
Abstract
Retrosynthesis is a procedure where a target molecule is transformed into potential reactants and thus the synthesis routes can be identified. Recently, computational approaches have been developed to accelerate the design of synthesis routes. In this paper,we develop a generative framework G2Retro for one-step retrosynthesis prediction. G2Retro imitates the reversed logic of synthetic reactions. It first predicts the reaction centers in the target molecules (products), identifies the synthons needed to assemble the products, and transforms these synthons into reactants. G2Retro defines a comprehensive set of reaction center types, and learns from the molecular graphs of the products to predict potential reaction centers. To complete synthons into reactants, G2Retro considers all the involved synthon structures and the product structures to identify the optimal completion paths, and accordingly attaches small substructures sequentially to the synthons. Here we show that G2Retro is able to better predict the reactants for given products in the benchmark dataset than the state-of-the-art methods.
Similar content being viewed by others

Introduction
Retrosynthesis is a procedure where a target molecule is transformed into potential reactants and thus the synthesis routes can be identified. One-step retrosynthesis, which transforms a molecule into the possible direct reactants that can be used to synthesize the molecule, serves as the foundation of multi-step synthesis planning1,2 that identifies a full synthesis route in which the target molecule can be made through a series of one-step synthesis reactions. In drug discovery, identifying feasible synthesis routes for drug-like molecules remains a factor that substantially challenges medicinal chemists in making the desired molecules experimentally3. An extensive, diverse library of high-quality synthesis routes for a given molecule has the potential to enable more feasible reaction solutions starting from commercially available, chemical building blocks, and to provide more options for operationally simple, high-yielding transformations using widely accessible reactants.
Current retrosynthesis planning is primarily conducted by synthetic and medicinal chemists based on their knowledge and experience. It has been long known that there exists substantial disagreement among chemists in assessing synthesisbilty and designing synthesis routes4,5,6,7. In addition, an ever-increasing number of new chemical reactions makes it highly challenging for a chemist to keep up to date. Therefore, a data-driven model that predicts synthetic reactions could provide a useful complement to chemist evaluations, and could provide a large pool of potential reactions that the chemists can consider. There exist proprietary synthesis reaction databases manually curated from the literature, including Reaxys8 and SciFinder9. Unfortunately, the high prices of these databases act to limit their accessibility in some academic and small biotech settings. Open-sourced synthesis reaction databases such as the Open Reaction Database10 are limited in the reactions they cover (e.g., majorities are United States Patent and Trademark Office (USPTO) public reactions11) and their search functionalities (e.g., via SMILES strings). Even with the aid of these databases, the development of new reactions and synthetic pathways for the preparation of challenging molecules remains non-trivial. In addition, database searches can be time-consuming with low throughput, particularly when without extensive domain knowledge to guide the process. Recent in silico retrosynthesis prediction methods using deep learning12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32 have enabled alternative computationally generative processes to accelerate the conventional paradigm. These deep-learning methods learn from string-based representations (SMILES) or graph representations of given molecules, and generate possible reactant structures that can be used to synthesize these molecules, leveraging the advancement of natural language processing33, graph neural networks34, variational auto-encoders35 and other techniques in deep learning. They have demonstrated strong potential to substantially accelerate and advance retrosynthesis analysis36. In this manuscript, we focus on the one-step retrosynthesis prediction, which predicts the possible direct reactants for the synthesis of the target molecules, and acts as the foundation of multi-step retrosynthesis analysis1.
We develop a semi-template-based method via deep learning for one-step retrosynthesis prediction, denoted as G2Retro. G2Retro imitates the reversed logic of synthetic reactions: it first predicts the reaction centers in the target molecules, identifies the synthons needed to assemble the final products, and transforms these synthons into reactants. Therefore, G2Retro follows the semi-template-based frame, as in the previous methods27,28,29,30. To predict reaction centers, G2Retro learns from the molecular graphs of the products via a customized graph representation learning37 and embedding approach (in “Molecule Representation Learning” Section), and uses the graph structures to predict potential reaction centers. G2Retro defines a comprehensive set of reaction center types, and for each reaction center type, uses the graph structures that are most relevant to that reaction center type (in “Reaction Center Identification” Section). G2Retro-B integrates information of synthetically accessible fragments in its molecule graph representation learning (in Supplementary Note 1).
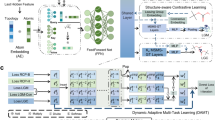
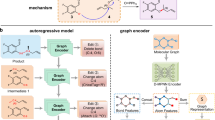
The predicted reaction centers by G2Retro split the products into synthons. To complete synthons into reactants, G2Retro considers all the involved synthon structures and the product structures to identify the optimal completion paths (in “Attachment Continuity Prediction (AACP)” Section), and accordingly attaches small substructures (i.e., bonds or rings) sequentially to the synthons until the extended synthon structures are predicted as possible reactants (in “Attachment Type Prediction (AATP)” Section). All the involved predictions in G2Retro and G2Retro-B are done via tailored neural networks. Note that G2Retro and G2Retro-B allow multiple reaction centers and multiple completion paths for each product to increase diversity in its predicted reactions. That is, the top predicted reaction centers (according to predicted likelihoods) are all tested in synthon completion to produce different reactions. Meanwhile, to avoid the exhaustive generation of all possible reactions from the top reaction centers, G2Retro prioritizes the most possible completion paths via a new beam search strategy (in “Inference” Section). An ensemble of G2Retro was also developed, denoted as G2Retro-ens, an ensemble of G2Retro, increases the pool of generated reactions by combining multiple G2Retro models and their predictions. Figure 1 presents an overview of G2Retro. A comprehensive review of existing retrosynthesis prediction methods and related fragment-based molecular generation methods is available in “Related Work” Section.

a G2Retro reaction center identification. G2Retro uses a graph message passing network (GMPN); G2Retro predicts three types of reaction centers: newly formed bonds (BF-center), bonds with type changes (BC-center), and atoms with leaving fragments (A-center); for BF-center, G2Retro also predicts bonds that have type changes induced by the newly formed bonds (BTCP); for all the reaction center types, G2Retro predicts atoms with charge changes (ACP). b G2Retro synthon completion. G2Retro uses GMPN to represent both the products and the synthons; G2Retro sequentially predicts whether a new substructure should be attached (AACP) and the type of the attachment (AATP); G2Retro adds predicted substructures until AACP predicts ‘stop’.
As a summary, G2Retro has the following advantages:
-
G2Retro follows a semi-template-based framework, predicts reaction centers of different types in products first, and then transforms the resulting synthons into reactants by adding substructures to the synthons. This process imitates the reversed logic of synthetic reactions and enables necessary interpretability as to which reaction centers are predicted by G2Retro, which reactants are generated from the reaction centers and the corresponding step-by-step generation process.
-
G2Retro defines a comprehensive set of reaction center types, covering 97.5% of the test data and conforming to synthetic chemistry knowledge. New customized neural networks are developed to predict each type of the reaction centers as well as their associated atom changes. Multiple reaction center candidates are considered for each product to enable diverse reactions generated from different reaction centers in the predicted reactions.
-
G2Retro develops a new fragment-based generation strategy compared to the previous semi-template-based methods27,28,29,30, to complete synthons into reactants by sequentially attaching substructures (i.e., bonds and rings) starting from the predicted reaction centers (in “Synthon Completion” Section). The prediction of these substructure attachments utilizes a holistic view of the most updated structures of the synthon to be completed, and the structures of the final product and other synthons.
-
G2Retro employs a new, effective beam search strategy compared to the previous semi-template-based methods27,28,29,30, that prioritizes the most possible reactants and the corresponding completion actions along the synthon completion paths. The beam search also allows multiple different reaction centers, enabling diversity in the completed reactants.
-
G2Retro and G2Retro-B are compared with nineteen baseline methods and demonstrate the state-of-the-art performance over the benchmark data (in “Overall Comparison” Section). Case studies show that G2Retro could propose diverse and reasonable synthesis routes with high predicted likelihoods that are not included in the benchmark data (in “Case Study” Section).
-
G2Retro-ens is an ensemble of G2Retro models and demonstrates strong performance on the benchmark data compared to two baseline methods with data augmentation (in “Performance of Ensemble-based Methods” Section).
Results
Overall comparison
Table 1 presents the overall comparison between G2Retro, G2Retro-B and the baseline methods on one-step retrosynthesis under two conditions, following the standard protocol in literature12,13,14,19,24,26,27,28,29,30: (1) when the reaction type is given a priori for both model training and inference (i.e., “Reaction type known”); and (2) when the reaction type is always unknown (i.e., “Reaction type unknown”). When the reaction type is known, G2Retro uses a one-hot encoder as an additional feature for each atom in product molecules indicating the reaction type. Particularly, for Semi-TB methods, the performance in Table 1 corresponds to the predictions out of the two steps, that is, the synthon completion is done according to a reaction center that is predicted from the reaction center prediction step. Following the prior work12,14,28,29, we used the top-k (k = 1,3,5,10) accuracy to evaluate the overall performance of all the methods. Top-k accuracy is the ratio of test products that have their ground truth correctly predicted among their top-k predictions. Higher top-k accuracy indicates better performance. Note that ground truth reactions are those included in the benchmark data. While there is always one ground-truth reaction for each product in the benchmark data, there may exist actually numerous feasible reactions for each product that are not included in the benchmark data. Therefore, reactants that are considered incorrect based on the benchmark data might still be plausible and included in other larger databases. Also, note that the top-k accuracies of all the baseline methods are the reported results in their original papers (issues related to the comparison among methods are discussed later). Details of baseline methods are available in “Baselines” Section.
Comparison with semi-template-based (Semi-TB) methods
When the reaction type is known, compared to other Semi-TB methods, G2Retro achieves the best performance on top-3 (84.2%), top-5 (88.5%) and top-10 (91.7%) accuracies, corresponding to 3.2%, 2.9%, and 3.4% improvement over those from the best baselines (81.6% for RetroPrime30 on top-3, 86.0% and 88.7% for G2G28 on top-5 and top-10) on these three metrics. In terms of top-1 accuracy, G2Retro-B achieves the third-best performance (63.6%) compared to those of RetroPrime (64.8%) and GraphRetro (63.9%) on this metric. While G2Retro underperforms RetroPrime on one metric, it is substantially better than RetroPrime on all the other metrics: G2Retro outperforms RetroPrime on top-3 accuracy at 3.2%, on top-5 accuracy at 4.1%, and on top-10 accuracy at 5.6%.
When the reaction type is unknown, a similar trend is observed: G2Retro-B outperforms all the Semi-TB baseline methods on all the top accuracy metrics, with 0.7% improvement over the best baseline GraphRetro on top-1 accuracy, and 4.7%, 9.7% and 13.9% improvement over those from the best baseline RetroPrime on top-3, top-5 and top-10 accuracies. G2Retro has a performance similar to that of G2Retro-B, with an even better top-3 performance 74.6% that is 5.4% improvement from that of RetroPrime.
Compared with the performance with known reaction types, all the methods including G2Retro and G2Retro-B have worse performance when the reaction types are unknown. It is well-known in synthetic chemistry that there are several well-characterized reaction types. These types have distinct patterns in their reactions and reaction centers. For example, acylation reactions are very common approaches to creating amide and sulfonamide linkages. They are known for their efficiency and high yields, especially when they involve acyl/sulfonyl halides38. The improved performance with known reaction types integrated into retrosynthesis model training demonstrates that leveraging a priori reaction type information could benefit retrosynthesis prediction in general. However, in real applications, reaction types are typically not available in retrosynthesis when only the target molecule is presented. The superior performance of G2Retro and G2Retro-B in “reaction type unknown” condition demonstrates their great utility in real applications.
As Table 1 shows, G2Retro and G2Retro-B can cover (i.e., can be applied to) 97.5% of the test reactions, which determines the upper bound of accuracy values, due to the definition of reaction centers (the rest 2.5% correspond to reactions with multiple newly formed or changed bonds). Among other Semi-TB methods, G2G and GraphRetro29 also have limited coverage on test set (97.9% for G2G and 95.0% for GraphRetro). RetroXpert27 has 100% coverage because its reactant SMILES generation from synthons recovers all possible reaction centers. RetroPrime30 also has 100% coverage due to its very comprehensive set of reaction centers. Although G2Retro and G2Retro-B cannot cover all possible cases in the test set, they still outperform other Semi-TB methods, measured over the entire test set. More discussion on the coverage of the two steps in Semi-TB methods is available in the Section “Individual Module Performance”.
GraphRetro and RetroPrime are two strong baselines. GraphRetro has good top-1 accuracies but much worse results on other top accuracy metrics. According to its authors29, GraphRetro tends to bias its beam search to the most possible reaction center. Thus, it may prioritize the most possible reactants from the most possible reaction center at the very top of its predictions. However, if the most possible reaction centers are not the ground truth, GraphRetro would totally miss the ground truth in its beam search, resulting in poor performance on other top accuracy metrics. In addition, such focused beam search limits the diversity of identified synthons, and thus the completed reactants. RetroPrime achieves the best top-1 accuracy with reaction type known. It uses augmented SMILES strings (i.e., each product has multiple, equivalent, non-canonical SMILES strings) in training the two sequence-to-sequence transformers. It is likely that top results in RetroPrime correspond to the ground truth but in different, augmented SMILES strings, and thus high top-1 accuracy but low and similar other top accuracies. These three Semi-TB baseline methods only perform well on one certain metric (in one certain condition), but do not show consistent optimality across many metrics or across the two conditions.
Compared to these baselines, G2Retro always achieves the best performance on all the top accuracy metrics (except on top-1 accuracy when reaction types are known). High top-k accuracies at all different k are desired as they indicate the holistically high ranking positions of the ground truth in the predicted reactions, and thus the capability of models in recovering knowledge from data. High top-k accuracies with k > 1 may signify plausible reactions not included in the dataset, as will be examined later in Section “Case Study”. This is because high top-k (k > 1) accuracy implies that there might be a few reactions different from the ground truth but are very possible and thus are ranked on top. Such results may enable the exploration of multiple synthesis routes and may be of synthetic value if specific coupling methods fail or if specific starting materials are unavailable. From the above two aspects, over all the metrics, G2Retro and G2Retro-B achieve the overall best performance compared to the three strong Semi-TB methods.
G2Retro-B performs slightly better than G2Retro when the reaction types are unknown, but worse than G2Retro when the reaction types are known. G2Retro-B integrates synthetically accessible fragments in atom embeddings (Eq. S3 in Supplementary Note 1). When the reaction types are unknown, the fragment information provides additional local contexts to atoms, which could facilitate better decisions on reaction center prediction and synthon completion. When the reaction types are known, atom embeddings directly integrate the reaction type information in G2Retro, which may outweigh the contextual information provided by the fragments, and thus G2Retro-B does not achieve additional performance improvement from G2Retro.
Comparison with template-free (TF) methods
G2Retro and G2Retro-B also demonstrate superior or competitive performance compared to TF methods on all the top accuracies. With reaction types known, G2Retro is the best on top-3, top-5 top-10 accuracies compared to all the template-free methods; with reaction types unknown, G2Retro-B is the best on top-3, top-5 and top-10 accuracies, and is the second best one on top-1 accuracy. For example, G2Retro is 4.9% better than the best TF method on top-3 accuracy (i.e., Retroformer25) with the reaction types unknown. Most TF methods such as Dual24 and Chemformer20 have the competitive performance on top-1 accuracy but relatively worse results on other top accuracy metrics. This could be due to that these TF methods with SMILES representations may fail to generate diverse or even many valid reactants with beam search39, leading to limited variation in their predicted results, and thus low and similar top-3, top-5 and top-10 accuracies. This lack of diversity and richness in the predictions, in addition to the lack of interpretability during the chemical sequence transformation process, could hinder the application of TF methods in retrosynthesis prediction. However, the prediction diversity and richness in G2Retro is enabled by the multiple possible reaction centers predicted by G2Retro and the corresponding completed reactants.
In terms of the coverage on the test set, all the SMILES-based TF methods can cover the entire test set, because all the reactions can be represented as SMILES string transformation. The graph-based TF method MEGAN26 also covers the entire test set due to its comprehensive set of graph edit actions. Compared to these TF methods, though without the full coverage on the test set, G2Retro and G2Retro-B model reactions through a two-step process of reaction center identification and synthon completion, allowing for the interpretability of reaction centers in the predicted reactants. Overall, G2Retro and G2Retro-B achieve even better performance than the methods with full coverage, measured on the entire test set.
Comparison with template-based (TB) methods
G2Retro and G2Retro-B achieve competitive performance with that from the TB methods. With reaction types known, G2Retro achieves either the second or the third on all the top accuracies; with reaction types unknown, G2Retro-B achieves the best performance on top-1 (54.1%), and either the second or the third on all the other top accuracies. For example, with reaction types unknown, G2Retro-B is the second best on top-3 accuracy, with 3.8% difference from the best performance of LocalRetro16; G2Retro-B slightly underperforms the second-best baseline MHNreact15 on top-10 (86.7% compared to 87.9% from MHNreact), but outperforms MHNreact on all the other metrics. LocalRetro is a very strong TB method. It extracted 731 templates from the benchmark training data, whereas other TB methods have much more templates (11,647 for GLN and 9162 for MHNreact). Therefore, LocalRetro could achieve better template selection over a small template set compared to others over much larger template sets. However, LocalRetro may suffer from scalability issues on large datasets because it scores all the reaction templates on all the potential reaction centers (i.e., all atoms and all bonds) in the product molecules. In general, all TB methods may not generalize well to reactions that are not covered by the templates29. In terms of coverage on the test set, Table 1 shows that the templates used in Retrosim, Neuralsym and MHNreact can cover the entire test set, while the templates used in GLN14 and LocalRetro cannot (93.3% for GLN and 98.1% for LocalRetro). Unlike TB methods, G2Retro does not use reaction templates, and only scores all the bonds and atoms once for reaction center identification, and thus is much more scalable in inference. It learns the patterns from training data and thus has a better chance to discover new patterns from the training data that are not covered by templates.
Individual module performance
Following the typical evaluation for Semi-TB methods as in literature29, Table 2 presents the individual performance of the two modules—reaction center identification and synthon completion in Semi-TB methods. In Table 2, for the reaction center identification module, the top-k accuracy measures the ratio of test products that have the ground-truth reaction center correctly predicted among the top-k predictions. In the synthon completion module, the synthon completion is done according to the ground-truth reaction center, not the predicted reaction center; the top-k accuracy measures the ratio of test products that have the ground-truth reactants correctly predicted among the top-k predictions. Please note that here “ground-truth” reaction center means the reaction center as appears in the benchmark data per our reaction center definition.
Comparison on reaction center identification
Among all the Semi-TB methods, the definitions of reaction centers vary. In G2G, reaction centers are referred to as the only one newly formed bond during the reaction, and reaction center identification predicts whether there is such a new bond (and its location) or not in the products as in a classification problem. This reaction center definition and classification can cover 97.9% of the test data (the rest 2.1% correspond to multiple newly formed bonds). GraphRetro defines the reaction center as the newly formed bond (BF-center as defined in Section “Reaction Centers with New Bond Formation” but without induced bond changes), the changed bond (BC-center as in “Reaction Centers with Bond Type Change”) and the single atom with changed hydrogen count (A-center as in “Reaction Centers with Single Atoms”), which in total covers 95.0% of the reactions in the test set. RetroPrime aims to identify all the atoms involved in the reactions as reaction centers, which covers all the reactions in the test set. G2Retro extends the definition of the reaction center in GraphRetro with induced bond type change and atom charge changes, covering 97.5% of the test set.
Due to the data leakage issue as revealed by Yan et al.27 (i.e., reaction center is given in both the training and test data), the reported G2G reaction center identification performance as cited in Table 2 is overestimated, but the updated results have not been provided in their Github. GraphRetro uses two functions, one for bonds and one for atoms, to predict reaction centers. While these functions are able to predict well when such bonds and atoms are truly reaction centers (i.e., performance in parentheses in Table 2), GraphRetro’s reaction center definition covers the least (95%) of the test set compared to the other methods, resulting in still low accuracies (i.e., performance outside parentheses) over the test set. RetroPrime has a very generic definition of reaction centers—any atoms involved in the reactions, and uses one unified model to predict these atoms. However, as these atoms may experience different changes (e.g., connected to or disconnected from other atoms), a unified model not customized to specific changes may not suffice, leading to overall relatively low accuracies compared to other methods, particularly when reaction types are unknown. G2Retro and G2Retro-B have the most comprehensive definition of reaction centers (Section “Reaction Center Identification”) with high coverage (97.5%) on the test set. In addition, G2Retro and G2Retro-B use a specific predictor for each of the reaction center types. Therefore, they achieve the best overall accuracy among the entire test set, as well as good performance over the reactions covered by its reaction center definition.
Comparison on synthon completion
To compare synthon completion performance, all the ground-truth reaction centers defined by different methods are given and used to start the completion processes. G2G predicts only bond establishment in its reaction center identification and thus has to deal with any associated changes such as bond type change in its synthon completion process, which complicates the synthon completion prediction. Therefore, its performance on synthon completion is the worst among all the methods.
GraphRetro formulates the synthon completion as a classification problem over all the subgraphs that can realize the difference between the synthons and reactants. Therefore, its synthon completion is not guaranteed to work for all possible products (e.g., 99.7% coverage over the test set), particularly if the needed subgraph is not included in the pre-defined vocabulary. Among all the products that GraphRetro can handle, its synthon completion performance is the best, due to that classification can be much easier than generation as all the other methods do. However, since GraphRetro does not do well in reaction center identification, overall, it does not outperform other methods in retrosynthesis prediction as Table 1 demonstrates. In addition, the synthon completion module of GraphRetro may fail to accurately estimate the likelihoods of leaving groups, due to the ignorance of overall structures of predicted reactants. Such inaccurate likelihood estimation may aggravate the bias of beam search and reduce the diversity of predicted reactants as discussed in GraphRetro29.
RetroPrime transforms the synthons to reactants using a Transformer, but similarly to G2G, also needs to deal with additional predictions such as bond type change. RetroPrime’s synthon completion performs reasonably well on top-1 accuracies. Together with its good top-1 accuracy on reaction center identification, RetroPrime achieves the best top-1 accuracy with reaction type known as demonstrated in Table 1. RetroPrime uses a rule to enumerate predicted reactants from the top-3 reaction centers, limiting the potential diversity of predicted reactants. On average, RetroPrime underperforms G2Retro, particularly on top-3 and top-5 accuracies in synthon completion.
G2Retro does not use BRICS fragments in synthon completion because the fragment information is not available for the substructures that will be attached to synthons. Compared to GraphRetro, G2Retro leverages a generative process to add substructures to synthons in synthon completion, which is inherently more difficult than classification as in GraphRetro but could be generalizable to new products and reactants. Meanwhile, G2Retro does not limit the number of reaction centers within the top-10 predicted reactants, and thus increases the diversity of predicted reactants.
Although G2Retro does not outperform GraphRetro in the synthon completion module alone, its generative process allows G2Retro to consider all the intermediate molecular structures and more accurately estimate the likelihood of each completion action, conditioned on the reaction centers and the corresponding synthons from its reaction center identification module (i.e., not the ground-truth reaction centers). Consequently, despite employing a beam search strategy similar to that of GraphRetro, the generative process of G2Retro could alleviate the bias of beam search on most possible reaction centers by accurately estimating the likelihood of the completed reactants. In contrast, GraphRetro may not generalize well, particularly given that GraphRetro’s reaction center identification does not perform well with respect to the ground-truth reaction centers (i.e., in the top panel of Table 2), but its synthon completion module is trained using the ground-truth reaction centers (i.e., in the bottom panel of Table 2).
Performance on different reaction types
Table 3 presents the top-k accuracy (k = 1,3,5,10) of the reactions of different types. This method appears to predict certain reaction types more accurately than others as shown in Table 3. This is likely due to the relative structural diversity among potential reactants, particularly for substrates that can all provide the same products. For example, in the case of oxidations, only a very limited set of substrates can be utilized to generate a ketone, most commonly the oxidation of an alcohol, although ketones can certainly be accessed through other types of reactions as well. This leads to the relatively higher accuracies of G2Retro on the reactions of oxidations (e.g., 62.2% top-1 accuracy with reaction type unknown). In terms of reductions, however, numerous substrates could be utilized to generate an amine, including reductions of amides, nitro groups, and nitriles to name a few. In addition, there are numerous methods to access the same amines through various structurally unique deprotection reactions. The number of methods available to access a specific functional group, therefore, may make it more difficult to accurately predict which method has been used for a specific molecule, leading to the lower accuracies on reactions of deprotections (e.g., 58.3% top-1 accuracy with reaction type known). This would certainly be the case in carbon-carbon bond forming reactions as well, which can be assembled in a number of ways from various substrates, potentially leading to a somewhat lower prediction success rate (e.g., 37.2% top-1 accuracy with reaction type unknown). In addition, as shown in our case studies, in molecules containing more than one functional group, there are often multiple ways in which that molecule can be assembled by targeting each individual functional group as the reaction center. This means that there are multiple valid reaction pathways which could be considered by synthetic chemists in order to most efficiently construct a molecule. Please note that G2Retro is designed to predict reactions that involve three types of reaction centers: (1) a single newly formed bond with induced changes in bond types; (2) a single changed bond; (3) a single atom with a fragment removed. As a result, G2Retro could not fully cover reaction types such as rearrangement, isomerization, cyclization and click reactions, which involve multiple changes in bond formation or atom detachment. This illustrates G2Retro’s limitation in handling all possible reaction types. It is worth noting that other semi-template-based methods such as G2G and GraphRetro, also share this limitation. Therefore, developing an effective semi-template-based method that overcomes this limitation could be an interesting future research direction.
Performance of ensemble-based methods
We also compared the performance of an ensemble of G2Retro, referred to as G2Retro-ens, with AT31 and R − SMILES32, both of which test each target product multiple times and are strong baselines. AT and R − SMILES represent each target molecule using multiple non-canonical but equivalent SMILES strings, and use the multiple SMILES strings during model training and testing. By combining the predictions from the multiple SMILES strings of the same target product, these methods have the choice to explore a larger reaction subspace seeded by the SMILES strings, and thus achieve better prediction performance. Compared to the SMILES strings, G2Retro uses molecular graph representations, and thus each molecule can only have a unique representation. Instead of augmenting molecule representations but still being able to explore a larger reaction subspace as AT and R − SMILES do, G2Retro-ens tests each molecule multiple times using multiple G2Retro models. Details of G2Retro-ens are available in the supplementary Note 2.
Table 4 presents the comparison among G2Retro-ens, AT and R − SMILES on top-k accuracy (k = 1,3,5,10) over all the reactions and the reactions covered by G2Retro, both with the reaction type unknown. Please note that the performance of AT and R − SMILES on reactions with known types is not available in the respective papers31,32, and the methods also cannot be easily extended to handle known reaction types. In Table 4, all the methods test each molecule 20 times, that is, AT and R − SMILES augment each target molecule with 20 SMILES strings, and G2Retro-ens uses an ensemble of 20 models to test each molecule. The results of AT and R − SMILES are calculated using the source code and data available from the respective papers. Table 4 shows that G2Retro-ens achieves competitive performance with the best baseline R − SMILES. Over all the reactions, G2Retro-ens achieves almost the best performance on top-1 (56.4%, compared to 56.5% for R − SMILES), and only slightly underperforms the best baseline R − SMILES on top-3, top-5 and top 10 (78.8% vs 79.2% on top-3; 85.2% vs 86.2% on top-5; 90.5% vs 91.0% on top-10). Over the reactions covered by G2Retro, G2Retro-ens outperforms the baseline R − SMILES on top-1 accuracy at 1.76%, on top-3 accuracy at 1.25%, on top-5 accuracy at 1.28%, and on top-10 accuracy at 1.53%. Compared to R − SMILES, which is an end-to-end black-box that directly transfers product SMILES string to reactant SMILES strings, G2Retro provides certain interpretability of the predicted reaction centers, and what reactants are generated from them. More details about the comparison on different reaction types and on reactions covered by G2Retro-ens are available in the supplementary Note 2.
Case study
G2Retro can predict multiple reactions for each product due to multiple predicted reaction centers. This variability could be useful for chemical synthesis in order to consider all possible reaction strategies. In order to illustrate the predictive power of G2Retro, we have highlighted the top-10 predicted reactants by G2Retro with reaction types unknown for four newly approved drug molecules in 2022, including Mitapivat, Tapinorf, Mavacamten, and Oteseconazole40. Among them, the predicted reactants for Mitapivat and Tapinorf are presented in Fig. 2aa and ba which will be discussed later; the results and the discussions for Mavacamten and Oteseconazole are available in Supplementary Figure 1, Supplementary Figure 2 and Supplementary Note 3. Note that these drugs are not included in our training, validation, or testing data. Therefore, how G2Retro works on these drugs truly indicates its predictive power for new molecules.

a Predicted reactions by G2Retro for Mitapivat; b Predicted reactions by G2Retro for Tapinorf. Numbers next to each atom are the indices of the atoms. Atoms with same indices in different subfigures are corresponding to each other. Atoms and bonds colored in red are leaving groups for synthon completion. Molecules with labels ending in (a) are product/target molecules; molecules with labels ending in (b) are the reactants reported in patents; molecules with labels ending in (c–l) are the top predicted reactants.
Mitapivat as in Fig. 2aa is a drug approved for hereditary hemolytic anemias in 202241. The synthetic route within the patent42 reporting the discovery of Mitapivat utilizes an amide coupling reaction to form the C2-N23 bond (Fig. 2ab). This is correctly predicted by G2Retro as the top-1 reaction (Fig. 2ac). As indicated by the top-5 reaction (Fig. 2ag), G2Retro also predicts that the amide coupling reaction could be performed with the carboxylate salt of one of the reactants, a useful reactant under the right pH conditions. G2Retro also predicts that the acyl chloride as the substrate in this transformation would also react with the amine group and produce the desired molecule (Fig. 2aj), In addition, G2Retro identifies the N7-S8 bond of sulfonamide linkage as the reaction center (e.g., Fig. 2ad, ae, af, ak, al). Most impressively, G2Retro predicts various S8 sulfonyl groups reacting with the N7 amine group, such as sulfonyl chloride (Fig. 2ad), sulfonyl fluoride (Fig. 2ae) and sulfonic acid (Fig. 2af), which are theoretically feasible for the formation of the N7-S8 bond. G2Retro also predicts that the N26-C27 bond could be the reaction center and formed by the N26 amine group reacting through a reductive amination with ketone in Fig. 2ah or through a nucleophilic substitution with the chloride in Fig. 2ai.
Tapinarof as in Fig. 2ba is a drug approved for plaque psoriasis and atopic dermatits43. The reported synthesis in patent44 constructs this drug by removing the protecting groups on O5 and O10 (Fig. 2bb). G2Retro correctly predicts the deprotection of the methyl groups on O5 (Fig. 2bc) or O10 (Fig. 2bd), which would work to produce the desired molecule, although the ground truth failed to be predicted due to the limitation of reaction centers. Similarly, G2Retro generates possible reactants that contain different types of protected alcohols, as seen with the methoxymethyl groups on O5 and O10 in Fig. 2bf and bi and the benzyl-protected O5 in Fig. 2bj. Most impressively, G2Retro also identifies the alkene linkage between C11 and C12 (Fig. 2be and bl) and the C-C bond between C7 and C11 (Fig. 2bg, bh, ad bk) as reaction centers with various coupling reactions. These coupling reactions include McMurry coupling45 (Fig. 2be), Wittig coupling46 (Fig. 2bl) and Suzuki coupling47 (Fig. 2bg and bh).
In addition, we also highlighted two molecules in the test set and their predicted reactions by G2Retro with reaction types unknown in Fig. 3a and b, respectively. The product in Fig. 3aa contains amide linkages and was assembled in the patent literature utilizing amide coupling reactions (ground truth in Fig. 3ab). G2Retro correctly predicted this coupling as the top-1 reaction for the construction of this molecule (Fig. 3ac). The other reactions predicted, however, are also very instructive into the strengths and limitations of G2Retro. In Fig. 3aa, the product has two amide groups in the side chain of the molecule. G2Retro identified both of these linkages as potential reaction centers (e.g., in Fig. 3ac between N5 and C6; in Fig. 3ag between N1 and C2). Typically, chemists would disconnect the molecule at the C6 amide carbonyl rather than C2 so that a fully elaborated side chain can be introduced to complete the molecule. This approach would generally be considered more efficient since its reaction introduces more complexity into the molecule in a single step and would therefore be predicted to limit the total number of steps necessary to construct the molecule. In some limited cases, however, it may be necessary to introduce the nitrogen at N1 last (e.g., in Fig. 3af–ah), so this should also be considered a feasible reaction. In addition to the typical amide coupling strategy, which takes place between an amine and a carboxylic acid, G2Retro also correctly identifies the reaction of the amine with an acid chloride to make the same bond (Fig. 3ad). Although this was not the strategy utilized in the ground-truth study, this strategy would certainly be expected to work in this case for construction of this molecule. The other common reaction that was predicted for this example was the nucleophilic addition of the N5 (or N1) amine into the C6 (or C2) carbonyl of an ester (N5-C6 - Fig. 3ae, ai, aj, ak, al and N1-C2 - Fig. 3ag and ah). This type of reaction, which is essentially a transamidation reaction, should also work to provide the product. Interestingly, however, G2Retro predicts several different esters as substrates for this transformation (Fig. 3ac, ae, ai, aj, ak and al). While these are different substrates, the variation of the ester side chain in these cases would not typically be considered as greatly different by a synthetic chemist unless steric or electronic contributions affect the reactivity/electrophilicity of the ester carbonyl.

a Predicted reactions by G2Retro for product “NC(=O)CNC(=O)C1CC12CCCCC2''; b Predicted reactions by G2Retro for product “CCOC(=O)c1csc(-c2ccc(F)cc2)c1''. Numbers next to each atom are the indices of the atoms. Atoms with same indices in different subfigures are corresponding to each other. Atoms and bonds colored in red are leaving groups for synthon completion. Molecules with labels ending in (a) are product/target molecules; molecules with labels ending in (b) are the ground-truth reactants in USPTO-50K; molecules with labels ending in (c–l) are the top predicted reactants.
Retrosynthesis of the product in Fig. 3b involves a C-C bond forming reaction between C9 and C10 (Fig. 3ba). The disconnection of the carbon-carbon bond between the two aromatic rings, a heteroaromatic thiophene and a benzene ring in this case, represents the most obvious disconnection in the molecule. In this case, the top-1 reaction (Fig. 3bc) predicted by G2Retro for this transformation is a Suzuki coupling47, a common metal-mediated coupling between a boronic acid reactant and a corresponding aryl halide. This common transformation is the same reaction observed in the ground truth (Fig. 3bb). Interestingly, G2Retro also identifies additional permutations of this Suzuki reaction through changing the nature of the aryl halide (Fig. 3bk and bl). Traditionally, aryl chlorides (Fig. 3bk) are less reactive than aryl bromides or iodides (Fig. 3bc and bl) for coupling reactions and in the past were considered unreactive in these reactions. Newer methods48 using specially designed ligands, however, have made the use of such chlorides possible. The other difference observed in the predicted Suzuki couplings is the use of a boronic ester (Fig. 3bg) vs a boronic acid (Fig. 3bc). Both boronic acids and boronic esters are common reagents for these transformations, with many being readily available from commercial sources. G2Retro also predicts that an esterification reaction at the C4 carboxylic acid would also work to produce the desired molecule (Fig. 3bd). While this is potentially not as synthetically useful for building the molecule, it is a reasonable transformation. Most impressively, G2Retro also predicts other coupling reactions49 for the biaryl coupling reaction. These other methods include an Ullmann-type coupling50 (Fig. 3be and bi) a Stille coupling51 (Fig. 3bf), and a Kumada coupling48,52 (Fig. 3bj). This versatility predicted in the top-10 reactions may be of synthetic value for substrates if specific coupling methods fail or if the functionality necessary for one type of coupling reaction is not able to be easily prepared.
The above examples indicate that the predicted reactions from G2Retro rather than the ground truth could be still possible and synthetically useful. Therefore, a more comprehensive evaluation strategy is needed not to miss those possible and potentially novel synthesis reactions.
Diversity on predicted reactions
Diversity in predicted reactions is always desired, as it has the potential to enable the exploration of multiple synthesis routes. G2Retro has the mechanisms to facilitate diverse predictions: The beam search strategy in G2Retro allows multiple reaction centers and multiple different attachments, and therefore potentially different scaffolds and structures in the predicted reactants.
To analyze the diversity of G2Retro results, we analyzed the reaction centers among the top-predicted reactions. We identified a set of products such that their third or fifth predicted reactions are the ground truth, referred to as having a hit at 3 or 5, respectively. Please note each predicted reaction was scored using the sum of the log-likelihoods of all the predictions along the transformation paths from the product to its reactants (please refer to Section “Inference”), and then ranked based on the score. Thus, the predicted reactions ranked above the ground truth have a higher likelihood than the ground truth. Given that G2Retro has demonstrated strong performance as in Table 1 in scoring and prioritizing the ground-truth reactions, we assume that its likelihood calculation is reliable and therefore, the reactions ranked above the ground truth might also be likely to occur.
Figure 4a and b presents the distribution of products with hits at 3 or 5 over the number of reaction centers among predicted reactions ranked above the ground truth. Figure 4a shows that more than 50% of the products with a hit at 3 have their top-3 reactions from two different reaction centers; about 20% of the products have their top-3 reactions from three different reaction centers. Figure 4b shows that for products with a hit at 5, almost 40% have two reaction centers, and another 40% have three reaction centers, among their top-5 predicted reactions; more than 10% have four reaction centers. Thus, Fig. 4a and b clearly demonstrate that the top predicted reactions were diverse, demonstrated by the different reaction centers they were derived from. Meanwhile, we acknowledge that the diverse, top predictions may still be errors and thus, more reliable wet-lab experimental validation is needed.

a Percentage of products (Product (%)) with the different number of predicted reaction centers and the third predicted reaction as the ground-truth reaction (i.e., hits at 3); b Percentage of products (Product (%)) with the different number of predicted reaction centers and the fifth predicted reaction as the ground-truth reaction (i.e., hits at 5); c Predicted reactions by G2Retro for product “CCOC(=O)Cn1ccc(NC(=O)c2ccc(Cl)s2)n”. Numbers next to each atom are the indices of the atoms. Atoms with the same indices in different subfigures correspond to each other. Different reaction centers are highlighted in different colors (blue, red and olive). Atoms and bonds colored in red are leaving groups for synthon completion. Molecules with labels ending in (a) are product/target molecules; molecules with labels ending in (b) are the ground-truth reactants in USPTO-50K; molecules with labels ending in (c–h) are the top predicted reactants.
Figure 4c presents an example of very diverse reactions with diverse reaction centers predicted by G2Retro. For the product in Fig. 4ca, G2Retro predicts three different reaction centers: an amide bond (between C12 and N11), a nitrogen-carbon bond (between N7 and C6) and ester (between O3 and C2). The patent reported that the target molecule was synthesized from a carboxylic acid derivative and an amine using amide coupling with a widely-used coupling reagent, EDC (Fig. 4cb). G2Retro predicted an acyl chloride-amine reactant pair as the top-1 result (Fig. 4cc), a potentially viable and even high yielding synthetic approach. It also predicts three reactant pairs from the other two reaction centers as possible routes within the top 4 (Fig. 4cd and cf at which involve alkylation reactions to form the C6-N7 bond; Fig. 4ce at which forms the ester linkage between O3 and C2).
We also analyzed the reaction diversity by comparing the number of reaction centers in products with high reaction diversity and low reaction diversity. For each product, the diversity of its predicted reactions is represented by the distribution of all pairwise similarities of its predicted reactions, that is, lower reaction similarities indicate higher reaction diversity. Please note that the reaction similarity is only applicable to two reactions that share the same product. Therefore, the product is not considered in the similarity calculation. Formally, for reaction R1: M1 + M2 → Mp and reaction R2: M3 + M4 → Mp, the similarity between R1 and R2 was calculated as follows,
where simm() is a similarity function over molecules, calculated using Tanimoto coefficient over 2,048-bit Morgan fingerprints of the molecules. For reaction R1: M1 → Mp and reaction R2: M2 + M3 → Mp, the similarity between them was calculated as follows,
where M2 + M3 denotes the composite molecule consisting of two disconnected components M2 and M3. For reaction R1: M1 → Mp and reaction R2: M2 → Mp, the similarity between them was calculated as follows,
We clustered the products according to their reaction similarity distributions using the K-means clustering algorithm in Euclidean distances. The clustering algorithm is presented in Supplementary Algorithm 1 in Supplementary Note 4. Figure 5a presents the clustering results for products that have their ground-truth reaction correctly predicted among the top-10 predictions. In Fig. 5a, the first four clusters have on average lower reaction similarities (on average 0.46 among the four clusters; 0.41, 0.45, 0.45, 0.49 in each of the clusters, respectively), and thus are referred to as high-reaction-diversity clusters (HRD); the other six clusters, referred to as low-reaction-diversity clusters (LRD), have relatively higher reaction similarities (on average 0.58 for among the six clusters; 0.52, 0.53, 0.58, 0.62, 0.67, 0.67 in each of the clusters, respectively).

a Clustering on test products based on similarities of their predicted reactions. The x-axis indicates the range of reaction similarities (e.g., the column between 0.1 and 0.2 indicates the range (0.1, 0.2]); the y-axis shows the cluster ID and the cluster size. Each row in the heatmap corresponds to the reaction similarity distribution of a product belonging to a specific cluster; each block in the row corresponds to the frequency of reaction similarities within each similarity range, and the block color represents the scale of the frequency (e.g., a darker color indicates a higher frequency value). The clusters are labeled as 'HRD' for high-reaction-diversity clusters with average low reaction similarities, and 'LRD' for low-reaction-diversity clusters with average high reaction similarities. b Test product distributions over the number of reaction centers of HRD products. c Test product distributions over the number of reaction centers of LRD products.
Figure 5b and c present the distributions of the number of reaction centers in the products of these two clusters. Comparing Fig. 5b and c, HRD products tend to have more reaction centers in their predicted reactions than those in LRD products, and the number of reaction centers correlates well with reaction diversity (-0.8486 between the average reaction similarities and the number of reaction centers). Particularly, the first cluster (in HRD), which has the highest reaction diversity (lowest reaction similarity), has on average 4.41 reaction centers in the top-10 predicted reactions of each product, compared to the average 3.92 reaction centers in the top-10 predicted reactions of each product in LRD clusters. The ninth and tenth clusters, which have the lowest reaction diversity, have on average 2.57 reaction centers. These results clearly show the diversity of G2Retro predictions.
Discussion
Comparison among template-based, template-free and semi-template-based methods
Template-based methods were first developed for retrosynthesis prediction. They match products into pre-defined templates that are extracted from training data or hand-crafted based on knowledge. A notable advantage of templates is that they can enable strong interpretability (e.g., each template may correspond to a certain reaction type, a chemical scaffold, or a reactivity pattern) and thus result in reactions that better conform to domain knowledge. They can also well fit the data if the templates are extracted from the data. However, they suffer from a lack of strong learning capabilities and a lack of generalizability, if the templates do not cover and cannot automatically discover novel reaction patterns.
Template-free methods largely leverage the technological advancement in Natural Language Processing (NLP), including large-scale language models such as Transformer and BART53, and also many pre-training techniques. Most of them formulate a reaction as a SMILES string translation problem. Rather than enumerating pre-defined patterns (i.e., templates) as template-based methods do, template-free methods are equipped with much stronger learning capabilities from SMILES strings and can represent latent reaction transformation patterns in an operable manner. However, template-free methods sacrifice their interpretability as it is non-retrieval to decipher why an atom (analogous to a token in NLP) is generated next along the SMILES strings, or what chemical knowledge the actions correspond to. In addition, as SMILES strings are a ‘flattened’ representation of molecular graphs according to the atom orderings from a graph traversal, template-free methods using SMILES strings only cannot fully leverage molecular structures, which ultimately determine molecule synthesizability and reaction types. To mitigate this issue, some template-free methods either enrich the product SMILES representation with molecular graph information19,23,25 or decode reactant SMILES strings from product molecular graphs21, which, however, require additional learning of the mapping from molecular graphs to SMILES and thus increase the learning complexity.
Semi-template-based methods, typically over molecular graphs, represent the most recent and also in general the best performing retrosynthesis prediction methods. They utilize the powerful graph representation learning paradigm to better capture molecule structures. They also take advantage of graph (variational) auto-encoder frameworks or sequential predictions to empower the models with generative ability. More importantly, semi-template-based methods have the mechanism to enable diversity among predicted reactions, by allowing multiple samplings from the latent space. Meanwhile, semi-template-based methods have two steps: (1) reaction center identification, and (2) synthon completion, better complying with how chemical reactions are understood and enabling certain interpretability of predicted reaction centers and derived reactants. G2Retro is a semi-template-based method and achieves superior performance to other methods, demonstrating it as a state-of-the-art method for retrosynthesis prediction.
Comparison issues among existing methods
In our study of the baseline methods, several issues were identified among existing methods that make comparison across different methods hard. In Table 1, RetroXpert’s results are from its updated GitHub54, as their results originally reported in their manuscript had a data leakage issue (all the reaction centers were implicitly given) and thus were overestimated27. G2G may also suffer from the data leakage issue as discussed in its github55, but G2G’s results were only available from its original paper, though likely overestimated. In addition, there have been some reproducibility issues with G2G56, as we also observed in our study. All the methods except Neuralsym, LV-Trans, Dual and Retroformer published their code and datasets. Among these methods, most template-free methods including SCROP, GET, Chemformer, TiedTransformer, GTA and AT used the same data split, which is, however, different from the benchmark data split used in the other methods. For example, the training set of these template-free methods has 40,029 reactions, while the training set of the other methods including G2Retro has 40,008 reactions. Even though all the methods adopted the same ratio (i.e., 80%/10%/10% for training/validation/test set) to split the benchmark dataset, their splits, particularly their test sets, are not identical, making it hard to compare these methods. In this manuscript, we adopted the data split used by the previous semi-template-based methods; for the template-free methods with different data splits, we still used the results reported by their authors. We believe reproducibility and unbiased comparison (e.g., on the same benchmark data and same splits, generating the same amount of results to compare) among all the retrosynthesis prediction methods are critical to moving this research forward. They require dedicated research, implementation and regulatory effort from the entire research community, for example, by following the Open Science Policy from the European Union57 and the Data Sharing Policy from the United States National Institute of Health58. Unfortunately, it is out of the scope of this manuscript.
Conclusions
G2Retro predicts reactions of given target molecules by predicting their reaction centers, and then completing the resulting synthons by attaching small substructures. Based on a comparison against twenty baseline methods over a benchmark dataset, G2Retro achieves the state-of-the-art performance under most metrics. The case studies show that G2Retro also enables diverse predictions. However, G2Retro still has several limitations. First, the three types of reaction centers in G2Retro still cannot cover all possible reaction center types (e.g., the reactions with multiple newly formed bonds). Therefore, a more comprehensive definition of reaction center types is still needed. G2Retro cannot cover bonds or rings that are attached at the reaction centers but do not appear in the training data either, as the substructures that G2Retro employs to complete synthons are extracted only from training data. In addition, the atom-mapping between products and reactants that is required by G2Retro (and required by many existing methods) to complete synthons is not always available or of high quality (it is available in USPTO-50K). To identify such mappings, it requires to calculate graph isomorphism, which is an NP-hard problem. Moreover, the sum of log-likelihoods of all the involved predictions (i.e., reaction center prediction, attached atom type prediction) that G2Retro uses to prioritize reactions, is not necessarily the same as the likelihood of the reactions, which could affect the quality of the prioritized reactions. We are also investigating a systemic evaluation and in vitro validation protocol, in addition to using top-k accuracy, as we discussed earlier. Multiple-step retrosynthesis could be possible by applying G2Retro multiple times iteratively, each time on a reactant as the target molecule. Connected after the deep generative models that have been developed to optimize small molecule structures and properties59,60 for lead optimization, G2Retro has a great potential to generate synthetic reactions for these in silico generated drug-like molecules, and thus substantially speed up the drug development process.
Methods
G2Retro is developed for the one-step retrosynthesis prediction problem, that is, given the target molecule (i.e., product), G2Retro identifies a set of reactants that can be used to synthesize the molecule through one synthetic reaction. Following the prior semi-template-based methods28,29, G2Retro generates reactants from products in two steps. In the first step, G2Retro identifies the reaction center from the target molecule using the center identification module. G2Retro defines the reaction centers as the single bond that is either newly formed or has the bond type changed, or the single atom with changed hydrogen count during the reaction. G2Retro also incorporates into the reaction center the bonds neighboring the reaction centers that have type changes induced by the newly formed bond, and the atoms with charge changes within the target molecule (more details in “Reaction Center Identification” Section). Given the reaction center, G2Retro converts the target molecule into a set of intermediate molecular structures referred to as synthons, which are incomplete molecules and will be completed into reactants. In the second step, G2Retro completes synthons into reactants by sequentially attaching bonds or rings in the synthon completion module. The intermediate molecular structures before being completed to reactants are referred to as updated synthons. Figure 1 presents the overall model architecture of G2Retro. All the algorithms are presented in Supplementary Note 5.
Molecule representations and notations
Supplementary Table 3 in Supplementary Note 6 presents the key notations used in this manuscript. A synthetic reaction involves a set of reactants {Mr} and a product molecule Mp that is synthesized from the reactants. Please note that we do not consider reagents or catalysts in this study. Each reactant Mr has a corresponding synthon Ms, representing the substructures of Mr that appear in Mp. We represent the product molecule Mp using a molecular graph \({{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}_{p}^{M}{{\mbox{}}}\), denoted as \({{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}_{p}^{M}{{\mbox{}}}=({{\mbox{}}}{{{{{{{\mathcal{A}}}}}}}}{{\mbox{}}},{{\mbox{}}}{{{{{{{\mathcal{B}}}}}}}}{{\mbox{}}})\), where \({{\mbox{}}}{{{{{{{\mathcal{A}}}}}}}}{{\mbox{}}}\) is the set of atoms {ai} in Mp, and \({{\mbox{}}}{{{{{{{\mathcal{B}}}}}}}}{{\mbox{}}}\) is the set of corresponding bonds {bij}, where bij connects atoms ai and aj. We also represent the set of the reactants {Mr} or the set of synthons {Ms} of Mp using only one molecular graph \({{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}_{r}^{M}{{\mbox{}}}\) or \({{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}_{s}^{M}{{\mbox{}}}\), respectively. Here, \({{{{{{{{\mathcal{G}}}}}}}}}_{r}^{M}\) and \({{{{{{{{\mathcal{G}}}}}}}}}_{s}^{M}\) could be disconnected with each connected component representing one reactant or one synthon.
For synthon completion, we define a substructure z as a bond (i.e., z = bij) or a ring structure (i.e., z = {bij∣ai, aj ∈ a single or polycyclic ring}) that is used to complete synthons into reactants. We construct a substructure vocabulary \({{\mbox{}}}{{{{{{{\mathcal{Z}}}}}}}}{{\mbox{}}}=\{{{\mbox{}}}z{{\mbox{}}}\}\) by comparing \({{{{{{{{\mathcal{G}}}}}}}}}_{r}\)’s and their corresponding \({{{{{{{{\mathcal{G}}}}}}}}}_{s}\)’s in the training data, and extracting all the possible substructures from their differences. In total, G2Retro extracted 83 substructures, covering all the reactions in the test data. Details about these substructures are available in Supplementary Fig. 3 and Supplementary Fig. 4 in Supplementary Note 7. Note that different from templates used in TB methods, the substructures G2Retro used are only bonds and rings, and multiple bonds and rings can be attached to complete a synthon. For simplicity, when no ambiguity arises, we omit the super/sub-scripts and use \({{{{{{{\mathcal{G}}}}}}}}\) to represent \({{{{{{{{\mathcal{G}}}}}}}}}^{M}\).
Molecule representation learning
G2Retro learns the atom representations over the molecular graph \({{{{{{{\mathcal{G}}}}}}}}\) using the same message passing networks (MPN) as in Chen et al.59 (Supplementary Algorithm 4 in Supplementary Note 5).
G2Retro first learns atom embeddings to capture the atom types and their local neighborhood structures by passing the messages along the bonds in the molecular graphs. Each bond bij is associated with two message vectors mij and mji. The message \({{{\mbox{}}}{{{{{{{\bf{m}}}}}}}}{{\mbox{}}}}_{ij}^{(t)}\) at t-th iteration encodes the messages passing from ai to aj, and is updated as follows,
where xi is the atom feature vector, including the atom type, valence, charge, the number of hydrogens, whether the atom is included in a ring and whether the ring is aromatic; xij is the bond feature vector, including the bond type, whether the bond is conjugated or aromatic, and whether the bond is in a ring; \({W}_{i}^{a}\)’s (i = 1,2,3,4) are the learnable parameter matrices; \({{{\mbox{}}}{{{{{{{\bf{m}}}}}}}}{{\mbox{}}}}_{ij}^{(0)}\) is initialized with the zero vector; \({{\mbox{}}}{{{{{{{\mathcal{N}}}}}}}}{{\mbox{}}}({{{\mbox{}}}a{{\mbox{}}}}_{i})\) is the set with all the neighbors of ai (i.e., atoms connected with ai); and ReLU is the activation function. The message \({{{\mbox{}}}{{{{{{{\bf{m}}}}}}}}{{\mbox{}}}}_{ij}^{(t)}\) captures the structure of t-hop neighbors passing through the bond bij to aj, by iteratively aggregating the neighborhood messages \({{{\mbox{}}}{{{{{{{\bf{m}}}}}}}}{{\mbox{}}}}_{ki}^{(t-1)}\). With the maximum ta iterations, G2Retro derives the atom embedding ai as follows,
where \({{{\mbox{}}}{{{{{{{\bf{m}}}}}}}}{{\mbox{}}}}_{ki}^{(1\cdots {t}_{a})}\) denotes the concatenation of \(\{{{{\mbox{}}}{{{{{{{\bf{m}}}}}}}}{{\mbox{}}}}_{ki}^{(t)}| t\in [1:{t}_{a}]\}\); \({U}_{i}^{a}\)’s (i = 1,2,3) are the learnable parameter matrices. The embedding of the molecular graph \({{{{{{{\mathcal{G}}}}}}}}\) is calculated by summing over all the atom embeddings as follows,
For Mp and Ms, their embeddings calculated from their moleculear graphs as above are denoted as hp and hs, respectively.
Reaction center identification
Given a product Mp, G2Retro defines three types of reaction centers in Mp (Supplementary Algorithm 3 in Supplementary Note 5).
-
1.
a new bond bij, referred to as bond formation center (BF-center), that is formed across the reactants during the reaction but does not exist in any of the reactants;
-
2.
an existing bond bij in a reactant, referred to as bond type change center (BC-center), whose type changes during the reaction due to the gain or loss of hydrogens, while no other changes (e.g., new bond formation) happen; and
-
3.
an atom in a reactant, referred to as atom reaction center (A-center), from which a fragment is removed during the reaction, without new bond formation or bond type changes.
The above three types of reaction centers cover 97.7% of the training set. The remaining 2.3% of the reactions in the training data involve multiple new bond formations or bond type changes, and will be left for future research. Note that with a single atom as the reaction center, the synthon is the product itself. We refer to all the transformations needed to change a product to synthons as product-synthon transformations, denoted as p2s-T (Supplementary Algorithm 5 in Supplementary Note 5).
Reaction centers with new bond formation (BF-center)
Following Somnath et al.29, G2Retro derives the bond representations as follows,
where Abs( ⋅ ) represents the absolute difference; \({U}_{i}^{b}\)’s (i = 1,2,3,4) are the learnable parameter matrices. G2Retro uses the sum and the absolute difference of embeddings of the connected atoms to capture the local neighborhood structure of bond bij. Meanwhile, the two terms are both permutation-invariant to the order of ai and aj, and together can differentiate the information in ai and aj. With the bond representation, G2Retro calculates a score for each bond bij as follows,
where hp is the representation of the product graph \({{{{{{{{\mathcal{G}}}}}}}}}_{p}\) calculated as in Eq. (6); qb is a learnable parameter vector and \({Q}_{1}^{b}\) and \({Q}_{2}^{b}\) are the learnable parameter matrices. G2Retro measures how likely bond bij is a BF-center using sb(bij) by looking at the bond itself (i.e., bij) and the structure of the entire product graph (i.e., hp). G2Retro scores each bond in Mp and selects the most possible BF-center candidates {bij} with the highest scores. G2Retro breaks each product at each possible BF-center into synthons, and thus can generate multiple possible reactions.
In synthetic reactions, the formation of new bonds could induce the changes of neighbor bonds. Therefore, G2Retro also predicts whether the types of bonds neighboring the BF-center are changed during the reaction, referred to as the BF-center induced bond type change prediction (BTCP). Given the BF-centerbij, the set of the bonds neighboring bij is referred to as the BF-center neighbor bonds, denoted as \({{{{{{{{\mathcal{C}}}}}}}}}_{{\mathsf{BF}}}\), that is:
Thus, G2Retro predicts a probability distribution \({{{{{{{{\bf{f}}}}}}}}}^{b}\in {{\mathbb{R}}}^{1\times 4}\) for each neighboring bond in \({{{{{{{{\mathcal{C}}}}}}}}}_{{\mathsf{BF}}}\), denoted as \({{{\mbox{}}}b{{\mbox{}}}}_{i/jk}\in {{\mbox{}}}{{{{{{{{\mathcal{C}}}}}}}}}_{{\mathsf{BF}}}{{\mbox{}}}\), as follows,
where \({V}_{i}^{b}\)’s (i = 1,2,3) are the learnable parameter matrices. The first element \({{{{{{{{\bf{f}}}}}}}}}_{1}^{b}\) in fb represents how likely the bi/jk type is changed during the reaction (It is determined as type change if \({{{{{{{{\bf{f}}}}}}}}}_{1}^{b}\) is not the maximum in fb), and the other three represent how likely the original bi/jk in the reactant is single, double or triple bond, respectively (these three elements are reset to 0 if bi/jk type is predicted unchanged). Here, G2Retro measures neighbor bond type change by looking at the neighbor bond itself (i.e., bi/jk), the BF-center (i.e., bij) and the overall product (i.e., hp). G2Retro updates the synthons \({{{{{{{{\mathcal{G}}}}}}}}}_{s}\) by changing the neighboring bonds of the BF-center to their predicted original types. The predicted changed neighbor bonds are denoted as \({{{\mbox{}}}{{{{{{{{\mathcal{C}}}}}}}}}_{{\mathsf{BF}}}{{\mbox{}}}}^{{\prime} }\).
Reaction centers with bond type change (BC-center)
If a reaction center is due to a bond type change without new bond formations, G2Retro calculates a score vector \({{{{{{{{\bf{s}}}}}}}}}^{c}\in {{\mathbb{R}}}^{1\times 3}\) for each bond bij in Mp as follows,
where \({Q}_{i}^{c}\)’s (i = 1,2,3) are the learnable parameter matrices. Each element in sc(bij), denoted as \({s}_{k}^{c}({{{\mbox{}}}b{{\mbox{}}}}_{ij})\) (k = 1, 2, 3), represents, if bij is the BC-center, the score of bij’s original type in \({{{{{{{{\mathcal{G}}}}}}}}}_{r}\) being single, double, and triple bond, respectively. The element in sc corresponding to bij’s type in \({{{{{{{{\mathcal{G}}}}}}}}}_{p}\) is reset to 0 (i.e., bij’s type has to be different in \({{{{{{{{\mathcal{G}}}}}}}}}_{r}\) compared to that in \({{{{{{{{\mathcal{G}}}}}}}}}_{p}\)). Thus, the most possible BC-center candidates {bij} and their possible original bond types scored by sc( ⋅ ) are selected. G2Retro then changes the corresponding bond type to construct the synthons.
Reaction centers with single atoms (A-center)
If a reaction center is only at a single atom with a fragment removed, G2Retro predicts a center score for each atom ai in Mp as follows,
where qa is a learnable parameter vector and \({Q}_{1}^{a}\) and \({Q}_{2}^{a}\) are the learnable parameter matrices. G2Retro selects the atoms {ai} in Mp with the highest scores as potential A-center’s. In synthon completion, new fragments will be attached at the atom reaction centers.
Atom charge prediction (ACP)
For all the atoms ai involved in the reaction center or BF-center changed neighbor bonds \({{{\mbox{}}}{{{{{{{{\mathcal{C}}}}}}}}}_{{\mathsf{BF}}}{{\mbox{}}}}^{{\prime} }\), G2Retro also predicts whether the charge of ai remains unchanged in reactants. G2Retro uses an embedding c to represent all the involved bond formations and changes in p2s-T. If the reaction center is predicted as a BF-center at bij, G2Retro calculates the embedding c as follows,
where \({{{\mbox{}}}{{{{{{{{\mathcal{C}}}}}}}}}_{{\mathsf{BF}}}{{\mbox{}}}}^{{\prime} }\) is a subset of \({{{{{{{{\mathcal{C}}}}}}}}}_{{\mathsf{BF}}}\) with all the bonds that changed types; \({{{{{{{{\bf{x}}}}}}}}}_{kl}^{{\prime} }\) is a 1 × 4 one-hot vector, in which \({{{{{{{{\bf{x}}}}}}}}}_{kl}^{{\prime} }(0)=1\) if bond bkl is the bond formation center (i.e., bkl = bij), or \({{{{{{{{\bf{x}}}}}}}}}_{kl}^{{\prime} }(i)=1\) (i = 1, 2, 3) if bkl type is changed from single, double or triple bond in reactants, respectively, during the reaction (i.e., bkl is in \({{{\mbox{}}}{{{{{{{{\mathcal{C}}}}}}}}}_{{\mathsf{BF}}}{{\mbox{}}}}^{{\prime} }({{{\mbox{}}}b{{\mbox{}}}}_{ij})\)); \({W}_{i}^{c}\)’s (i = 1, 2, 3) are the learnable parameter matrices.
If the reaction center is predicted as a BC-center at bij, c is calculated as follows,
where \({{{{{{{{\bf{x}}}}}}}}}_{ij}^{{\prime} }(0)=0\) and \({{{{{{{{\bf{x}}}}}}}}}_{ij}^{{\prime} }(i)=1\) (i = 1, 2, 3) if bkl type is changed from single, double or triple bond in reactants, respectively, during the reaction. If the reaction center is an A-center, no p2s-T are needed and thus c = 0.
With the embedding c for p2s-T, G2Retro calculates the probabilities that ai will have charge changes during the reaction as follows,
where \({V}_{1}^{c}\) and \({V}_{2}^{c}\) are the learnable parameter matrices; \({{{{{{{{\bf{f}}}}}}}}}^{c}\in {{\mathbb{R}}}^{1\times 3}\) is a vector representing the probabilities of accepting one electron, donating one electron or no electron change during the reaction. The option corresponding to the maximum value in fc is selected and will be applied to update synthon charges accordingly. G2Retro considers at most one electron change since this is the case for all the reactions in the benchmark data.
Reaction center identification module training
With the scores for three types of reaction centers, G2Retro minimizes the following cross entropy loss to learn the above scoring functions (i.e., Eqs. (8), (11) and (12)),
where y* (x = a, b, c) is the label indicating whether the corresponding candidate is the ground-truth reaction center of type * (y* = 1) or not (y* = 0); \({{\mathbb{I}}}_{k}(x)\) is an indicator function (\({{\mathbb{I}}}_{k}(x)=1\) if x = k, 0 otherwise), and thus \({{\mathbb{I}}}_{k}({y}_{ij}^{c})\) indicates whether the ground-truth bond type of bij is k or not (k = 1, 2, 3 indicating single, double or triple bond); and l*( ⋅ ) (* = a, b)/\(({l}_{k}^{c}(\cdot ))\) is the probability calculated by normalizing the score s*( ⋅ )/\({s}_{k}^{c}\), that is, \({l}^{* }(x)=\exp ({s}^{* }(x))/\Delta\), where \(\Delta ={\sum }_{{{{\mbox{}}}b{{\mbox{}}}}_{ij}\in {{\mbox{}}}{{{{{{{\mathcal{B}}}}}}}}{{\mbox{}}}}(\exp ({s}^{b}({{{\mbox{}}}b{{\mbox{}}}}_{ij}))+\mathop{\sum }\nolimits_{k = 1}^{3}\exp ({s}_{k}^{c}({{{\mbox{}}}b{{\mbox{}}}}_{ij})))+{\sum }_{{{{\mbox{}}}a{{\mbox{}}}}_{i}\in {{\mbox{}}}{{{{{{{\mathcal{A}}}}}}}}{{\mbox{}}}}\exp ({s}^{a}({{{\mbox{}}}a{{\mbox{}}}}_{i}))\) (\({l}_{k}^{c}(x)=\exp ({s}_{k}^{c}(x))/\Delta\)). Similarly, G2Retro also learns the predictor fb( ⋅ ) for neighbor bond changes (Eq. (10)) and fc( ⋅ ) for atom charge changes (Eq. (15)) by minimizing their respective cross entropy loss \({{{{{{{{\mathcal{L}}}}}}}}}^{b}\) and \({{{{{{{{\mathcal{L}}}}}}}}}^{c}\). Therefore, the center identification module learns the predictors by solving the following optimization problem:
where Θ is the set of all the parameters in the prediction functions. We used Adam algorithm to solve the optimization problem and do the same for the other training objectives.
Synthon completion
Once the reaction centers are identified and all the product-synthon transformations (p2s-T) are conducted to generate synthons from products, G2Retro completes the synthons into the reactants by sequentially attaching substructures (Supplementary Algorithm 6 in Supplementary Note 5). All the actions involved in this process are referred to as synthon-reactant transformations. During the completion process, any intermediate molecules {M*} are represented as molecular graph \(\{{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}\}\). At step t, we denote the atom in the intermediate molecular graph \({{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}}^{(t)}\) (\({{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}}^{(0)}={{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}_{s}{{\mbox{}}}\)) that new substructures will be attached to as a(t), and denote the substructure attached to a(t) as z(t), resulting in \({{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}}^{(t+1)}\).
Atom attachment prediction
The algorithm for atom attachment prediction is presented in Supplementary Algorithm 8 in Supplementary Note 5. G2Retro first predicts whether further attachment should be added to a(t) or should stop at a(t), referred to as the atom attachment continuity prediction (AACP), with the probability calculated as follows,
where
In Eq. (18), a(t) is the embedding of a(t) calculated over the graph \({{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}}^{(t)}\) (Eq. (5)); hs is the representation for all the synthons as in Eq. (19); \({V}_{i}^{o}\)’s (i = 1,2,3) are the learnable parameter matrices; σ is the sigmoid function. In Eq. (19), G2Retro calculates the representations by applying MPN over the graph \({{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}_{s}{{\mbox{}}}\) that could be disconnected, and the resulted representation is equivalent to applying MPN over each \({{{{{{{{\mathcal{G}}}}}}}}}_{s}\)’s connected component independently and then summing over their representations. G2Retro intuitively measures “how likely” the atom has a new substructure attached to it by looking at the atom itself (i.e., a(t)), all the synthons (i.e., hs), and the product (i.e., hp). Note that in Eq. (18), BRICS fragment information (i.e., \({{{\mbox{}}}{{{{{{{\bf{a}}}}}}}}{{\mbox{}}}}^{{\prime} }\) as in Eq. S3 in Supplementary Note 1) is not used because the fragments for the substructures that will be attached to a(t) will not be available until the substructures are determined.
If a(t) is predicted to attach with a new substructure, G2Retro predicts the type of the new substructure, referred to as the atom attachment type prediction (AATP), with the probabilities of all the substructure types in the vocabulary \({{{{{{{\mathcal{Z}}}}}}}}\), calculated as follows,
where \({V}_{i}^{{{\mbox{}}}z{{\mbox{}}}}\)’s (i = 1,2,3) are the learnable parameter matrices. Higher probability for a substructure type z indicates that z is more likely to be selected as z(t). The atoms a ∈ z(t) in the attached substructure are stored for further attachment, that is, they, together with any newly added atoms along the iterative process, will become a(T) (T = t + 1, t + 2, ⋯ ) in a depth-first order in the retrospective reactant graphs. G2Retro stops the entire synthon completion process after all the atoms in the reaction centers and the newly added atoms are predicted to have no more substructures to be attached.
Synthon completion model training
G2Retro trains the synthon completion module using the teacher forcing strategy, and attaches the ground-truth fragments instead of the prediction results to the intermediate molecules during training. G2Retro learns the predictors fo( ⋅ ) (Eq. (18)) and fz( ⋅ ) (Eq. (20)) by minimizing their cross entropy losses \({{{{{{{{\mathcal{L}}}}}}}}}^{o}\) and \({{{{{{{{\mathcal{L}}}}}}}}}^{{{\mbox{}}}z{{\mbox{}}}}\) as follows:
where Φ is the set of parameters.
Inference
The algorithm for G2Retro inference is presented in Supplementary Algorithm 2 in Supplementary Note 5.
Top-K reaction center selection
During the inference, G2Retro generates a ranked list of candidate reactant graphs \(\{{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}_{r}{{\mbox{}}}\}\) (note that each reactant graph can be disconnected with multiple connected components each representing a reactant). With a beam size K, for each product, G2Retro first selects the top-K most possible reaction centers from each reaction center type (BF-center, BC-center and A-center), and then selects the top-K most possible reaction centers from all the 3K candidates based on their corresponding scores (i.e., sb as in Eq. (8) for BF-center, sc as in Eq. (11) for BC-center, and sa as in Equation (12) for A-center). Then G2Retro converts the product graph \({{{{{{{{\mathcal{G}}}}}}}}}_{p}\) into the top-K synthon graphs \({\{{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}_{s,i}{{\mbox{}}}\}}_{i = 1}^{K}\) accordingly. Different reaction centers lead to diverse synthons. For these synthon graphs, neighbor bond type change is predicted when necessary; atom charge change is predicted for all the atoms involved in reaction centers and their neighboring bonds \({{{{{{{{\mathcal{C}}}}}}}}}_{{\mathsf{BF}}}\) for BF-center’s. All the bond type changes and atom charge changes are predicted as those with the highest probabilities as in Eq. (10) and Eq. (15), respectively.
Top-N reactant graph generation
Once the top-K reaction centers for each product are selected and their synthon graphs are generated, G2Retro completes the synthon graphs \({\{{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}_{s,i}{{\mbox{}}}\}}_{i = 1}^{K}\) into reactant graphs. During the completion, G2Retro scores each possible reactant graph and uses their final scores to select the top-N reactant graphs, and thus top-N most possible synthetic reactions, for each product. Since during synthon completion, the attachment substructure type prediction (Eq. (20)) gives a distribution of all possible attachment substructures; by using top possible substructures, each synthon and its intermediate graphs can be extended to multiple different intermediate graphs, leading to exponentially many reactant graphs and diversity in the predicted reactions. The intermediate graphs are denoted as \({\{{{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}}_{ij}^{(t)}\}}_{i = 1}^{K}\), where \({{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}}_{ij}^{(t)}\) is for the j-th possible intermediate graph of the i-th synthon graph \({{{{{{{{\mathcal{G}}}}}}}}}_{s,i}\) at step t. However, to fully generate all the possible completed reactant graphs, excessive computation is demanded. Instead, G2Retro applies a greedy beam search strategy (Supplementary Algorithm 7 in Supplementary Note 5) to only explore the most possible top reactant graph completion paths.
In the beam search strategy, G2Retro scores each intermediate graph \({{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}}_{ij}^{(t)}\) using a score \({{{\mbox{}}}s{{\mbox{}}}}_{ij}^{(t)}\), which is calculated as the sum over all the log-likelihoods of all the predictions along the completion path from \({{{{{{{{\mathcal{G}}}}}}}}}_{s}\) up to \({{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}}_{ij}^{(t)}\); \({{{\mbox{}}}s{{\mbox{}}}}_{ij}^{(0)}\) is initialized as the sum of the log-likelihoods of all the predictions from \({{{{{{{{\mathcal{G}}}}}}}}}_{p}\) to \({{{{{{{{\mathcal{G}}}}}}}}}_{s}\). At each step t (t≤30), each intermediate graph \({{{\mbox{}}}{{{{{{{{\mathcal{G}}}}}}}}}^{* }{{\mbox{}}}}_{ij}^{(t)}\) is extended to at most N+1 intermediate graph candidates. These N+1 candidates include the one that is predicted to stop at the atom that new substructures could be attached to (i.e., as a(t) in Eq. (18); this intermediate graph could be further completed at other atoms) in this step, and at most N candidates with the top-N predicted substructures attached (Eq. (20)). Among all the candidates generated from all the intermediate graphs at step t, the top-N scored ones will be further forwarded into the next completion step t+1. In case some of the top-N graphs are fully completed, the remaining will go through the next steps. This process will be ended until the number of all the completed reactant graphs at different steps reaches or goes above N. Then, among all the incomplete graphs at the last step, the intermediate graphs with log-likelihood values higher than the N-th largest score in all the completed ones will continue to complete as above. The entire process will end until no more intermediate graphs are qualified to further completion. Among all the completed graphs, the top-N graphs are selected as the generated reactants.
Related work
Deep-learning-based retrosynthesis prediction methods are typically categorized into three classes: template based (TB), template free (TF) and semi-template based (Semi-TB).
Template-based methods
Template-based methods formulate the retrosynthesis problem as a selection problem over a set of reaction templates. These templates can be either hand-crafted by experts61 or automatically extracted from known reactions in databases12,13,14,15,16. Szymkuc et al.61 provided a review on using reaction templates coded by human experts for synthetic planning. However, these rules may not cover a large set of reactions due to the limitation of human annotation capacity. Recent template-based methods extract reaction templates automatically from databases. With the reaction templates available, Coley et al.12 (Retrosim) selected the reaction templates that the corresponding reactions in the database have the products most similar with the target molecules, in order to synthesize the target molecules. Dai et al.14 learned the joint probabilities of templates matched in the product molecules and all its possible reactants using two energy functions, one for reaction template scoring and the other for reactant scoring conditioned on templates. Seidl et al.15 (MHNreact) learned to associate the target molecule with the relevant reaction templates using a modern Hopfield network. Chen et al.16 (LocalRetro) scored the suitability of all the reaction templates at all the potential reaction centers (atoms and bonds) in the target molecule. The use of templates provides interpretability toward the reasoning behind the generated reactions. However, these templates also limit the template-based methods to the reactions only covered by the templates.
Template-free methods
Template-free methods directly learn to transform the product into the reactants without using the reaction templates17,18,19,20,21,22,24,25,31. Most template-free methods utilize the sequence representations of molecules (SMILES) and formulate the transformation between the product and its corresponding reactants as a sequence-to-sequence problem. Many SMILES-based methods use Transformer33, a language model with attention mechanisms to model the relationship across tokens. Transformer follows the encoder-decoder architecture, which encodes the product SMILES string into a latent vector and then decodes the vector into the reactant SMILES strings. For example, Kim et al.22 (TiedTransformer) learned the transformation from a product to its reactants using two coupled Transformers with shared parameters, one for the forward product prediction (synthesis) and the other for the backward reactant prediction (retrosynthesis). During the inference, they leveraged both the forward and backward models to find the best reactions. Sun et al.24 (Dual) transformed a product to its reactants using an energy-based framework. They also leveraged the duality of the forward and backward models by training them together and selected the best reactions with the highest energy value from the two models. Tetko et al.31 (AT) learned to transform a product into its reactants using a Transformer trained on a dataset augmented with various non-canonical SMILES representations of each molecule. In AT, each target molecule was tested multiple times using different SMILES string representations. Zhong et al.32 (R − SMILES) aligned the product and reactant SMILES strings to minimize their edit distance, and trained a transformer to decode the reactant SMILES strings from the products. They also augmented the training dataset and tested each target molecule multiple times as in AT. In addition to SMILES-based template-free methods, Sacha et al.26 (MEGAN) formulated retrosynthesis as a graph editing process from a product to its reactants. These graph edits include the change in the atom properties or the bond types, or the addition of the new atoms or the benzene rings into the synthons. These template-free methods are independent of reaction templates, and thus they may have better generalizability to unknown reactions compared to template-based methods. However, template-free methods lack interpretability toward the reasoning behind their end-to-end predictions. SMILES-based template-free methods also suffer from the validity issue that the generated sequences may fail to follow the grammar of SMILES strings or violate chemical rules17.
Semi-template-based methods
Semi-template-based methods26,27,28,29,30 do not use reaction templates, or they do not directly transform a product into its reactants. Instead, semi-template-based methods follow a two-step workflow utilizing atom-mappings: (1) they first identify the reaction centers and transform the product into synthons (intermediate molecules) using the reaction centers; and then (2) they complete the synthons into the reactants. Shi et al.28 (G2G) first predicted reaction centers as bonds that can be used to split the product into the synthons, and then utilized a variational autoencoder35 to complete synthons into reactants by sequentially adding new bonds or new atoms. Somnath et al.29 (GraphRetro) predicted the bonds with changed bond types or the atoms with changed hydrogen count as the reaction centers, and then completed the synthons by selecting the pre-extracted subgraphs that realize the difference between synthons and reactants. Wang et al.30 (RetroPrime) formulated the reaction center identification and synthon completion problems as two sequence-to-sequence problems (i.e., product to synthon, and synthon to reactant), and trained two Transformers for these problems, respectively. The prediction of reaction centers first in the above methods allows better interpretability toward the reasoning behind the generation process. The two-step workflow also empowers these methods to diversify their generated reactants by allowing multiple different reaction center predictions forwarded into their synthon completion step.
G2Retro also identifies the reaction centers and then completes the synthons into the reactants in a sequential way as G2G does. However, G2Retro is different from G2G. G2Retro can cover multiple types of reaction centers while G2G takes only the newly formed bonds as the reaction center, which leads to lower coverage of G2G on the dataset. During synthon completion, G2Retro attaches substructures (e.g., rings and bonds) instead of single atoms as in G2G, into synthons to simplify the completion process. In addition and more importantly, G2Retro uses other synthons of the same reaction and also the product to complete a synthon, and thus the synthon completion is more contextualized for the product, while G2G does not consider other synthons.
Fragment-based molecule generation
Following the idea of fragment-based drug design62,63, fragment-based molecule generation methods have been developed. For example, Jin et al.64 first decomposed a molecular graph into a junction tree of chemical substructures, and then used a variational autoencoder over the junction trees and its chemical substructures to generate and assemble new molecules (JT-VAE). Podda et al.65 encoded and decoded a sequence of fragments via a variational autoencoder, and generated new molecules by connecting fragments generated from the autoencoder. Chen et al.59 optimized a molecule by removing and attaching substructures in a starting molecule. G2Retro generates reactants from synthons also by attaching new substructures. However, the generation strategy in G2Retro is fundamentally different from that in the previous fragment-based molecule generation methods. During synthon completion, G2Retro does not encode the synthons using their substructures as what JT-VAE and Modof do. It does not either encode or decode the substructures that are to be attached to the synthons. Instead, G2Retro attaches the substructures to a specific, identified atom in the molecular graph of the synthons. Therefore, G2Retro can directly attach a substructure to the predicted reaction centers.
Data preprocessing and experimental settings
We used the benchmark dataset provided by Yan et al.27. This dataset, also referred to as USPTO-50K, contains 50K chemical reactions that are randomly sampled from a large dataset collected by Lowe11 from US patents published between 1976 and September 2016. Each reaction in the large dataset is atom-mapped so that each atom in the product is uniquely mapped to an atom in the reactants. The 50K reactions in USPTO-50K are classified into 10 reaction types by Schneider et al.66. To avoid the information leakage issue27 (e.g., reaction center is given in both the training and test data), all the product SMILES strings in USPTO-50K are canonicalized. We used exactly the same training/validation/test data splits of USPTO-50K as in the previous methods12,27, which contain 40K/5K/5K reactions, respectively. Table 5 presents the data statistics. We trained G2Retro models on the 40K training data, with parameters tuned on the 5K validation data, and tested on the 5K test data. For reproducibility purposes, details about model training and parameter tuning are provided in Supplementary Note 8.
Baselines
We compared G2Retro with the state-of-the-art baseline methods for the one-step retrosynthesis problem, including five template-based (TB) methods, ten template-free (TF) methods and five semi-template-based (Semi-TB) methods. Inspired by the recent success of using fragments in other tasks67, we further extended G2Retro into G2Retro-B by incorporating the fragments generated from the breaking retrosynthetically interesting chemical substructures (BRICS) fragmentation algorithm68. Details of G2Retro-B are available in Supplementary Note 1. The experimental setting for G2Retro-B is identical to that of G2Retro.
Template-based baseline methods The five TB baseline methods include Retrosim, Neuralsym, GLN, MHNreact and LocalRetro. These methods first mine reaction templates from training data and apply only these templates to construct reactants from the target molecule.
-
Retrosim12 selects the templates of reactions that produce molecules most similar to the target molecule.
-
Neuralsym13 predicts suitable templates using product fingerprints through a multi-layer perceptron.
-
GLN14 predicts reactions using two energy functions, one for template scoring and the other for reactant scoring conditioned on templates.
-
MHNreact15 learns the associations between molecules and reaction templates using modern Hopfield networks, and selects templates based on the associations.
-
LocalRetro16 selects templates against each atom and each bond using classifiers.
Template-free baseline methods The ten TF baseline methods all use Transformer over SMILES string representations of products and/or reactants.
-
SCROP17 maps the SMILES strings of products to the SMILES strings of reactants using a Transformer, and then corrects syntax errors (e.g., mismatch of parentheses in SMILES strings) to ensure valid reactant SMILES strings.
-
LV-Trans18 pre-trains a vanilla Transformer using reactions generated from templates, and then fine-tunes the Transformer with a multinomial latent variable representing reaction types.
-
GET19 trains standard Transformer encoders and decoders using the combined atom representations learned from molecular graphs and from SMILES strings.
-
Chemformer20 translates product SMILES strings into reactant SMILES strings using Transformer, which is pre-trained on an independent dataset to recover masked SMILES strings (i.e., with some atoms masked out) or to normalize augmented SMILES strings (i.e., multiple, equivalent non-canonical SMILES strings for each SMILES string).
-
Graph2SMILES21 encodes molecular graphs using graph neural networks with attention mechanisms, and decodes the reactant SMILES strings from the graph representations using a Transformer decoder.
-
TiedTransformer22 uses two Transformers with shared parameters to learn the transformation from products to reactants and vice versa, respectively, and selects the best reactions using the likelihood values from these two Transformers.
-
GTA23 enhances a Transformer with truncated attention connections regulated by molecular graph structures.
-
Dual24 uses an energy-based model with two Transformers to learn the transformation from product SMILES strings to reactants’ SMILES strings and vice versa, and selects the best reactions using the energy.
-
Retroformer25 predicts the reaction center region using a reaction center detection module, and uses the embedding of predicted centers as a condition to transform via Transformer the product into the reactants in SMILES. Although Retroformer predicts the reaction center, it does not split products into synthons using the reaction center, and thus does not follow a two-step, semi-template-based framework.
-
MEGAN26 transforms the product molecular graphs into the corresponding reactant graphs using a sequence of graph edits (e.g., change atom charges, add a new bond) that are learned from products and their reactants in the training set.
Semi-template-based methods
The five Semi-TB baseline methods all use molecular graph representations. Most of them explicitly predict reaction centers first.
-
RetroPrime30 trains two Transformers independently to predict the transformation from the product to its synthons and from the synthons to the reactants, respectively.
-
RetroXpert27 predicts reaction centers on molecular graphs via a graph attention network, and transforms resulting synthons to reactants using a Transformer.
-
G2G28 predicts reaction centers on molecular graphs via a graph neural network, and completes synthons into reactants through sequential additions of new atoms or bonds using the latent variables sampled from the latent space of a variational graph autoencoder.
-
GraphRetro29 predicts reaction centers via a message passing neural network over molecular graphs, and completes synthons by selecting the subgraphs in a vocabulary that realize the difference between the synthons and reactants.
Data availability
The data used in this paper are available publicly69 at the link https://doi.org/10.5281/zenodo.7839013 and the link https://github.com/ninglab/G2Retro.
Code availability
The code for G2Retro, G2Retro-B and G2Retro-ens is available publicly69 at the link https://doi.org/10.5281/zenodo.7839013 and the link https://github.com/ninglab/G2Retro. A web portal for G2Retro is available at the link http://go.osu.edu/G2Retro.
References
Segler, M. H. S., Preuss, M. & Waller, M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555, 604–610 (2018).
Chen, B., Li, C., Dai, H. & Song, L. Retro*: Learning retrosynthetic planning with neural guided A* search. In (eds III, H. D. & Singh, A.) Proceedings of the 37th International Conference on Machine Learning, vol. 119, 1608–1616 (PMLR, 2020).
Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018).
Lajiness, M. S., Maggiora, G. M. & Shanmugasundaram, V. Assessment of the consistency of medicinal chemists in reviewing sets of compounds. J. Med. Chem. 47, 4891–4896 (2004).
Huang, Q., Li, L.-L. & Yang, S.-Y. RASA: A rapid retrosynthesis-based scoring method for the assessment of synthetic accessibility of drug-like molecules. J. Chem. Inf. Model. 51, 2768–2777 (2011).
Takaoka, Y. et al. Development of a method for evaluating drug-likeness and ease of synthesis using a data set in which compounds are assigned scores based on chemists’ intuition. J. Chem. Inf. Comput. Sci. 43, 1269–1275 (2003).
Kutchukian, P. S. et al. Inside the mind of a medicinal chemist: The role of human bias in compound prioritization during drug discovery. PLoS ONE 7, e48476 (2012).
Reaxys. Reaxys is a registered trademark of relx intellectual properties sa used under license.Https://www.reaxys.com. Accessed: 2022-05-22.
Gabrielson, S. W. SciFinder. J. Med. Libr. Assoc. 106, 588–590 (2018).
Kearnes, S. M. et al. The open reaction database. J. Am. Chem. Soc. 143, 18820–18826 (2021).
Lowe, D. M. Chemical reactions from us patents (1976-sep2016). https://doi.org/10.6084/m9.figshare.5104873.v1 Accessed: 2022-11-06.
Coley, C. W., Rogers, L., Green, W. H. & Jensen, K. F. Computer-assisted retrosynthesis based on molecular similarity. ACS Cent. Sci. 3, 1237–1245 (2017).
Segler, M. H. S. & Waller, M. P. Neural-symbolic machine learning for retrosynthesis and reaction prediction. Chem. Eur. J. 23, 5966–5971 (2017).
Dai, H., Li, C., Coley, C., Dai, B. & Song, L. Retrosynthesis prediction with conditional graph logic network. In (eds Wallach, H. et al.) Advances in Neural Information Processing Systems, vol. 32, 8872–8882 (Curran Associates, Inc., 2019).
Seidl, P. et al. Improving few- and zero-shot reaction template prediction using modern hopfield networks. J. Chem. Inf. Model. 62, 2111–2120 (2022).
Chen, S. & Jung, Y. Deep retrosynthetic reaction prediction using local reactivity and global attention. JACS Au 1, 1612–1620 (2021).
Zheng, S., Rao, J., Zhang, Z., Xu, J. & Yang, Y. Predicting retrosynthetic reactions using self-corrected transformer neural networks. J. Chem. Inf. Model. 60, 47–55 (2019).
Chen, B., Shen, T., Jaakkola, T. S. & Barzilay, R. Learning to make generalizable and diverse predictions for retrosynthesis (2019). arXiv:1910.09688v1.
Mao, K. et al. Molecular graph enhanced transformer for retrosynthesis prediction. Neurocomputing 457, 193–202 (2021).
Irwin, R., Dimitriadis, S., He, J. & Bjerrum, E. J. Chemformer: a pre-trained transformer for computational chemistry. Mach. Learn.: Sci. Technol. 3, 015022 (2022).
Tu, Z. & Coley, C. W. Permutation invariant graph-to-sequence model for template-free retrosynthesis and reaction prediction. J. Chem. Inf. Model. 62, 3503–3513 (2022).
Kim, E., Lee, D., Kwon, Y., Park, M. S. & Choi, Y.-S. Valid, plausible, and diverse retrosynthesis using tied two-way transformers with latent variables. J. Chem. Inf. Model. 61, 123–133 (2021).
Seo, S.-W. et al. GTA: Graph truncated attention for retrosynthesis. Proc. AAAI Conf. Artif. Intell. 35, 531–539 (2021).
Sun, R., Dai, H., Li, L., Kearnes, S. & Dai, B. Towards understanding retrosynthesis by energy-based models. In (eds Ranzato, M., Beygelzimer, A., Dauphin, Y., Liang, P. & Vaughan, J. W.) Advances in Neural Information Processing Systems, vol. 34, 10186–10194 (Curran Associates, Inc., 2021).
Wan, Y., Hsieh, C.-Y., Liao, B. & Zhang, S. Retroformer: Pushing the limits of end-to-end retrosynthesis transformer. In (eds Chaudhuri, K. et al.) Proceedings of the 39th International Conference on Machine Learning, vol. 162, 22475–22490 (PMLR, 2022).
Sacha, M. et al. Molecule edit graph attention network: Modeling chemical reactions as sequences of graph edits. J. Chem. Inf. Model. 61, 3273–3284 (2021).
Yan, C. et al. Retroxpert: Decompose retrosynthesis prediction like a chemist. In (eds Larochelle, H., Ranzato, M., Hadsell, R., Balcan, M. F. & Lin, H.) Advances in Neural Information Processing Systems, vol. 33, 11248–11258 (Curran Associates, Inc., 2020).
Shi, C., Xu, M., Guo, H., Zhang, M. & Tang, J. A graph to graphs framework for retrosynthesis prediction. In (eds III, H. D. & Singh, A.) Proceedings of the 37th International Conference on Machine Learning, vol. 119, 8818–8827 (PMLR, 2020).
Somnath, V. R., Bunne, C., Coley, C., Krause, A. & Barzilay, R. Learning graph models for retrosynthesis prediction. In (eds Ranzato, M., Beygelzimer, A., Dauphin, Y., Liang, P. & Vaughan, J. W.) Advances in Neural Information Processing Systems, vol. 34, 9405–9415 (Curran Associates, Inc., 2021).
Wang, X. et al. RetroPrime: A diverse, plausible and transformer-based method for single-step retrosynthesis predictions. Chem. Eng. J. 420, 129845 (2021).
Tetko, I. V., Karpov, P., Deursen, R. V. & Godin, G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 11, 5575 (2020).
Zhong, Z. et al. Root-aligned SMILES: a tight representation for chemical reaction prediction. Chem. Sci. 13, 9023–9034 (2022).
Vaswani, A. et al. Attention is all you need. In (eds Guyon, I. et al.) Advances in Neural Information Processing Systems, vol. 30 (Curran Associates, Inc., 2017).
Kipf, T. N. & Welling, M. Semi-supervised classification with graph convolutional networks. In International Conference on Learning Representations (OpenReview.net, 2017).
Kingma, D. P. & Welling, M. Auto-encoding variational bayes. In International Conference on Learning Representations (OpenReview.net, 2014).
Venkatasubramanian, V. & Mann, V. Artificial intelligence in reaction prediction and chemical synthesis. Curr. Opin. Chem. Eng. 36, 100749 (2022).
Chen, F., Wang, Y.-C., Wang, B. & Kuo, C.-C. J. Graph representation learning: a survey. APSIPA Trans. Signal Inf. Process. 9, e15 (2020).
Brown, D. G. & Bostrom, J. Analysis of past and present synthetic methodologies on medicinal chemistry: Where have all the new reactions gone? J. Med. Chem. 59, 4443–4458 (2015).
Vijayakumar, A. K. et al. Diverse beam search: Decoding diverse solutions from neural sequence models (2016). arXiv:1610.02424v2.
Novel drug approvals for 2022. https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2022 Accessed: 2023-04-28.
Al-Samkari, H. & van Beers, E. J. Mitapivat, a novel pyruvate kinase activator, for the treatment of hereditary hemolytic anemias. Ther. Adv. Hematol. 12, 204062072110660 (2021).
Sizemore, J. P., Guo, L., Mirmehrabi, M. & Su, Y. Crystalline forms of n-(4-(4-(cyclopropylmethyl) piperazine-1-carbonyl)phenyl)quinoline-8-sulfonamide. World Intellectual Property Organization WO/2019/104134A1 (2019).
Keam, S. J. Tapinarof cream 1%: First approval. Drugs 82, 1221–1228 (2022).
Chen, G., Webster, J., Li, J., Hu, K. & Zhu, J. Anti-inflammatory and psoriasis treatment and protein kinase inhibition by hydroxyltilbenes and novel stilbene derivatives and analogues. World Intellectual Property Organization WO/2001/042231A2 (2001).
Duan, X.-F., Zeng, J., Lü, J.-W. & Zhang, Z.-B. Insights into the general and efficient cross McMurry reactions between ketones. J. Org. Chem. 71, 9873–9876 (2006).
Robiette, R., Richardson, J., Aggarwal, V. K. & Harvey, J. N. Reactivity and selectivity in the wittig reaction: a computational study. J. Am. Chem. Soc. 128, 2394–2409 (2006).
Miyaura, N. & Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 95, 2457–2483 (1995).
LeBlond, C. R., Andrews, A. T., Sun, Y. & Sowa, J. R. Activation of aryl chlorides for suzuki cross-coupling by ligandless, heterogeneous palladium. Org. Lett. 3, 1555–1557 (2001).
Yin, L. & Liebscher, J. Carbon-carbon coupling reactions catalyzed by heterogeneous palladium catalysts. Chem. Rev. 107, 133–173 (2006).
Fanta, P. E. The ullmann synthesis of biaryls. Synthesis 1974, 9–21 (1974).
Stille, J. K. The palladium-catalyzed cross-coupling reactions of organotin reagents with organic electrophiles[new synthetic methods(58)]. Angew. Chem., Int. Ed. Engl. 25, 508–524 (1986).
Tamao, K., Sumitani, K. & Kumada, M. Selective carbon-carbon bond formation by cross-coupling of grignard reagents with organic halides. catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 94, 4374–4376 (1972).
Lewis, M. et al. BART: Denoising sequence-to-sequence pre-training for natural language generation, translation, and comprehension. In Proceedings of the 58th Annual Meeting of the Association for Computational Linguistics (Association for Computational Linguistics, 2020).
Yan, C. et al. Retroxpert. https://github.com/uta-smile/RetroXpert Accessed: 2022-10-01.
Somnath, V. R.https://github.com/uta-smile/RetroXpert/issues/15#issuecomment-864845942 Accessed: 2022-06-01.
DimGorr. https://github.com/DeepGraphLearning/torchdrug/issues/131 Accessed: 2022-10-01.
Open science. https://research-and-innovation.ec.europa.eu/strategy/strategy-2020-2024/our-digital-future/open-science_en Accessed: 2023-04-28.
Data management. https://sharing.nih.gov/data-management-and-sharing-policy/data-management Accessed: 2023-04-28.
Chen, Z., Min, M. R., Parthasarathy, S. & Ning, X. A deep generative model for molecule optimization via one fragment modification. Nat. Mach. Intell. 3, 1040–1049 (2021).
Jin, W., Barzilay, D. & Jaakkola, T. Hierarchical generation of molecular graphs using structural motifs. In (eds III, H. D. & Singh, A.) Proceedings of the 37th International Conference on Machine Learning, vol. 119, 4839–4848 (PMLR, 2020).
Szymkuć, S. et al. Computer-assisted synthetic planning: The end of the beginning. Angew. Chem., Int. Ed. Engl. 55, 5904–5937 (2016).
Murray, C. & Rees, D. The rise of fragment-based drug discovery. Nat. Chem. 1, 187–92 (2009).
Hajduk, P. J. & Greer, J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discov. 6, 211–219 (2007).
Jin, W., Barzilay, R. & Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In (eds Dy, J. & Krause, A.) Proceedings of the 35th International Conference on Machine Learning, vol. 80, 2323-2332 (PMLR, 2018).
Podda, M., Bacciu, D. & Micheli, A. A deep generative model for fragment-based molecule generation. In (eds Chiappa, S. & Calandra, R.) Proceedings of the Twenty Third International Conference on Artificial Intelligence and Statistics, vol. 108, 2240–2250 (PMLR, 2020).
Schneider, N., Stiefl, N. & Landrum, G. A. What’s what: The (nearly) definitive guide to reaction role assignment. J. Chem. Inf. Model. 56, 2336–2346 (2016).
Zhang, Z., Liu, Q., Wang, H., Lu, C. & Lee, C.-K. Motif-based graph self-supervised learning for molecular property prediction. In (eds Ranzato, M., Beygelzimer, A., Dauphin, Y., Liang, P. & Vaughan, J. W.) Advances in Neural Information Processing Systems, vol. 34, 15870–15882 (Curran Associates, Inc., 2021).
Degen, J., Wegscheid-Gerlach, C., Zaliani, A. & Rarey, M. On the art of compiling and using ‘drug-like’ chemical fragment spaces. ChemMedChem 3, 1503–1507 (2008).
Code for G2Retro as a two-step graph generative models for retrosynthesis prediction. https://doi.org/10.5281/zenodo.7839013 (2023).
Acknowledgements
This project was made possible, in part, by support from the National Science Foundation grant nos. IIS-2133650 (X.N.), and The Ohio State University President’s Research Excellence program (X.N., H.S.). Any opinions, findings and conclusions or recommendations expressed in this paper are those of the authors and do not necessarily reflect the views of the funding agency. We thank Dr. Michael A. Walters for his constructive comments.
Author information
Authors and Affiliations
Contributions
X.N. conceived the research. X.N. and H.S. obtained funding for the research. Z.C. and X.N. designed the research. Z.C. and X.N. conducted the research, including data curation, formal analysis, methodology design and implementation, result analysis and visualization. Z.C. and X.N. drafted the original paper. O.R.A. and J.R.F. provided comments on case studies. H.S. provided comments on the original paper. Z.C. and X.N. conducted the paper editing and revision. All authors reviewed the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Z., Ayinde, O.R., Fuchs, J.R. et al. G2Retro as a two-step graph generative models for retrosynthesis prediction. Commun Chem 6, 102 (2023). https://doi.org/10.1038/s42004-023-00897-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-023-00897-3
This article is cited by
-
Retrosynthesis prediction using an end-to-end graph generative architecture for molecular graph editing
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
