregated Au20(TBBT)16 (624 ns) (Supplementary Figs. 7 and 8).

a Normalized PL of solid Au20-AC and Au20(TBBT)16. Inset: The crystal of Au20-AC under visible light (top) and ultraviolet light (bottom), respectively. b SWV of 0.015 mM Au20-AC and Au20(TBBT)16 in 1:1 TOL: ACN with 0.1 M TBAP. Pt disk was used as working electrode, Pt foil and SCE were used as counter and reference electrode, respectively.
Cyclic voltammetry (CV) studies of Au20-AC and Au20(TBBT)16 were carried out by scanning between −1.7 and 1.2 V vs. SCE at a scan rate of 0.1 V s−1 (Supplementary Figs. 9 and S10). The square wave voltammetry (SWV) of Au20-AC and Au20(TBBT)16 was tested in a 1:1 solution of toluene (TOL)/acetonitrile (ACN) with 0.1 M tetrabutylammonium perchlorate (TBAP) as the electrolyte. As shown in Fig. 2b, two quasi-reversible oxidation waves at EOx = 0.71 V and 1.01 V were observed (Fig. 2b, red line in top panel). These correspond to the Au20-AC being oxidized consecutively to [Au20-AC]1+ and then [Au20-AC]2+. Au20-AC shows one multi-electron reduction wave at ERe = −1.19 V, which indicates an irreversible reduction process in Au20-AC. Two oxidation peaks of Au20(TBBT)16 at 0.74 V and 1.02 V and a multi-electron reduction peak at −1.13 V were observed (Fig. 2b, blue line in down panel). All these redox processes of Au20(TBBT)16 are quasi-reversible. The potential differences between the first reduction and oxidation peak of Au20-AC and Au20(TBBT)16 were 2.10 and 2.07 V. The surface ligands and motifs of Au20-AC lowered its reduction and oxidation potentials compared to those of Au20(TBBT)16. The propensity of these nanoclusters to gain and lose electrons is related to their HOMO–LUMO gap and ligand-metal and metal-metal electronic coupling/interaction properties41.
To rationalize the electrochemical properties of the gold clusters, DFT/PBE calculations were performed—the HOMO energies of Au20-AC and Au20(TBBT)16 were calculated to be −4.23 eV and −4.41 eV, respectively (Supplementary Fig. 11). This is consistent with the lower oxidation potential of Au20-AC compared to Au20(TBBT)16 (0.71 V vs 0.74 V); since the oxidation process involves removing an electron from the HOMO energy level, the Au20-AC cluster with the higher HOMO energy level will lose an electron more easily compared to Au20(TBBT)16, whose lower HOMO energy level (−4.41 eV) is consistent with its higher relative oxidation potential (0.74 V). On the contrary, the LUMO energy level will acquire an electron during reduction, thus the Au20-AC cluster with a higher LUMO energy level (−2.38 eV) will have more difficulty in obtaining an electron, resulting in a relatively low reduction potential (−1.19 V). The Au20(TBBT)16 cluster with a lower LUMO energy level (−2.74 eV) will have a relatively high reduction potential (−1.13 V). The HOMO-LUMO gaps of the Au20-AC and Au20(TBBT)16 clusters are 1.85 eV and 1.67 eV, respectively. The DFT calculations are in good agreement with the experiments.
ECL of Au20 isomers in solution state and DFT theoretical analysis
The ECLs of both nanoclusters in solution state was studied in TOL/ACN (1:1) with Pt mesh and applied potential of between −1.6 V and 1.1 V. Figure 3a shows the step ECLs of both Au20 nanoclusters with negative and positive potentials alternately applied for three cycles. Overall, the ECL of Au20-AC is stronger than that of Au20(TBBT)16. Au20-AC shows intense ECL at −1.6 V (onset of 15 and 25 s) but weak ECL at 1.2 V (onset of 10, 20 and 30 s). In addition, the self-annihilation ECL of Au20-AC and Au20(TBBT)16 were explored in potential scanning experiment. As shown in Fig. 3b, both cathodic and anodic ECL were observed in the 3rd cycle scanning potential for Au20-AC and the onset potential of reductive ECL of Au20-AC is at −1.0 V that is consistent with the onset potential of reduction peak in CV curve. No cathodic ECL signal was observed in the potential sweep of the first cycle because no cationic radicals were generated at this time (Supplementary Fig. 12). The ECL intensity of Au20-AC under consecutive potential scans decreased slightly with relative standard deviation (RSD) of 7.52% (Supplementary Fig. 13). While Au20(TBBT)16 display quite weak self-annihilation ECL at all potentials (Fig. 3b, blue curve).

a Potential step self-annihilation ECL of Au20-AC and Au20(TBBT)16. The concentration of nanocluster is 0.015 mM in Tol/ACN (1:1) with 0.1 M TBAP electrolyte. The potential was set between −1.6 and 1.1 V and stepped cyclically, each potential was held for 5 s, and no potential was applied in the first and final 5 s. Pt mesh was used as working electrode, Pt foil and SCE were used as counter and reference electrode, respectively. b Self-annihilation ECL-voltage curves from −1.6 V to 1.1 V of Au20-AC and Au20(TBBT)16 in solution state. Initial scan to negative potential direction. The 3rd cycle data was shown. c, d Theoretical calculation of structures of different valence states of Au20-AC and Au20(TBBT)16 radical. The corresponding bond length is shown in Supplementary Table 2.
To explore the influence factors of ECL performance of such two Au20 nanoclusters, we performed DFT calculations and studied the metal core and motif effects on their ECLs. As shown in Fig. 3c, d and Supplementary Table 2, both the Au2-Au3 and Au5-Au6 bond lengths in the Au7 kernel of the [Au20-AC]- anion were longer than those in the neutral Au20-AC cluster (3.281 Å vs 2.810 Å and 3.050 Å vs 2.819 Å, respectively), reflecting their lower bond energies. There was no significant difference in the averaged Au-S distances in the core-shell and motif, indicating bare influence of different thiols and motifs on ECL performance (Supplementary Fig. 14 and Table 3). These bond length analyses and structural representations (Fig. 3c, d) indicate that the metal core of [Au20-AC]- is distorted, reducing the overall stability of the [Au20-AC]- and precluding its ability to react with cluster cations to form a strong anodic ECL signal (Fig. 3a, onset of 10, 20 and 30 s). The low stability of [Au20-AC]- from theoretical simulations is consistent with the result obtained from voltammetric curves that [Au20-AC]- is irreversible during the reduction process (Fig. 2b, top panel). However, more stable cluster radical cations can react with newly formed radical anions to form stronger cathodic ECL signals during the application of negative potential (Fig. 3a, onset of 15 and 25 s).
A possible self-annihilation ECL mechanism of the Au20 clusters is proposed in Supplementary Equation (1-4) (Supplementary Information Section 3); Au20 and Au20* denote the ground- and excited-states of the Au20 (including Au20-AC and Au20(TBBT)16) clusters, respectively. In the self-annihilation ECL process, negatively charged Au20−∙ and positively charged Au20+∙ cluster radicals are generated by electrode electron transfer reaction (Supplementary Equation (1) and (2)). Electrons or holes are transferred between cluster and the electrode surface. The generated radical cation and anion react and produce excited Au20* (Supplementary Equation (3)), which relax to the ground state and release the energy via photon emission (Supplementary Equation (4)). The self-annihilation ECL intensity is dependent on the stabilities of the Au20−∙ and Au20+∙ intermediates that are generated on the electrode upon application of a potential. Simultaneously, the transfer rate of the electrons and holes, the reaction capability of radical anions and cations, and the radiative efficiency of the excited state will affect the ECL intensity. Although the metastability of anodic and cationic radicals of Au20(TBBT)16 cluster were not observed in the DFT theoretical simulation, its ECL signal was barely observed in potential and scanning experiments. Thus, we studied the emission capability of the excited states of Au20-AC and Au20(TBBT)16. As shown in Supplementary Fig. 15 and Table 4, both excited nanoclusters displayed comparable emission capability. We speculate that the limited transfer rate of the electrons and holes and weak reaction capability between radical anions and cations may be responsible for the weak self-annihilation of Au20(TBBT)16.
ECL efficiency of Au20-AC and Au20(TBBT)16 in solution state
The ECL efficiency of Au20-AC and Au20(TBBT)16 is assessed by comparing to Ru(bpy)32+-TPrA under the same measurement conditions. Figure 4a shows the step ECLs of Au20-AC, Au20(TBBT)16 and Ru(bpy)32+ in solution state in the prescnece of 5 mM TPrA as coreactant, in which “catalytic route” reactions occured42. The ECL efficiencies of Au20-AC and Au20(TBBT)16 were evaluated by Ru(bpy)32+/TPrA standard (Supplementary Fig. 16 and Table 5). The ECL efficiency of Au20-AC/TPrA is more than 7 times higher than that of Ru(bpy)32+/TPrA. Intense coreactant ECL of Au20-AC allow its ECL spectrum to be collected. As shown the inset in Fig. 4a, Au20-AC display an ECL emission band centered at 830 nm. In ECL-voltage curves, the ECL signal of Au20-AC began to appear at about 0.5 V, and reached maximum at 1.1 V, while the signal of Au20(TBBT)16 is still very weak (Fig. 4b).

Coreactant ECL of Au20-AC, Au20(TBBT)16 and Ru(bpy)32+ in solution state. Coreactant ECL experiments were executed in 1:1 TOL: ACN with 0.1 M TBAP and 5 mM TPrA. Pt mesh was used as working electrode, Pt foil and SCE were used as counter and reference electrodes, respectively. a Potential step coreactant ECLs of Au20-AC, Au20(TBBT)16 and Ru(bpy)32+ in solution state. Inset is the spectrum of Au20-AC in solution state. The electrode potential was held for 2 s at denoted potentials in each step over three cycles. No potential was applied in the first and final 2 s. b ECL-voltage curves of Au20-AC, Au20(TBBT)16 and Ru(bpy)32+ in solution state. Potential scan rate is 0.1 V/s. The 3rd cycle data was shown.
Solid-state ECL of Au20-AC and Au20(TBBT)16 in coreaction pathway
Au20-AC and Au20(TBBT)16 displayed red emission in solid state after light irradiation due to the AIE effect. With this in hand, we studied their SSECL. The SSECL of both nanoclusters was studied by separately loading 15 μg of each onto a GCE and allowing the electrode surface to dry in air. The electrochemical impedance spectroscopy (EIS) of both nanoclusters showed that their resistances are comparable (Supplementary Fig. 17), indicating a similar amount of both nanoclusters on electrode. The SSECL was conducted in 0.01 M phosphate buffer solution (PBS, pH = 7.5) in the presence of 0.1 M KCl as an electrolyte. As shown in Fig. 5, the similar oxidation potential of the cluster and coreactant is favorable to the ECL generation. Au20-AC showed the strongest emission at 1.1 V (Fig. 5a), but Au20(TBBT)16 showed the strongest emission at 0.9 V in forward scan (Fig. 5b). The relatively strong SSECL of Au20(TBBT)16 may be due to the motif aggregation effect. The long eight-membered ring motif of Au20(TBBT)16 induces intense SSECL. The SSECLs of both Au20 nanoclusters are much higher than that of Au25(SC2H4Ph)18 whose sweeping potential ECL cannot be detected, and step ECL is quite weak (Supplementary Fig. 18). Both RIR and coreactant effects contribute significantly to the intense ECL performance. Intense SSECLs allow the spectra of Au20-AC and Au20(TBBT)16 to be collected. The ECL spectra of Au20-AC and Au20(TBBT)16 at various potentials are presented in Fig. 5c, d. No emission peak shift was observed for either Au20 cluster. All the peaks of Au20-AC were centered at λmax = 860 nm, and the strongest emission of Au20(TBBT)16 was at λmax = 770 nm. Therefore, the ECL emission of Au20-AC was 15 nm red-shifted compared to its PL emission, but the ECL emission of Au20(TBBT)16 was 20 nm blue-shifted compared to its PL emission. This slight difference between PL and ECL might reflect differences in their excited states43.

a, b SSECL-voltage curves of Au20-AC (red line) and Au20(TBBT)16 (blue line) in the presence of 5 mM TPrA. Coreactant SSECL experiments were carried out in 0.01 M PBS with 0.1 M KCl. 15 μg Au20-AC and Au20(TBBT)16 were coated on GCE, Pt foil and SCE were used as counter and reference electrode. The potential was set from -0.1 to 1.1 V at 0.1 V/s. The 3rd cycle data was shown. c, d The SSECL spectra of Au20-AC and Au20(TBBT)16 in the forward scan (pink) and reverse scan (blue). The potential was set from −0.1 to 1.1 V at 0.1 V/s with one spectrum collected every 1 s.
Conclusions
The structurally predicted Au20(SAdm)12(CHT)4 (Au20-AC) nanocluster has been prepared and its electrochemiluminescence performance compared with that of its isomer Au20(TBBT)16. Au20-AC showed a strongly cathodic and weakly anodic ECL via a self-annihilation pathway in solution state. By comparing the averaged Au-Au and Au-S distances in various gold cluster radicals, DFT simulation revealed the impact of the gold core on the electrochemiluminescence of Au20-AC and its isomer Au20(TBBT)16. The [Au20-AC]-∙ with a deformed Au7 kernel are metastable, which results in its weakly anodic ECL. Solid Au20-AC and Au20(TBBT)16 both showed intense ECL in the presence of the TPrA co-reactant, and therefore are suitable for developing solid-state, anti-quenching ECL luminophores. Coreactant SSECL spectra were also collected, and their intense signals enabled ECL and PL to be compared. Further work to explore and address the interactions between the emitter, electrode, and solution interfaces are underway. This work provides insights into the relationship between the structure and ECL properties of atomic precision metal nanoclusters, and is expected to pave the way for new emitters that can be used for biosensing and immunoassays.
Materials and methods
Synthesis
Au20-AC is prepared by ligand etching using Au18(CHT)14 as a precursor. Au18(CHT)14 nanoclusters were synthesized according to the method reported in the literature39.
Step 1: Synthesis of Au18(CHT)14. 150 mg HAuCl4·3H2O and 260 mg L‐glutathione were mixed with methanol for 15 min and then stirred at a lower speed. After 15 min, the mixture was diluted with a large amount (20 mL) of methanol, with high-speed stirring. After 15 min, a freshly prepared solution of sodium cyanoborohydride (NaBH3CN, 63 mg in 5 ml methanol) was added. After 6 h, the methanol solution was dried and 20 mL H2O and 20 mL dichloromethane (CH2Cl2) added. Excess cyclohexanethiol (1 mL) was added at 40 °C, to accomplish the two-phase ligand exchange. After stirring for 10 hours, the dichloromethane phase was dried and the Au18(SC6H11)14 residue thoroughly washed with methanol.
Step 2: Synthesis of Au20(SAdm)12(CHT)4. 100 mg (598.80 μmol) 1-Adamantanethiol (HSAdm) was added to a solution of Au18(SC6H11)14 (~10 mg, 1.94 μmol) in CH2Cl2 at 40 °C. After mixing overnight, the mixture was pipetted onto a TLC plate which was separated in a developing tank (CH2Cl2/Hex =1:4 v/v) (Supplementary Fig. 1). Bands corresponding to the different nanoclusters were removed and dissolved in CH2Cl2. Rhombic crystals of Au20(SAdm)12(CHT)4 were obtained within 3 days via layer diffusion of methanol into a CH2Cl2 solution of the nanoclusters. The yield was about 10.2% based on the Au element (calculated from HAuCl4·3H2O).
Electrochemical measurements
Electrochemical experiments were performed on a CHI 660e. A platinum mesh was used as the working electrode. A Pt foil and saturated calomel electrode (SCE) served as the counter and reference electrodes, respectively. The concentration of the samples was ~0.015 mM with 0.1 M TBAP in 10 ml toluene/acetonitrile (Volume ratio 1:1), and the solution was purged with argon for 15 min before experiments. All data were collected at room temperature.
ECL experiments were also performed with a three-electrode system in a quartz cuvette. A platinum mesh as the working electrode, which can provide a larger specific surface area compared to platinum disk electrodes. The dimensions of the platinum mesh electrodes used in the experiments are all 10 × 10 mm, and the effective surface area is 92.9 mm2 according to the calculation formula \(I=2.69\times {10}^{5}A{D}^{1/2}{n}^{3/2}{v}^{1/2}c\)44. The cuvettes were aligned at a fixed position with respect to the camera for consistency. 15 μg samples were coated on glassy carbon electrode (GCE) for SSECL. A saturated calomel electrode (SCE) served as the reference electrode and a Pt foil as the counter electrode. The emission intensity was recorded with an Andor iDUS CCD camera (model No: DU401A-BR-DD). The camera was externally triggered by the potentiostat (Gamry Reference 600+) for synchronization. ECL spectra were collected with an Andor spectrograph (Kymera 193i). The sample solution was purged for about 15 min with argon prior to the measurements.
Data availability
The data that support the findings of this study, including supplementary method, figures, and tables, are available in its Supplementary Information files. Other relevant data are available from the corresponding author upon reasonable request. The X-ray crystallographic coordinates for structures of Au20(SAdm)12(CHT)4 in this article can be found from Supplementary Data 1 or at the Cambridge Crystallographic Data Centre (CCDC: www.ccdc.cam.ac.uk) under accession number 2120320.
References
Ding, Z. et al. Electrochemistry and electrogenerated chemiluminescence from silicon nanocrystal quantum dots. Science 296, 1293–1297 (2002).
Li, L., Chen, Y. & Zhu, J. J. Recent advances in electrochemiluminescence analysis. Anal. Chem. 89, 358–371 (2017).
Liu, Z., Qi, W. & Xu, G. Recent advances in electrochemiluminescence. Chem. Soc. Rev. 44, 3117–3142 (2015).
Miao, W. Electrogenerated chemiluminescence and its biorelated applications. Chem. Rev. 108, 2506–2553 (2008).
Zhang, Y. et al. Switching the photoluminescence and electrochemiluminescence of liposoluble porphyrin in aqueous phase by molecular. Regul. Angew. Chem. Int. Ed. 59, 23261–23267 (2020).
Han, Z. et al. Electrochemiluminescence platforms based on small water-insoluble organic molecules for ultrasensitive aqueous-phase detection. Angew. Chem. Int. Ed. 58, 5915–5919 (2019).
Tan, X., Zhang, B. & Zou, G. Z. Electrochemistry and electrochemiluminescence of organometal halide perovskite nanocrystals in aqueous medium. J. Am. Chem. Soc. 139, 8772–8776 (2017).
Zhu, D. et al. Dual intrareticular oxidation of mixed-ligand metal-organic frameworks for stepwise electrochemiluminescence. J. Am. Chem. Soc. 143, 3049–3053 (2021).
Murray, R. W. Nanoelectrochemistry: metal nanoparticles, nanoelectrodes, and nanopores. Chem. Rev. 108, 2688–2720 (2008).
Carrara, S., Aliprandi, A., Hogan, C. F. & De Cola, L. Aggregation-induced electrochemiluminescence of platinum(II) complexes. J. Am. Chem. Soc. 139, 14605–14610 (2017).
Wei, X. et al. Aggregation-induced electrochemiluminescence of carboranyl carbazoles in aqueous media. Angew. Chem. Int. Ed. 58, 3162–3166 (2019).
Peng, H. et al. Dual enhancement of gold nanocluster electrochemiluminescence: electrocatalytic excitation and aggregation-induced emission. Angew. Chem. Int. Ed. 59, 9982–9985 (2020).
Kwak, K. & Lee, D. Electrochemistry of atomically precise metal nanoclusters. Acc. Chem. Res 52, 12–22 (2019).
Swanick, K. N., Hesari, M., Workentin, M. S. & Ding, Z. Interrogating near-infrared electrogenerated chemiluminescence of Au25(SC2H4Ph)18+ clusters. J. Am. Chem. Soc. 134, 15205–15208 (2012).
Hesari, M., Workentin, M. S. & Ding, Z. Highly efficient electrogenerated chemiluminescence of Au38 nanoclusters. ACS Nano 8, 8543–8553 (2014).
Hesari, M. & Ding, Z. A grand avenue to Au nanocluster electrochemiluminescence. Acc. Chem. Res 50, 218–230 (2017).
Tian, R. et al. Localization of Au nanoclusters on layered double hydroxides nanosheets: confinement-induced emission enhancement and temperature-responsive luminescence. Adv. Funct. Mater. 25, 5006–5015 (2015).
Jin, R. C., Zeng, C. J., Zhou, M. & Chen, Y. X. Atomically precise colloidal metal nanoclusters and nanoparticles: fundamentals and opportunities. Chem. Rev. 116, 10346–10413 (2016).
Liu, H. et al. Atomic-precision gold clusters for NIR-II imaging. Adv. Mater. 31, e1901015 (2019).
Kang, X. & Zhu, M. Tailoring the photoluminescence of atomically precise nanoclusters. Chem. Soc. Rev. 48, 2422–2457 (2019).
Hesari, M., Ding, Z. F. & Workentin, M. S. Electrogenerated chemiluminescence of monodisperse Au144(SC2H4Ph)60 clusters. Organometallics 33, 4888–4892 (2014).
Chakraborty, I. & Pradeep, T. Atomically precise clusters of noble metals: emerging link between atoms and nanoparticles. Chem. Rev. 117, 8208–8271 (2017).
Tian, S. et al. Structural isomerism in gold nanoparticles revealed by X-ray crystallography. [Corrected]. Nat. Commun. 6, 8667 (2015).
Chen, Y. et al. Isomerism in Au28(SR)20 nanocluster and stable structures. J. Am. Chem. Soc. 138, 1482–1485 (2016).
Xia, N. et al. Structural oscillation revealed in gold nanoparticles. J. Am. Chem. Soc. 142, 12140–12145 (2020).
Qin, Z. et al. Atomically precise nanoclusters with reversible isomeric transformation for rotary nanomotors. Nat. Commun. 11, 6019 (2020).
Hesari, M. & Ding, Z. Identifying highly photoelectrochemical active sites of two Au21 nanocluster isomers toward bright near-infrared electrochemiluminescence. J. Am. Chem. Soc. 143, 19474–19485 (2021).
Mei, J., Leung, N. L. C., Kwok, R. T. K., Lam, J. W. Y. & Tang, B. Z. Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 115, 11718–11940 (2015).
Luo, J. et al. Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem. Commun. 18, 1740–1741 (2001).
Wei, X., Zhu, M. J., Yan, H., Lu, C. & Xu, J. J. Recent advances in aggregation-induced electrochemiluminescence. Chemistry 25, 12671–12683 (2019).
Wong, J. M. et al. Revealing crystallization-induced blue-shift emission of a di-boron complex by enhanced photoluminescence and electrochemiluminescence. Angew. Chem. Int. Ed. 59, 17461–17466 (2020).
Wu, Z. et al. Unraveling the impact of gold(I)-thiolate motifs on the aggregation-induced emission of gold nanoclusters. Angew. Chem. Int. Ed. 59, 9934–9939 (2020).
Luo, Z. T. et al. From aggregation-induced emission of Au(I)-thiolate complexes to ultrabright Au(0)@Au(I)-thiolate core-shell nanoclusters. J. Am. Chem. Soc. 134, 16662–16670 (2012).
Wang, P. et al. A revisit to the structure of Au20(SCH2CH2Ph)16: a cubic nanocrystal-like gold kernel. Nanoscale 10, 10357–10364 (2018).
Zeng, C., Liu, C., Chen, Y., Rosi, N. L. & Jin, R. Gold-thiolate ring as a protecting motif in the Au20(SR)16 nanocluster and implications. J. Am. Chem. Soc. 136, 11922–11925 (2014).
Liao, Y. H. et al. In-situ produced ascorbic acid as coreactant for an ultrasensitive solid-state tris(2,2 ‘-bipyridyl) ruthenium(II) electrochemiluminescence aptasensor. Biosens. Bioelectron. 26, 4815–4818 (2011).
Jin, Z. C. et al. Electroactive metal-organic frameworks as emitters for self-enhanced electrochemiluminescence in aqueous medium. Angew. Chem. Int. Ed. 59, 10446–10450 (2020).
Zhang, H. et al. Bacteria photosensitized by intracellular gold nanoclusters for solar fuel production. Nat. Nanotechnol. 13, 900 (2018).
Chen, S. et al. The structure and optical properties of the [Au18(SR)14] nanocluster. Angew. Chem. Int. Ed. 54, 3145–3149 (2015).
Li, Y. et al. Double-helical assembly of heterodimeric nanoclusters into supercrystals. Nature 594, 380–384 (2021).
Lee, D., Donkers, R. L., DeSimone, J. M. & Murray, R. W. Voltammetry and electron-transfer dynamics in a molecular melt of a 1.2 nm metal quantum dot. J. Am. Chem. Soc. 125, 1182–1183 (2003).
Miao, W., Choi, J.-P. & Bard, A. J. Electrogenerated Chemiluminescence 69: The Tris(2,2‘-bipyridine)ruthenium(II), (Ru(bpy)32+)/Tri-n-propylamine (TPrA) System RevisitedA New Route Involving TPrA•+ Cation Radicals. J. Am. Chem. Soc. 124, 14478–14485 (2002).
Chen, S. et al. Near infrared electrochemiluminescence of rod-shape 25-atom AuAg nanoclusters that is hundreds-fold stronger than that of Ru(bpy)3 standard. J. Am. Chem. Soc. 141, 9603–9609 (2019).
Song, X. et al. Peptide-based electrochemiluminescence biosensors using silver nanoclusters as signal probes and Pd-Cu2O hybrid nanoconcaves as coreactant promoters for immunoassays. Anal. Chem. 93, 13045–13053 (2021).
Acknowledgements
We acknowledge the finical support of the NSFC (Grant Nos. 21631001, 22004001), the Ministry of Education, the Ministry of Education and Education Department of Anhui Province (2008085QB84), and the University Synergy Innovation Program of Anhui Province (GXXT-2020-053). Y.P. acknowledges financial support from the NSFC (91961121).
Author information
Authors and Affiliations
Contributions
S.C., Y.L., and K.K. synthesized the samples and carried out the experimental tests. B.Y., X.W., and L.J., synthesized the samples. P.W. and Y.P. performed the DFT calculations. S.C. and Y.L. solved the crystal structures. S.C., P.W., Y.P., and M.Z. designed the study. S.C., Y.L., and P.W. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Zhifeng Ding, Jun-Jie Zhu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, S., Liu, Y., Kuang, K. et al. Impact of the metal core on the electrochemiluminescence of a pair of atomically precise Au20 nanocluster isomers. Commun Chem 6, 105 (2023). https://doi.org/10.1038/s42004-023-00907-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-023-00907-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
