
Abstract
Details on the stereo-dynamic topology of chemi-ionizations highlight the role of the centrifugal barrier of colliding reactants: it acts as a selector of the orbital quantum number effective for reaction in a state-to-state treatment. Here, an accurate internally consistent formulation of the Optical interaction potentials, obtained by the combined analysis of scattering and spectroscopic experimental findings, casts light on structure, energy and angular momentum couplings of the precursor (pre-reactive) state controlling the stereo-dynamics of prototypical chemi-ionization reactions. The closest approach (turning point) of reagents, is found to control the relative weight of two different reaction mechanisms: (i) A direct mechanism stimulated by exchange chemical forces mainly acting at short separation distances and high collision energy; (ii) An indirect mechanism, caused by the combination of weak chemical and physical forces dominant at larger distances, mainly probed at low collision energy, that can be triggered by a virtual photon exchange between reagents.
Similar content being viewed by others
Introduction
The aim of the present work is to characterize basic details on the topology of the stereo-dynamics controlling chemi-ionization reactions, an important class of bimolecular processes, triggered by electronically excited species, occurring in flames, plasmas, planetary atmospheres, and interstellar medium.
Since long time the characterization of the stereo-dynamics of elementary chemical-physical processes represents a hot topic of research (see for instance refs. 1,2,3,4,5,6,7,8,9,10,11,12,13 and references therein). Presently, this target can be pursued by investigating in detail, under controlled conditions, some prototypical cases as those presented in this study. In particular, the focus is on barrier-less chemi-ionization reactions in order to provide unique-direct information on basic quantities determining the topology of their stereo-dynamics. The precursor state (PS), formed by collisions of reagents, plays here an important role being coincident with the reaction transition state. All the features of such a state, as well as its structure and stability, are directly or indirectly controlled by intermolecular forces operative in each collision complex.
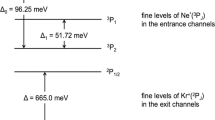
Chemi-ionization reactions (CHEMI) are bimolecular processes involving as reagents a highly energetic X* species, electronically excited in a metastable state, interacting with another partner atomic or molecular M (see for instance refs. 7,8,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30 and references therein). Therefore, CHEMI can be so schematized:
Where [X···M]+ is an intermediate ionic collision complex formed by a spontaneous autoionization of the [X···M]* neutral excited PS which is the reaction transition state (TS) of such processes. Recently, it has been shown that the [X···M]* TS can evolve towards the formation of the final ions via two microscopic reaction mechanisms23,24,25: (i) An exchange mechanism (oxidative-chemical), hereinafter referred to as direct mechanism (DM). It is triggered by strong chemical forces that act mainly at short separation distances, inducing an electron transfer between the reactants through a prototype oxidation process; (ii) A radiative (optical-physical) mechanism, henceforth referred to in the text as an indirect mechanism (IM). It is caused by the combination of weak chemical and physical forces dominant at intermediate/larger distances and which provokes an electron ejection via a pure photoionization event determined by the exchange of a virtual photon between the reacting partners29. The occurrence of the two mechanisms has been suggested several years ago28,29 (see also ref. 30 and references therein) but only recently the nature and selectivity of intermolecular forces involved have been completely addressed23,24,25.
It is relevant to note that because of an external electronic cloud polarization, the PS [X···M]* formed at short separation distances tends to assume the configuration of an internal ionic core surrounded by a Rydberg electron. Therefore, DM can be considered as a particular case of ion chemistry, triggered by non-resonant charge transfer coupling entrance and exit channels25.
Usually, X* can be formed by the collision of X, in its ground electronic state, with electrons or cosmic rays, and it exhibits a lifetime sufficiently long to permit, in a bulk, several collisions with M, until it disappears for inelastic and/or reactive collisions. CHEMI involves X* and M as neutral reagents and the associated XM+, the parent M+ (with its fragmentation species) plus electrons as ionic products. They are barrier-less processes with stereo-dynamics controlled by a PS, in this case, coincident with the reaction TS. Therefore, they represent prototypes of elementary processes triggered by pronounced electronic rearrangements occurring in PS.
CHEMI are of great interest for fundamental and applied research7,8,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30 since they participate to the balance of elementary processes in several environments, where X* easily forms. They happen both at very low-temperature T (under conditions of the cold chemistry)18,21, as in interstellar medium, at intermediate T values, as in planetary atmospheres25, and at high T, as in flames and plasmas (see refs. 24,25 and references therein). CHEMI has been the target of several experimental and theoretical investigations13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32. Most of the experimental studies, carried out in the gas phase with the molecular beam (MB) technique, resolved the reactivity dependence on the stereo-dynamics of single collision events.
As mentioned above, rationalizing a huge number of experimental data from various laboratories on CHEMI promoted by Ne*(3P2,0) metastable atoms, we were able to generalize23,24,25 that, CHEMI occur via the DM, triggered by strong chemical forces, and/or via IM, controlled by the balance of weak intermolecular forces, having both chemical and physical nature. As stressed above, DM confines CHEMI in the framework of elementary exothermic oxidation reactions, while IM, usually stimulated by a virtual photon exchange between reagents, makes the reaction like an elementary photo-ionization process24,25,30. The relative role of DM/IM is found to be directly dependent on the separation distance R mainly probed by reagents in the entrance channels24,25.
The present study exploits an accurate formulation of the intermolecular interaction potentials, previously obtained23,24,25,33 and suggested by the complex phenomenology of open-shell P atoms, investigated by advanced experimental and theoretical methods, by the behaviour of ion-neutral systems, coupled by charge transfer, and by the spectroscopic properties of excimers. The obtained formulation has been tested on experimental findings of CHEMI, investigated in our and other laboratories by coupling scattering and spectroscopic techniques7,8,23,24,25,34,35. In order to emphasize innovative aspects of the reaction stereo-dynamics, we have found it convenient to refer to particular geometries of PS (or pre-reactive state), that open specific reaction channels. However, in the analysis and tests on the experimental findings, the full space of the relative geometries of reagents has been considered. In addition, in the case of NH3 CHEMI the two considered geometries are the most relevant ones promoting, within selected angular cones, the formation of NH3+ ionic product in the ground (X) and in the first excited (A) electronic states7,8,35. The acceptance of angular cones is discussed in detail in ref. 8. Therefore, the selection of individual reaction channels, triggered by specific cuts of the multi-dimensional interaction potential has been useful to properly address the following aspects of general interest:
-
How structure and energetic of PS (or TS), formed via collision of reagents, depending on the critical balance between the anisotropic interaction potential V, the collision energy, Ecoll, and the orbital angular momentum quantum number, ℓ, accompanying any scattering event. In a classical picture ℓ corresponds to the impact parameter b.
-
The role of the closest approach distance Rc (turning point), associated to each collision event, that limits the range of intermolecular distances R affecting the process7,8,30. Around Rc the valence orbital overlap is highest, the system inverts its relative motion, the radial velocity tends to zero and here the system spends most part of its collision time. Therefore, for each ℓ/b value, CHEMI has the highest probability to occur in the neighbourhood of Rc.
-
The change of the relative role of DM and IM that provides basic details on the dependence of the microscopic evolution on Ecoll and ℓ/b.
-
The limitations of theoretical models, exclusively based on the capture by long-range attractions, that are omitting the selectivity of phenomena triggered by forces emerging at shorter distances.
-
The criteria to be adopted to properly assess the orientation degree of polar reagents in the electric field gradient associated to the anisotropic intermolecular forces, controlling the collision dynamics.
Thanks to the above, to the best of our knowledge, we are able to perform the first original attempt to point out how the chemical reactivity depends on the angular momentum quantum number of the intermediate collision complex (precursor or pre-reactive state) leading to the reaction. In particular, we are able to clarify the way in which the centrifugal barrier of the colliding reagents strongly acts as a sort of selector of the orbital quantum number effective for the reaction. In our knowledge, this is the first attempt to depict the topology of the stereo-dynamics of a chemical reaction by a state-to-state treatment.
The results of present study, casting light on basic details of the topology of CHEMI stereo-dynamics, appear of great interest for the present as for many other processes, where they are operative but are usually obscured by other effects. In several cases such details are difficult to obtain directly, being mixed with other effects, as the averaging role by many body and multiple collision events, the masking contributions of the solvent, present in several reaction environments. Furthermore, obtained results suggest some limitations of the so-called capture models, usually exploited to describe many other barrier-less processes, like ion–molecule reactions, occurring in interstellar medium, planetary atmospheres, and plasmas.
Moreover, the different structure of the most stable PS respect to TS opposing the lowest energy barrier to reagents-products passage, makes the rationalization more difficult. Note that any speculation on CHEMI stereo-dynamics presented in this paper exploits a semi-classical treatment of the collision dynamics25,30 whose general aspects are given in the Supplementary Methods section of SI. Such treatment is quantitative for collision events occurring with relative kinetic energy confined in the thermal and hyper-thermal range of values and provides results that, at a semi-quantitative level, are proper to emphasize important selectivity of the scattering also under sub-thermal conditions. The treatment becomes less accurate when resonances, due to quantum effects, are observed, that effectively manifest when light reagents collide at low kinetic energy. This is confirmed by the observation of resonances in He*(3S1) + NH3 CHEMI20,36 due to orbiting effects, emphasized by the reduced mass of the system, that indeed disappear in the Ne*(3P) + NH3 CHEMI37.
In summary, in the next sections the internally consistent formulation of the multidimensional Optical potential, that triggers CHEMI for three different systems, is used to cast light on fundamental details of their reaction stereo-dynamics, as those determining different value and energy dependence of total ionization cross sections. Moreover, the use of specific cuts of the potential, controlling the formation of the precursor (pre-reactive) state with particular energy and structure, is basic to emphasize the different role of two elementary reaction mechanisms with its dependence on collision energy, spin-orbit level of Ne*(3P2,0) reagent, and orbital angular momentum (impact parameter) accompanying any two-body collision event. Such a difference, basic for the stereo-dynamics topology, casts light also on some limitations of the traditional capture models (for the reactivity at low temperature), that exclusively exploit the selectivity of the long-range forces combined with the effect of the centrifugal barrier.
Results and discussion
Observables and selectivity of the collision dynamics
Penning ionization electron spectra (PIES), total (σ), and partial (σp) ionization cross sections, measured as a function of Ecoll, and branching ratios (BRs) are available for a variety of CHEMI, investigated with MB technique. It has been demonstrated that PIES represents a sort of TS spectroscopy31,32, while σ and σp, with their dependence on Ecoll, more directly probe the features of the interaction potential affecting the dynamics in the entrance channels.
As nuclear processes, also CHEMI are assumed to be driven by an Optical potential30, whose real part V controls the dynamical evolution of the reagents during each collision event, while the imaginary component Γ defines the reactivity, that is the probability of passage from neutral entrance to ionic exit channels for each configuration of the collision complex. Recently, the Optical potentials were formulated, in an internally consistent way and in analytical form, for Ne*(3PJ) + Ar25, Ne*(3PJ) + N234, and Ne*(3P) + NH37,35. Their formulation, including angular and radial dependences for both real and imaginary components, permits to control the stereo-dynamics of such prototype CHEMI at increasing complexity of the M reagent, passing from an atom to a polyatomic molecule. More specifically, the proposed Optical potentials, reproducing most of experimental findings, obtained in our and in other laboratories, are here exploited to cast light on the basic points of general interest for the reaction dynamics stressed above.
For Ne*(3PJ) + Ar, Ne*(3PJ) + N2 the adopted treatment25,34 resolves the dependence of the reactivity on the spin-orbit levels J of the reagent Ne* that correlate with different symmetries of PS. For Ne*(3P) + NH335 the lack of experimental information suggests to consider only the average effect on J. However, in the latter case the measure at a defined Ecoll of the PIES38, with the shift of the main peaks position respect to that associated to pure photo-ionization spectra, provides a direct test of PS stability. Specifically, the analysis of PIES led also to separate reaction channels leading to NH3+ in its ground X and in the first excited electronic state A, with the subsequent fragmentation in NH2+ + H.
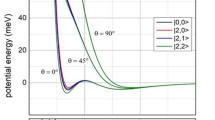
Figure 1 reports σ, calculated with the proposed Optical potentials, as a function of Ecoll which extends from sub-thermal up to hyper-thermal conditions: The real part of interaction potentials used for cross-section σ calculations are shown in Fig. 2.

They are calculated as a function of the collision energy using the Optical potentials formulated in refs. 7,8,25,34,35. For Ar and N2 the different reactivity of Ne*(3P2) reagent (dashed lines) with respect to that of Ne*(3P0) (full lines), with its dependence on the collision energy, is also resolved, which appears to be consistent with the experimental findings40. For NH3 the dependence of the reactivity on the precursor state geometry, opening the formation of NH3+ in its ground (full line) and in its first excited electronic state (dashed line), is shown. θ and φ are the polar angles as defined in the inset of the Figure.

Dependence on the intermolecular distance R of the total potential energy\(\,{V}_{t}\,\)given as sum of the real component V and of the centrifugal contribution VC, defined as \(\,{V}_{C}=\frac{{\hslash }^{2}}{2\mu }\cdot \frac{\ell (\ell +1)}{{R}^{2}}\), where μ is the reduced mass of each system. θ and ϕ are the polar orientation angles of the molecule, as defined in Fig. 1 for NH37,8,34,35. The Ecoll value has been fixed to 1.0 meV and represented by a horizontal dashed line. For both Ne*-Ar (a) and Ne*-N2 (b) the V component is that asymptotically correlating with Ne(3P0) fine level of the reagent25,34, while for Ne*-NH3 (c, d), the plotted V components account for the formation of the precursor state in two basic geometries, opening different CHEMI channels35.
In the case of ammonia, calculated σ has been separated in partial σp components, promoted by the most representative configurations of the PS opening the transition to the ionic product in the two different electronic states7,34. Actually, as detailed in ref. 8, the most quantitative treatment of CHEMI involving polar hydrogenated molecules, as ammonia, should include the average on the acceptance angular cone of each reaction channel. The adopted simplified treatment is useful to better emphasize the change of stereo-dynamics operative along the two different reaction channels.
For the three CHEMI, plotted results, with their different absolute value and dependence on Ecoll, are consistent with experimental results of Perugia8,39, Lausanne37, Eindhoven40, and Pittsburgh41 experiments. In particular, for the comparison with Perugia experiments see Fig. 3 of ref. 8 and Fig. 4 of ref. 34, with Eindhoven experiments, see again Fig. 4 in ref. 34, with Pittsburgh experiments, see Fig. 4 in ref. 25, and, finally, with Lausanne experiments see comments to Fig. 5 in ref. 35. Obtained results suggest that the stereo-dynamics in the three cases must be rather different, with a change also within the two channels effective for ammonia. Pronounced differences are exhibited also by CHEMI of Ar and N2, although in the entrance channels the long-range attraction is practically the same39. Such differences arise from the change of the Optical potential at intermediate and short separation distances and, consequently, of structure and reactivity of TS.
Cross-section values, reported in Fig. 1, also emphasize the higher reactivity, predicted for Ne*(3P0) in collision with N2 and Ar with respect to that of Ne*(3P2), emerging at high collision energies and confirmed by the Eindhoven experiments40. Here, we exploit specific cuts extracted from the multidimensional formulation of V and Γ components, to investigate new aspects of the stereo-dynamics topology more directly.
Figure 2 provides a comparison, on quantitative ground, of the interaction controlling the approach of reagents of investigated CHEMI. Selected V components, that refer to specific cuts of the multidimensional potential energy surfaces, are useful to especially show the dependence on the intermolecular distance R of the total potential \({V}_{t}\), defined as \({V}_{t}=V+{V}_{c}\), where the centrifugal potential \({{V}}_{c}\)is calculated using quantized ℓ values. Plotted results emphasize the capture efficiency of the reagents approaching at fixed Ecoll in the selected channels. From Fig. 2 it emerges that the centrifugal barrier strongly limits the value of the orbital angular momentum of the collision complex, defined by ℓ, leading to the reaction. At the low Ecoll considered in such Figure, the centrifugal barrier becomes a sort of selector of orbital angular momentum effective for the reaction, permitting or hindering an approach of reagents sufficiently close to be effective for reactive events. This is clearer in the case of ammonia, where all reactive events, leading to the formation of NH3+(X), are controlled by low values of ℓ that determine distances Rc very similar and confined at low values (Rc~2.4 Å). If ℓ gets over an upper limit, Rc jumps at much higher values (Rc > 10 Å) where the reaction probability vanishes, being the reagents too distant, and the intermolecular forces null. In addition, the Rc values (Rc > 4 Å) obtained for Ar and N2 confirm the selective role of the centrifugal barrier and suggest that the chemical interaction components at such R values play a marginal role, even at low ℓ values. For ammonia, the stereo-dynamics in the channel leading to the formation of NH3+(A) shows a behavior intermediate between the cases analyzed above.
The semi-classical relation, \(b\simeq \frac{\ell }{k}\), where k is the wave number associated to the collision event, allows to better characterize important details concerning the dependence of Rc on b or on ℓ. Such dependence arises from a critical balance between \({{V}}_{t}=V+{V}_{c}\) and Ecoll.
Figure 3 quantifies the effects of the centrifugal component of the interaction, highlighted in Fig. 2, on the reaction dynamics of the investigated systems.

The turning point (Rc) dependence on the impact parameter b, or, in quantum picture on \(\ell \), evaluated for the four systems (Ne*+NH3 producing NH3+(A) electronic excited ions (a), Ne*+NH3 producing NH3+(X) electronic ground state ions (b), Ne*+Ar (c) and Ne*+N2 (d) at five Ecoll values (see text). The ket indicates the selected atomic state of Ne*(3P0) reagent.
Specifically, Fig. 3 plots the Rc dependence on the impact parameter b, evaluated, for the four cuts of V, at five Ecoll values that cover 4 orders of magnitude. At the lowest collision energies, plotted data confirm in all cases the selective role of the centrifugal barrier, which generates Rc values confined in well-separated ranges, where the reaction probability is completely different. At the highest collision energies, a unique extended range of Rc values becomes effective.
Figure 3 shows that CHEMI of ammonia, leading to the formation of the molecular ion in the ground electronic state NH3+(X), is exclusively controlled by short-range forces for \({{E}}_{{coll}}\)≤ 100 meV. In particular, the reaction effectively occurs when b assumes small and intermediate values and Rc is confined at short range. Therefore, while the collision dynamics of reagents is fully controlled by the market capture from the strong attraction, determined by the combination of attractive electrostatic, induction, and dispersion components, the reactivity fully depends on the strength of chemical forces operative at short distances. An opposite behavior is exhibited by CHEMI of Ar and N2 at very low \({E}_{{coll}}\), since their dynamics and reactivity are fully affected by the balance of weak forces operative at intermediate and long range, as further detailed in the next section. An intermediate behavior is confirmed for the reaction channel of ammonia leading to the NH3+(A) formation.
Further selectivity
Additional details on the topology of the reaction stereo-dynamics, are suggested by the Fig. 4, where, in the upper panel, are plotted the Γ components, promoting DM and IM mechanisms, referred to the reaction channels investigated in this study (see Figs. 2 and 3). Such channels lead to the formation of product ions in the ground electronic state that for Ar+(2P3/2), an open shell atomic ion, is defined by Ω = 1/2 (Ω is the quantum number representing the absolute projection of J vector along R).

a Comparison between the different dependence on intermolecular distance R of Γ components determining direct mechanism (DM) and indirect mechanism (IM) in the selected channels of CHEMI. The overall ionization width for each investigated system is approximately given by the sum of the two correlated Γ-functions plotted35. b Branching ratios, defined as ΓDM/(ΓDM + ΓIM) and, ΓIM/(ΓDM + ΓIM) are here plotted as a function of R. c The behavior of the Cx marker tracing coefficient for Ne*(3P2,0) + N2 CHEMI involving the precursor state in a collinear configuration. d A schematic cartoon illustrates the DM and IM mechanisms for the same reaction. DM, triggered by strong chemical forces, occurs with a direct electron transfer between valence orbital of reagents. IM, stimulated by anisotropic weak interactions as polarization, spin-orbit, and Coriolis contributions, involves a changing of the atomic/molecular alignment and a mixing of atomic levels and molecular states of different symmetry23,29,41,47. IM can promote, in addition to a stimulated electron transfer, also a virtual photon exchange between reagents giving to a photo-ionization.
The relative role of DM and IM mechanisms, defined as branching ratio (BR), is reported as a function of R in the intermediate part of the same Figure. Reported BR quantifies the relative role of DM and IM in the vicinity of Rc values that represent, as seen before, the distances most effective for CHEMI.
The Optical potentials formulation provides also the \({C}_{x}\) coefficient, which represents an important marker-tracing of the electronic evolution of the reagents as a function of their separation distance24,25,34. In particular, \({{C}}_{x}\)quantifies the Σ molecular character degree assumed by PS formed via the collision of reagents. Note that a pure Σ state is a true molecular state defined by the quantum number Λ = 0 (see also below). Therefore, the \({{C}}_{x}\) value indicates if PS is confined in the structure of two separated partners (quantified by the asymptotic 1/3 and 2/3 values), in that of a weakly interacting adduct (where \({{C}}_{x}\) starts to vary) or in that of a true molecule with a defined electronic symmetry (where \({{C}}_{x}\)assumes 1 or 0 as constant value), or in the transition between limit structures (where \({{C}}_{x}\) strongly varies). Such a detailed analysis has been carried out only for CHEMI of Ar and N2, for which the different reactivity of Ne* reagent in 3P0 and 3P2 fine levels has been resolved (see Fig. 1). The\(\,{C}_{x}\) coefficient, plotted in the lower panel of Fig. 4, relates to the collinear configuration of Ne* + N2 reaction: it exhibits a behavior like that operative in Ne* + Ar. The two different lines refer, respectively, to the |1/2,1/2> state of the internal ionic state of the adduct, that asymptotically correlates with Ne*(3P0) reagent, and to the |3/2,1/2> internal ionic state of the same adduct that asymptotically correlates with Ne*(3P2) reagent. The two complementary behaviors suggest that, while PS formed by the Ne*(3P0) reagent at short range becomes a linear tri-atomic molecule with a Σ symmetry \(({C}_{x}=1),\)the one formed by Ne*(3P2) becomes a molecule with a Π symmetry \((\Lambda =1,{{C}}_{x}=0).\)This means that in the two different symmetries the half-filled orbital of the internal ionic core Ne+ is aligned parallel and perpendicular to the separation distance, and this causes a reactivity more efficient in the first case when chemical forces, whose strength strongly varies with R (depending on the overlap integral of valence orbital), come into play. Also note that at intermediate and large separation distance the two symmetries are mixed by the spin-orbit coupling, associated to the open shell nature of interacting partners, leading to the formation of atomic J fine levels.
Moreover, the bottom of Fig. 4 provides a cartoon of DM and IM promoting CHEMI of N2, whose relative importance strongly varies with R. In particular, DM, triggered by chemical forces operative at short R, requires a proper symmetry of the reagents molecular orbital in order to favor the direct electron transfer. On the other hand, IM, stimulated by weak intermolecular forces of chemical and physical nature, dominates at larger R, where a mixing of atomic/molecular states of different symmetry, promoted by polarization, spin-orbit, and Coriolis couplings (the latter related to centrifugal effects accompanying scattering events) manifests. It has been demonstrated24,25,34 that effective Γ components are just a combination of \({\varGamma }_{{DM}}\) and \({\varGamma }_{{IM}}\)weighted on the \({{C}}_{x}\)marker-tracing coefficient plotted in the same Figure.
Additional information is suggested in Fig. 5, especially if analyzed together with the results in Figs. 2–4. In particular, the relative contribution of two reaction mechanisms can be obtained by combining the results of the turning points in Fig. 5 with the radial dependence DM and IM given in Fig. 4. Moreover, the strong attraction in the entrance channel and the centrifugal barrier, that acts as a selector of b or values effective for the reaction (see Fig. 2), confirm that CHEMI of NH3, leading to the formation of NH3+ in the ground electronic state, is controlled exclusively by DM for Ecoll from 0.1 meV up to a value of 100 meV (see also Fig. 2, for Ecoll = 1 meV). For higher Ecoll, IM plays some role only at large \({{{\mathcal{l}}}}\)or \(b\) values. Instead, for the same reaction, the channel leading to the formation of NH3+ in the first excited electronic state is dominated by DM only for Ecoll up to few meV. For Ecoll ≥ 10 meV the reaction is determined by the balance of DM and IM and the DM/IM relative role increases with Ecoll. Therefore, IM plays some role only in a limited window of intermediate Ecoll values. All these details are suggested by combining results in Figs. 3–5.

Comparison between Rc (a), the reaction probability P(b) (b), and the ionization cross section contribution πb2P(b) (c) evaluated as a function of ℓ/b, at fixed \({E}_{{coll}}\,\)= 1.0 meV. Note that for such \({E}_{{coll}}\) value the semiclassical relation \(b\simeq \frac{\ell }{k}\) provides b(Å) \(\simeq \frac{\ell }{2.097}\) for ammonia, b(Å) \(\simeq \frac{\ell }{2.526}\) for Ar and b(Å) \(\simeq \frac{\ell }{2.362}\) for N2. For the reaction of ammonia, only the channel leading to the formation of NH3+ in the ground state X is considered.
CHEMI of Ar behaves in an opposite way, that is the IM dominates at low collision energy, because of a less pronounced role of attraction components (see Fig. 1), here mostly controlled by dispersion forces. From Figs. 3 and 5 it appears that at Ecoll < 10 meV the reaction exclusively occurs via IM, since the trapping effect of weak intermolecular forces is overcome at short range by the repulsion of size and of the centrifugal barrier. Consequently, the reagents remain always confined at large R. The transition to DM gradually occurs with the increase of Ecoll that permits a smooth passage to probing shorter Rc. This passage becomes evident for Ecoll ≥ 100 meV.
In the Ne*(3P0) + N2 reaction the approach of reagents is affected by two potential wells of limited depth (4–5 meV, see Fig. 2): the first one manifests at about R = 5.5 Å, being mostly determined by the dispersion attraction; the second one occurs at about R = 3.3 Å and arises from the combined effect of the floppy electronic cloud polarization of external atomic orbital 3 s and of the interaction between the disclosed atomic internal ionic core and the permanent electric quadrupole of the anisotropic N2 molecule. IM exclusively drives the reaction for Ecoll comparable/lower than 1 meV: because of the centrifugal barrier, the system probes exclusively the potential well at larger separation distance where IM dominates (see Figs. 3 and 5). DM promptly emerges at collision energy larger than 1 meV and becomes dominant already at Ecoll = 10 meV: here, the access to the second potential well, located at shorter R, is allowed (see Figs. 2 and 3). As for the other CHEMI, the role of IM becomes less relevant further increasing Ecoll.
Finally, the analytical formulation of the anisotropic V casts lights also on the natural molecular orientation (polarization) in the intermolecular electric field gradient with its effect on the reaction stereo-dynamics. The possibility of molecular polarization, during collisions driven by anisotropic intermolecular potentials, is usually evaluated exploiting the ratio between the average molecular rotation time, \({\tau }_{{rot}}\), and the collision time, \({\tau }_{{coll}}\)(see for instance ref. 37). If \({\tau }_{{rot}}\)«\({\tau }_{{coll}}\) the colliding system has time sufficient to assume the most stable configurations, while, when \({\tau }_{{rot}} > > {\tau }_{{coll}}\), the molecule is seen under sudden conditions by the other colliding partner.
In general, \({\tau }_{{coll}}\) should be evaluated as
where
defines the radial velocity, \({R}_{{in}}\)represents an initial reference distance and \({R}_{c}\) is the closest approach distance (turning point). In the case of collisions driven by high anisotropic interactions, \({v}_{r}\)can undergo the levelling effect of \(V\left(R\right)\) associated to the most attractive configurations. Under such conditions, \({R}_{{in}}\) can be taken as the distance where the interaction anisotropy is comparable with \({E}_{{rot}}\) and \({\tau }_{{rot}}\) can be substituted by \({\tau }_{{pend}}\) related to the formed libration-pendular states of the collision complex. Therefore, molecules rotationally relaxed, flying at low velocity and driven by strongly anisotropic interactions, in their collisions they are forced to assume the most stable configurations, undergoing the levelling effect of \(V\left(R\right).\)
This treatment accounts for some findings of CHEMI of ammonia, where the BR between channels leading to NH3+ in its ground X and in first excited electronic state A remains practically constant at a value 0.2-0.3 for Ecoll varying from 0.01up to 10 meV37 and increases only for highest Ecoll, that is when the levelling effect of the V attraction on \({v}_{r}\) vanishes38. Note that in the first case, molecules rotationally relaxed in seeded beams (with an average rotational period of some ps) have been used in low collision experiments37, while in the second case38 higher collision energies are probed with molecules in effusive beams, populating excited rotational levels.
In conclusion, for CHEMI this study emphasizes the important role of PS features, where the distance value of the closest approach Rc, with its dependence on the orbital angular momentum value, is crucial to define the relative role of DM and IM. This target has been achieved by exploiting a full, proper, and internally consistent formulation of the intermolecular interaction in some prototypical systems.
On the general ground, obtained results emphasize that at distances shorter than the centrifugal barrier location usually operate other selectivity, originated by the balance of short- and long-range intermolecular forces of different origin, not taken into account by traditional capture models, traditional Langevin42 and Langevin modified models (see also ref. 37). Some important results, concerning shortcomings of capture calculations by long-range dispersion forces have been recently reported for He*(3S,1S) + Li CHEMI43. The cases investigated in the present study are more complex, since simultaneously involving, as reagents, an open-shell P atom, with a high electron affinity ionic core, and molecular partners, with a permanent electric multipole. Related long-range forces arise from the balance between dispersion and several other anisotropic interaction components.
For CHEMI a key quantity is Rc with its neighborhood, for other processes is Rc, and its comparison which crossing points, between potential energy surfaces having different symmetry and/or spin multiplicity, opening non-adiabatic transition towards specific exit channels44. All these details, combined with the role of natural molecular orientation originated by anisotropic intermolecular forces, are crucial to rationalize hot topics, as Arrhenius and anti-Arrhenius behavior of the chemical reactivity, especially affecting the cold chemistry45.
Finally, a few years ago important stereo-dynamical effects were observed for He*(3P2) + H2 CHEMI under sub-thermal conditions46. Such effects have been ascribed to long- and short-range anisotropic interactions, controlled by the outer-occupied (strongly expanded) 2p orbital of He* and isotropic ground rotational state of para H2 and by the two helicity states of ortho H2 in its ground rotational state. The role of van der Waals and electrostatic effects (due to the quadrupole-quadrupole component) has been properly discussed. Present results cast light on the stereo-dynamics of Ne*(3P2,0) + atom, molecule CHEMI where the internal ionic core 2p5 of Ne* is isoelectronic of F atom with an increased electron affinity, while the outer electron is confined in the symmetric 3s atomic orbital. As a consequence, while the long-range forces are here essentially isotropic, most part of the anisotropy arises at an intermediate and short separation distance, where the PS is formed and the orientation of ionic core with respect to the atomic-molecular partner plays a crucial role.
Methods
Experimental technique
The experimental data discussed in the paper, have been measured by the MB technique in various laboratories but in all cases working in single collision conditions and at high resolution in angle and velocity22,38,40,41 (see also Supplementary References). A scheme of our MB machine available in the Perugia laboratory is given in Supplementary Fig. 1. The main characteristics of the experimental device are mentioned in the Supplementary Methods: further details can be found in a previous paper23 and in Supplementary References.
Cross section calculations
To assess all probed scattering properties, the semiclassical method, applicable until the de Broglie wave length of the system assumes values shorter or comparable to 1 Å and described elsewhere24,25,30, has been used. The ionization cross sections are calculated using the Optical Model (see below) exploiting Supplementary Equations 1–5 discussed in the Supplementary Methods.
Optical potential formulation
The Optical Potential Model is defined in Supplementary Equation 1. Its real part is provided by a generalization of open-shell atom interactions discussed in the Supplementary Methods and expressed analytically by Supplementary Equations 6–9. The adopted potential formulation takes into account of a non-covalent component of the interaction as well as of an anisotropic term that includes also contributions of a chemical origin (see the case of Ne*-NH3 CHEMI of Fig. 2). In the case of the imaginary part of the Optical potential, the formulation has been discussed in previous papers24,25,34,35 and same crucial details are emphasized in the present internally consistent analysis of the three different CHEMI dynamics (see, in particular, Fig. 4).
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information and from the corresponding author upon reasonable request.
References
Bernstein, R. B., Herschbach, D. R. & Levine, R. D. Dynamical aspects of stereochemistry. J. Phys. Chem. 91, 5365–5377 (1987).
Zare, R. N. Angular Momentum: Understanding Spatial Aspects in Chemistry and Physics, (Wiley-Interscience, New York, 1988).
Zare, R. N. Laser control of chemical reactions. Science 279, 1875–1879 (1998).
Wang, F., Lin, J.-S. & Liu, K. Steric control of the reaction of CH stretch–excited CHD3 with chlorine atom. Science 331, 900–903 (2011).
Chang, Y.-P. et al. Specific Chemical reactivities of spatially separated 3-Aminophenol conformers with Cold Ca+ ions. Science 342, 98–101 (2013).
Rösch, D., Willitsch, S., Chang, Y.-P. & Küpper, J. Chemical reactions of conformationally selected 3-aminophenol molecules in a beam with Coulomb-crystallized Ca+ ions. J. Chem. Phys. 140, 124202 (2014).
Falcinelli, S., Bartocci, A., Cavalli, S., Pirani, F. & Vecchiocattivi, F. Stereodynamics in the collisional autoionization of water, ammonia, and hydrogen sulfide with metastable rare gas atoms: competition between intermolecular halogen and hydrogen bonds. Chem. Eur. J. 22, 764–771 (2016).
Falcinelli, S., Rosi, M., Cavalli, S., Pirani, F. & Vecchiocattivi, F. Stereoselectivity in Autoionization reactions of hydrogenated molecules by metastable noble gas atoms: the role of the electronic coupling. Chem. Eur. J. 22, 12518–12526 (2016).
Jiang, B., Yang, M., Xie, D. & Guo, H. Quantum dynamics of polyatomic dissociative chemisorption on transition metal surfaces: mode specificity and bond selectivity. Chem. Soc. Rev. 45, 3621–3640 (2016).
Perreault, W. E., Mukherjee, N. & Zare, R. N. Quantum control of molecular collisions at 1 kelvin. Science 358, 356–359 (2017).
Chadwick, H. & Beck, R. D. Quantum state–resolved studies of chemisorption reactions. Annu. Rev. Phys. Chem. 68, 39–61 (2017).
Kilaj, A. et al. Conformer-specific polar cycloaddition of dibromobutadiene with trapped propene ions. Nat. Com. 12, 6047 (2021).
Falcinelli, S., Vecchiocattivi, F. & Pirani, F. Electronic rearrangements and angular momentum couplings in quantum state-to-state channels of prototype oxidation processes. J. Phys. Chem. A 125, 1461–1467 (2021).
Sugden, T. M. Excited species in flames. Annu. Rev. Phys. Chem. 13, 369–390 (1962).
Nakamura, H. Theoretical considerations on penning ionization processes. J. Chem. Phys. Jpn. 26, 1473–1479 (1969).
Miller, W. H. Theory of penning ionization. I. Atoms. J. Chem. Phys. 52, 3563–3572 (1970).
Niehaus, A. Penning Ionization. Ber. Bunsenges. Phys. Chem. 77, 632–640 (1983).
Morgner, H. & Niehaus, A. Experimental and theoretical study of the Penning ionization of H atoms by He metastables. J. Phys. B: Mol. Phys. 12, 1805–1820 (1979).
Arango, C. A., Shapiro, M. & Brumer, P. Cold atomic collisions: Coherent control of penning and associative ionization. Phys. Rev. Lett. 97, 193202 (2006).
Henson, A. B., Gersten, S., Shagam, Y., Narevicius, J. & Narevicius, E. Observation of resonances in penning ionization reactions at sub-Kelvin temperatures in merged beams. Science 338, 234–238 (2012).
Dulieu, O. and Osterwalder, A. Cold Chemistry. Molecular Scattering and Reactivity Near Absolute Zero (Royal Society of Chemistry, Cambridge, 2018).
Gordon, S. D. S. et al. Quantum-state-controlled channel branching in cold Ne(3P2)+Ar chemi-ionization. Nat. Chem. 10, 1190–1195 (2018).
Falcinelli, S., Vecchiocattivi, F. & Pirani, F. Adiabatic and nonadiabatic effects in the transition states of state-to-state autoionization processes. Phys. Rev. Lett. 121, 163403 (2018).
Falcinelli, S., Vecchiocattivi, F. & Pirani, F. General treatment for stereo-dynamics of state-to-state chemi-ionization reactions. Commun. Chem. 3, 64 (2020).
Falcinelli, S., Farrar, J. M., Vecchiocattivi, F. & Pirani, F. Quantum-state controlled reaction channels in chemiionization processes: radiative (optical - physical) and exchange (oxidative- chemical) mechanisms. Acc. Chem. Res. 53, 2248–2260 (2020).
Paliwal, P. et al. Determining the nature of quantum resonances by probing elastic and reactive scattering in cold collisions. Nat. Chem. 13, 94–98 (2021).
Ploenes, L., Straňák, P., Gao, H., Küpper, J. & Willitsch, S. A novel crossed-molecular beam experiment for investigating reactions of state- and conformationally selected strong-field-seeking molecules. Mol. Phys. 119, e1965234 (2021).
Hotop, H. & Niehaus, A. Reaction of excited atoms and molecules with atoms and molecules. Z. Phys. 228, 68–88 (1969).
Miller, W. H. & Morgner, H. A unified treatment of Penning ionization and excitation transfer. J. Chem. Phys. 67, 4923 (1977).
Siska, P. E. Molecular-beam studies of Penning ionization. Rev. Mod. Phys. 65, 337–412 (1993).
Hotop, H., Lorenzen, J. & Zatrow, A. Penning ionization electron spectrometry with state-selected, thermal-energy neon metastable atoms Ne(3P2), Ne(3P0) + Ar, Kr, Xe, Hg*. J. Electron Spectrosc. Relat. Phenom. 23, 347–365 (1981).
Benz, A. & Morgner, H. Transition state spectroscopy with electrons. Mol. Phys. 57, 319–336 (1986).
Dehmer, P. M. Rydberg states of van der Waals molecules—A comparison with Rydberg states of atoms and chemically bonded species. Comments . Mol. Phys. 13, 205–227 (1983).
Falcinelli, S., Vecchiocattivi, F. & Pirani, F. Selectivity of weak intermolecular forces and precursor state of elementary oxidation reactions, a new insight on Ne* + N2 chemiionization. Sci. Rep. 11, 19105 (2021).
Falcinelli, S., Vecchiocattivi, F., Farrar, J. M. & Pirani, F. Chemi-ionization reactions and basic stereodynamical effects in collisions of atom-molecule reagents. J. Phys. Chem. A 125, 3307–3315 (2021).
Jankunas, J., Jachymski, K., Hapka, M. & Osterwalder, A. Observation of orbiting resonances in He(3S1) + NH3 Penning ionization. J. Chem. Phys. 142, 164305 (2015).
Jankunas, J., Bertsche, B., Jachymski, K., Hapka, M. & Osterwalder, A. Dynamics of gas-phase Ne*+NH3 and Ne*+ND3 Penning ionization at low temperatures. J. Chem. Phys. 140, 244302 (2014).
Falcinelli, S., Bartocci, A., Cavalli, S., Pirani, F. & Vecchiocattivi, F. The stereo-dynamics of collisional autoionization of ammonia by helium and neon metastable excited atoms through molecular beam scattering experiments. J. Chem. Phys. 143, 164306 (2015).
Baudon, J. et al. Optical potential for Ne*(3P2,0) - Ar, N2 interactions. J. Chem. Phys. 95, 1801–1807 (1991).
Van den Berg, F. T. M., Schonenberg, J. H. M. & Beijerinck, H. C. W. Ionization of small molecules by state selected Ne*(3P0, 3P2) metastable atoms in the 0.06<E<6 eV energy range. Chem. Phys. 115, 359–379 (1987).
Gregor, R. W. & Siska, P. E. Differential elastic scattering of Ne*(3s3P2,0) by Ar, Kr, and Xe: Optical potentials and their orbital interpretation. J. Chem. Phys. 74, 1078–1092 (1981).
Langevin, M. P. Une formule fondamentale de théorie cinétique. Ann. Chim. Phys. 5, 245–288 (1905).
Dulitz, K. et al. Suppressing of Penning ionization by orbital angular momentum conservation. Phys. Rev. A 102, 022818 (2020).
Tsikritea, A., Jake, A., Diprose, J. A., Softley, T. P. & Heazlewood, B. R. Capture theory models: An overview of their development, experimental verification, and applications to ion–molecule reactions. J. Chem. Phys. 157, 060901 (2022).
Ascenzi, D. et al. Destruction of dimethyl ether and methyl formate by collisions with He+. Astron. Astrophys. 625, A72 (2019).
Shagam, Y. et al. Molecular hydrogen interacts more strongly when rotationally excited at low temperatures leading to faster reactions. Nat. Chem. 7, 921–926 (2015).
Aquilanti, V., Candori, R. & Pirani, F. Molecular beam studies of weak interactions for open shell atoms: The ground and the lowest excited states of rare gas oxides. J. Chem. Phys. 89, 6157–6164 (1989).
Acknowledgements
This work was supported and financed with the Fondo Ricerca di Base, 2019, dell’Università degli Studi di Perugia (Project Titled: Produzione di metano per reazione di idrogenazione di CO2 con e senza catalisi in fase solida mediante l’uso di energie rinnovabili). Support from Italian MIUR and University of Perugia (Italy) is acknowledged within the program Dipartimenti di Eccellenza 2018-2022.
Author information
Authors and Affiliations
Contributions
S.F., F.V., and F.P. conceived and designed the study. S.F. performed experiments. S.F., F.V., and F.P. developed the proposed model with related calculations and analysed the experimental data. All authors participated in the writing and editing the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Falcinelli, S., Vecchiocattivi, F. & Pirani, F. The topology of the reaction stereo-dynamics in chemi-ionizations. Commun Chem 6, 30 (2023). https://doi.org/10.1038/s42004-023-00830-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-023-00830-8
This article is cited by
-
Intermolecular interactions and the weakly bound precursor states of elementary physicochemical processes
Rendiconti Lincei. Scienze Fisiche e Naturali (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
