
Abstract
Owing to the versatile synthetic utility of its carbon-carbon double bond, low-cost industrial chemical 3,3,3-trifluoropropene (TFP) represents one of the most straightforward and cost-efficient precursors to prepare trifluoromethylated compounds. However, only limited methods for the efficient transformations of TFP have been reported so far. Here, we report a nickel-catalyzed dicarbofunctionalization of TFP. The reaction uses inexpensive NiCl2·6H2O as the catalyst and 4,4’-biMeO-bpy and PCy2Ph as the ligands, allowing the alkyl-arylation of TFP with a variety of tertiary alkyl iodides and arylzinc reagents in high efficiency. This nickel-catalyzed process overcomes the previous challenges by suppressing β-H and β-F eliminations from TFP, rendering this strategy effective for the transformations of TFP into medicinal interest trifluoromethylated compounds.
Similar content being viewed by others
Introduction
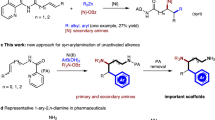
3,3,3-Trifluoropropene (TFP) is an low-cost industrial chemical used for the production of fluorinated polymers1,2,3,4,5 and refrigerants6,7. For practical applications, the functionalizations of the carbon-carbon double bond (C = C) in TFP are straightforward and cost-efficient strategies to prepare diversified trifluoromethylated compounds that are of great interests in pharmaceuticals and agrochemicals, due to the unique properties of the trifluoromethyl (CF3) group8,9,10,11. However, only limited methods for the transformations of TFP have been reported so far. Generally, these methods mainly focus either on the mono-functionalization of TFP, such as hydrofunctionalization12,13,14,15,16, or on the simple functionalization of TFP that produces limited types of products, such as polymerization3,5,17,18 and expoxidation19,20 (Fig. 1a). From the standpoint of synthetic efficiency, the dicarbofunctionalization of TFP would be a promising strategy to prepare trifluoromethylated compounds with multiple substitutes, enabling efficient construction of molecular complexity and structural diversity by forming two new C-C bonds in a one-pot reaction.

a Traditional transformations of TFP. b Previous work, metal-catalyzed transformation of TFP. c This work, Ni-catalyzed dicarbofunctionalization of TFP.
Recently, the nickel-catalyzed three-component dicarbofunctionalization of alkenes has received increasing attention21,22,23,24,25,26. However, the application of this process to the dicarbofunctionalization of TFP remains challenging, due to the undesired β-hydride (β-H)27,28,29 and β-fluoride (β-F)29,30,31 eliminations from the alkyl nickel intermediate (Fig. 1b). One general and effective strategy to suppress the β-H elimination in this process is using chelating group to form a stable nickellacycle intermediate32,33,34,35,36. While, the absence of chelating group on TFP renders it difficult to apply this strategy. It has also been demonstrated that the metal-mediated transformation of TFP is prone to defluorination driven by the formation of a strong metal-fluoride (M-F) bond37,38,39,40,41,42. To date, only a few examples of nickel-catalyzed transformations of TFP derivatives have been reported29,30,31, but they all undergo a defluorinative process that leads to the formation of gem-difluoroalkenes. As such, the development of a new catalytic system to overcome these challenges is imperative.
In our efforts to develop nickel-catalyzed dicarbofunctionalization of alkenes, we have established a nickel-catalyzed tandem radical process for dicarbofunctionalization of enamides through a chelation assisted strategy32. In this catalytic process, a highly active nickel(III) species bearing a chelated substrate was proposed as the key intermediate to suppress the β-hydride elimination. We envisioned the feasibility of nickel-catalyzed dicarbofunctionalization of TFP via a radical pathway in the absence of chelating group. Our design is based on the hypothesis that the nickel(III) species may enable the rate of reductive elimination step faster than those of undesired β-H and β-F eliminations. Following this hypothesis (Fig. 1c)43,44,45, the reaction may be induced by a nickel(I) complex [Nu-NiI(Ln)] (A) that can be derived from the transmetallation of a nucleophile with a nickel(I) species. Complex A reacts with an aliphatic electrophile to generate an alkyl radical (I) and nickel(II) [Nu-NiII(Ln)X] (B) via a single electron transfer (SET) pathway. Subsequently, I undergoes a radical addition with TFP to form a new alkyl radical (II), followed by combination with B to produce the key intermediate [Nu-NiIII(Ln)(X)-alkyl] (C). Finally, reductive elimination of C delivers the dicarbofunctionalized product.
Herein, we report a nickel catalyzed dicarbofunctionalization of TFP. This nickel-catalyzed process allows alkyl-arylation of TFP with a variety of arylzinc reagents and tertiary alkyl iodides, providing a series of trifluoromethyl-substituted secondary alkanes with moderate to good yields. The advantage of this protocol is the rapid construction of complex trifluoromethylated compounds by using inexpensive industrial chemical TFP and low-cost nickel catalyst. The successful suppression of defluorinative side reaction in this approach paves a new way to practical application of TFP, and even more fluorinated olefins, in the synthesis of trifluoromethylated compounds of medicinal interest.
Results and discussion
Optimization of Ni-catalyzed alkyl-arylation of TFP
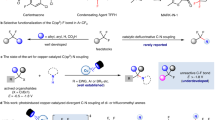
To test our hypothesis, tertiary alkyl iodide 2a and arylzinc reagent 3a were chosen as the model substrates for dicarbofunctionalization of TFP 1 (Table 1, see also Supplementary Tables 1–9). We found that the combination of NiCl2·DME (5 mol%) with bipyridine (bpy, L1) in DMA at 25 °C afforded dicarbofunctionalized product 4a in 60% yield along with small amount of Heck-type side product 6a (11% yield) (entry 1). Notably, no defluorinated side product 7a was observed under these reaction conditions. Further examination of different ligands showed that electron-rich ligand 4,4’-diMeO-bpy (L3) could improve the yield to 65% (entry 3); while, other ligands, such as electron-deficient and bulky bpy-based ligands as well as 1,10-phenanthroline derivatives, led to lower yields with observation of 7a in some cases (entries 2 and 4, see also Supplementary Table 1). Switching NiCl2·DME to monodentate phosphine ligand chelated nickel(II) salts benefited the reaction efficiency (entries 5–8). NiCl2(PPh3)2 provided 4a in 68% yield (entry 5) and a higher yield (75%) was obtained with [trans-NiCl2(PCy2Ph)2] (Ni-2)46 as the catalyst (entry 7). Most importantly, Ni-2 completely suppressed the formation of side products 6a and 7a. Control experiments showed that the absence of L3 only led to gem-difluoroalkene 7a and no product was obtained without nickel catalyst (entries 8–9). These results clearly demonstrate that nickel and ligand are essential in promoting the reaction, and the use of L3 benefits the inhibition of β-F elimination. Encouraged by these results, we found that the combination of inexpensive NiCl2·6H2O (5 mol%) with PCy2Ph (P1) (5 mol%) could afford 4a in 82% isolated yield (entry 12). Every possible permutation of a metal from Ni(NO3)2·6H2O and NiCl2·6H2O with a ligand from PCyPh2 and PCy3 was also applicable to the reaction, but relatively lower yields were obtained (entries 10, 11, 13 and 14). The absence of P1 dramatically decreased the yield (52%) (entry 15). We also tested pyridine-based co-ligands, such as 4-dimethylaminopyridine (DMAP) and 4-trifluoromethylpyridine, but they all provided lower yields (see also Supplementary Table 8). Interestingly, the use of dimethoxyethane (DME) as the cosolvent (DMA/DME = 1:2, v/v) without phosphine ligand benefited the reaction efficiency as well, but a lower yield (73%) was obtained (entry 16, see also Supplementary Table 9). These results demonstrate that the additional monodentate phosphine ligand is critical to the reaction efficiency. This bidentate ligand plus monodentate ligand strategy ([2 + 1] ligand system)45,47,48 provides a good opportunity to fine tune the reaction conditions without need to prepare nickel complex, thus demonstrating the synthetic simplicity of this catalytic system.
Ni-catalyzed alkyl-arylation of TFP
With the optimized reaction conditions in hand, the substrate scope of the reaction was examined. We found that the arylzinc reagent bearing an electron-withdrawing group, such as 4-fluorophenylzinc, provided a low yield (41%). This limitation can be addressed by using (t-Bu2MeP)∙HBF4 (P2) as the alternative ligand, providing corresponding product 4d in 58% isolated yield (Supplementary Table 10). P2 was also applicable to the model reaction, with 75% yield obtained (Supplementary Table 7). Similarly, the use of DME as the cosolvent (DMA/DME = 1/3, v/v) without P2 could also improve the yield (from 41% to 50%), but with lower activity than P2 (Supplementary Table 11). Thereby, both phosphine ligands P1 and P2 were chosen as the co-ligands for the reaction and the representative results were illustrated in Fig. 2.

aReaction conditions (unless otherwise specified): 1 (0.8 mmol, 2.0 equiv), 2 (0.4 mmol, 1.0 equiv), 3 (1.5 equiv in THF), and DMA (2 mL). Isolated yields are given. The phosphine ligands used for the substrates were enclosed in the parentheses. bReaction was conducted at 0 °C. cReaction was conducted without L3 and phosphine ligand, and 5 mol% NiCl2·DME was used. a Substrate scope of arylzinc reagents with 2a. b Substrate scope of alkyl iodides.
Generally, both electron-rich and electron-deficient arylzinc reagents were suitable substrates, providing the corresponding products 4 with high efficiency (Fig. 2a, Supplementary Fig. 2). Specifically, P1 was more suitable for electron-neutral substrates, and P2 was more suitable for the substrates bearing electron-withdrawing or electron-donating groups. However, this feature was not absolute, in some cases, P1 provided higher yields for electron-rich and electron-deficient arylzinc reagents (4f, 4g, 4m, 4n). The reaction exhibited good functional groups tolerance. Various important functional moieties, such as ester (4h, 4i), morpholine (4j), thioether (4l), 1,3-dioxolane (4m), and even aryl chloride (4n), were all compatible with the reaction conditions. Heteroarylzinc reagents including dibenzothiophene (4q) and indole (4r) derived substrates were also applicable to the reaction, demonstrating the generality of this approach.
In addition to arylzinc reagents, a series of tertiary alkyl iodides were also tested (Fig. 2b, Supplementary Fig. 1). tert-Butyl iodide (5a) and the substrate bearing a long carbon-chain, such as n-octyl (5b), underwent the reaction smoothly. Even cyclopropyl-containing substrate could also produce corresponding product 5c. Unfortunately, 5c was unstable upon purification with silica gel chromatography, and a ring-opening product 5c’ was obtained with dichloromethane as the eluent. The aliphatic chain bearing benzoyl (5d), cyano (5e), aryl chloride (5f), thiophene (5g), and alkyl bromide (5h) moieties showed good tolerance to the reaction. Remarkably, the pharmaceutical derived substrates, such as indomethacin- and gemfibrozil-containing tertiary alkyl iodides were competent coupling partners, providing 5i and 5j in synthetically useful yields. The alkylzinc reagent was also tested, leading to 5l in 26% yield. However, tertiary acyclic bromides and secondary alkyl halides, including benzylic iodides and bromides, were not applicable to the reaction.
To demonstrate the synthetic utility of this approach further, pyrroloindoline derivative 5k was prepared. As shown in Fig. 3a, treatment of TFP with tertiary benzyl bromide 2k and arylzinc reagent 3g afforded 5k in 51% yield. Since pyrroloindoline derivatives show significant bioactivities as a cholinesterase inhibitor17 and CF3 has important applications in medicinal chemistry3, the present nickel-catalyzed process may offer good potential for discovery of new bioactive compounds. The reaction can also be scaled up, as demonstrated by the gram-scale synthesis of 4a with 2.5 mol% catalyst loading, which provided a comparable yield (50% isolated yield) with the small-scale reaction (55% yield, determined by 19F NMR) (Fig. 3b). Although a lower yield was obtained, this approach trades off the overall yield loss from the multi-step synthesis of 4a. Deprotection of 4a provided free amine 8 in good yield, which can serve as a versatile building block for various transformations (Fig. 3c). For instance, condensation of 8 with a series of carboxylic acids, including azetidine- (9a) and cyclopropane-containing substrates (9b) and adapalene (9c) used for the treatment of acne vulgaris, provided the corresponding products 10a–10c in high efficiency. Because of its synthetic simplicity that can rapidly construct complex trifluoromethylated compounds by forging two new C-C bonds in one-pot reaction, this approach provides a facile route for application of TFP in medicinal chemistry.

a Synthesis of pyrroloindoline derivative. b Gram-scale synthesis of 4a. c Transformations of compound 4a.
To gain mechanistic insight into the reaction, several experiments were conducted (Fig. 4). Radical inhibition experiments with 1,4-dinitrobenzene and TEMPO as the radical inhibitors49,50 indicated that a radical was involved in the reaction (Fig. 4a). A radical trapping product 11 by TEMPO in the reaction, observed by LC-MS (Supplementary Fig. 3), further confirmed that an alkyl radical was involved in the reaction. We also prepared arylnickel(II) complex [p-tBu-C6H4-NiII(L2)Br] (B1) by reaction of Ni(COD)2 with 1-chloro-4-(1,1-dimethylethyl)benzene to identify the role of phosphine ligand and the active arylnickel species in the reaction (Fig. 4b)51. We used L2 instead of L3 as the supporting ligand because of difficulty in accessing [p-tBu-C6H4-NiII(L3)Br] (B1’). Given that nickel(II) can be reduced into nickel(I) by zinc52,53 several control experiments by using zinc to reduce B1 were also conducted (Fig. 4c). We found that the absence of phosphine ligand with or without reductant zinc afforded higher yields of 4c (57% without Zn; 59% with Zn) than those with P2 (32% without Zn; 19% with Zn) (Fig. 4c). Particularly, using zinc without P2 provided 4c in a yield three times higher than that with P2. These results are in sharp contrast to the catalytic reactions. We surmised that the phosphine ligand is soft and more susceptible to leaving the hard nickel center, compared with the hard bpy-based ligands, which may benefit the formation of an active precatalyst or protect the active nickel species from decomposition54. Here the phosphine ligand is likely to play a similar coordination role as DME, even with higher activity. The exact role of the phosphine ligand still remains elusive at this stage. Comparable yields of 4c were obtained in the absence of P2 with or without zinc (59% with Zn; 57% without Zn), indicating that the arylnickel(II) species is likely involved in the reaction. The involvement of this nickel species in the catalytic cycle are also supported by several facts: the catalytic reaction using L2 as the ligand provided 4c efficiently (Fig. 4d), and complex B1 could serve as a good catalyst for the reaction of 1 with 2a and 3a under standard reaction conditions, in which 3–7% yield of 4c that originated from B1 was observed (Fig. 4e). Since all the control experiments illustrated in Fig. 4c provided biaryl side product 4c’, we surmised that a diarylnickel(II) complex B2 and a NiCl2 were formed between two arylnickel(II) species, which underwent reductive elimination to produce the biaryl product and a Ni(0) (Fig. 5a)55. Comproportionation of Ni(0) with NiCl2 would generate a NiICl species56. The resulting active nickel(I) species underwent SET reduction of tertiary alkyl iodide 2a to afford alkyl radical I-1, which was subsequently trapped by TFP to produce a new alkyl radical II-1. Combination of arylnickle(II) complex B1 with the newly formed alkyl radical II-1 would generate a key intermediate aryl(alkyl)nickel(III) species C1. Finally, reductive elimination of Cl provided product 4c and regenerate Ni(I)X.

a Radical inhibition experiments with 1,4-dinitrobenzene and TEMPO. b Synthesis of arylnickel(II) complex B1. c Stoichiometric reactions of B1 with TFP and 2a. d Catalytic reaction using L2 as the ligand. e Standard reaction catalyzed by complex B1.

a Proposed mechanism for the formation of 4c from the reaction of B1 with 1 and 2a. b Proposed mechanism for the reaction.
On the basis of aforementioned results and previous reports23,34, a possible reaction mechanism was proposed (Fig. 5b). The reaction was initiated by the SET reduction of alkyl iodide with Ni(I)X to generate an alkyl radical (I) and Ni(II)X2 species, in which Ni(I)X was generated by the comproportionation of Ni(II)X2 with in-situ generated Ni(0)56. Subsequently, alkyl radical (I) was trapped by TFP to form a new alkyl radical (II). Meanwhile, transmetallation of arylzinc reagent with Ni(II)X2 provided arylnickel(II) complex B. Combination of B with II produced the key intermediate nickel(III) species C, which underwent reductive elimination to afford the dicarbofunctionalized product and regenerate Ni(I)X. However, the possible pathway illustrated in Fig. 1c still cannot be ruled out.
In conclusion, we have developed a nickel-catalyzed dicarbofunctionalization of industrial chemical TFP. The use of [2 + 1] ligand system overcomes the previous challenges by suppressing β-H and β-F eliminations of TFP, allowing alkyl-arylation of TFP with a variety of tertiary alkyl iodides and arylzinc reagents in high efficiency. The reaction exhibits good functional group tolerance, including pharmaceuticals, providing an efficient route for application of TFP in rapid synthesis of complex trifluoromethylated compounds that are of interests in medicinal chemistry. Most importantly, the successful suppression of β-F elimination side reaction paves a new way for diversified transformation of TFP, which may prompt the research on using industry relevant fluoroolefins as the starting materials for the synthesis of valuable fluorinated structures.
Methods
General procedure for the Ni-catalyzed alkylarylation of 3,3,3-trifluoropropene 1 with tertiary alkyl Iodides 2 and arylzinc reagents 3
To a 25 mL of Schlenck tube were added 4,4’-diMeO-2,2’-bpy (6 mol%), NiCl2 ∙ 6H2O (5 mol%) and the monodentate phosphine ligand (PCy2Ph or PtBu2Me ∙ HBF4, 5 mol%). The tube was evacuated and backfilled with argon for 3 times, then tertiary alkyl iodide 2 (0.4 mmol, 1.0 equiv) and TFP solution (1 M in DMA, 0.8 mmol, 2.0 equiv) were added under Ar. The resulting mixture was stirred for 20 min at room temperature, and the corresponding arylzinc reagent 3 (0.6 mmol, 1.5 equiv) was added slowly within a period of 5 min, and the tube was sealed with a Teflon cap. After stirring for 12 h at room temperature, the reaction mixture was quenched with aqueous NH4Cl solution and diluted with EtOAc. The reaction mixture was filtered through a pad of Celite, and the filtrate was extracted with EtOAc and washed with brine. The organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified with silica gel chromatography to give the corresponding products 4 or 5. Isolated yields are based on the average of two runs under identical conditions.
Data availability
References
Goldschmidt, A. The Polymerization of 3,3,3-Trifluoropropene and 2-Methyl-3,3,3-trifluoropropene. J. Am. Chem. Soc. 73, 2940 (1951).
Kostov, G., Ameduri, B. & Brandstadter, S. M. Radical telomerization of 3,3,3-trifluoropropene with diethyl hydrogen phosphonate: Characterization of the first telomeric adducts and assessment of the transfer constants. J. Fluor. Chem. 128, 910–918 (2007).
Kostov, G. K., Améduri, B. & Brandstadter, S. Telomerization of 3,3,3-Trifluoroprop-1-ene and Functionalization of Its Telomers. Collect. Czech. Chem. Commun. 73, 1747–1763 (2008).
Bruno, A. Controlled Radical (Co)polymerization of Fluoromonomers. Macromolecules 43, 10163–10184 (2010).
Soulestin, T., Ladmiral, V., Dos Santos, F. D. & Améduri, B. Vinylidene fluoride- and trifluoroethylene-containing fluorinated electroactive copolymers. How does chemistry impact properties? Prog. Polym. Sci. 72, 16–60 (2017).
Lai, N. A. Thermodynamic properties of HFO-1243zf and their application in study on a refrigeration cycle. Appl. Therm. Eng. 70, 1–6 (2014).
Gil, B. & Kasperski, J. Efficiency evaluation of the ejector cooling cycle using a new generation of HFO/HCFO refrigerant as a R134a replacement. Energies 11, 2136 (2018).
Hagmann, W. K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 51, 4359–4369 (2008).
Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 61, 5822–5880 (2018).
Ogawa, Y., Tokunaga, E., Kobayashi, O., Hirai, K. & Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 23, 101467 (2020).
Inoue, M., Sumii, Y. & Shibata, N. Contribution of organofluorine compounds to pharmaceuticals. ACS Omega 5, 10633–10640 (2020).
Tarrant, P., Dyckes, G. W., Dunmire, R. & Butler, G. B. The preparation of some fluoroalkylmethyldichlorosilanes and their hydrolysis products1. J. Am. Chem. Soc. 79, 6536–6540 (1957).
Ojima, I., Fuchikami, T. & Yatabe, M. The reactions of hydrosilanes with trifluoropropene and pentafluorostyrene catalyzed by ruthenium, rhodium and palladium complexes. J. Organomet. Chem. 260, 335–346 (1984).
Brown, H. C., Chen, G.-M., Jennings, M. P. & Ramachandran, P. V. Markovnikov hydroboration of perfluoroalkylethylenes. Angew. Chem. Int. Ed. 38, 2052–2054 (1999).
Shibahara, F., Nozaki, K. & Hiyama, T. Solvent-free asymmetric olefin hydroformylation catalyzed by highly cross-linked polystyrene-supported (R,S)-BINAPHOS−Rh(I) complex. J. Am. Chem. Soc. 125, 8555–8560 (2003).
Fanfoni, L., Diab, L., Smejkal, T. & Breit, B. Efficient synthesis of new fluorinated building blocks by means of hydroformylation. Chimia 68, 371–377 (2014).
Brown, D. W. & Wall, L. A. The radiation-induced copolymerization of tetrafluoroethylene and 3,3,3-trifluoropropene under pressure. J. Polym. Sci. A‐1 Polym. Chem. 6, 1367–1379 (1968).
Souzy, R., Ameduri, B. & Boutevin, B. Synthesis and (co)polymerization of monofluoro, difluoro, trifluorostyrene and ((trifluorovinyl)oxy)benzene. Prog. Polym. Sci. 29, 75–106 (2004).
Ramachandran, P. V. & Padiya, K. J. Preparative-scale synthesis of 3,3,3-trifluoropropene oxide. J. Fluor. Chem. 128, 1255–1259 (2007).
Katagiri, T. & Uneyama, K. A chemistry of 2,3-epoxy-1,1,1-trifluoropropane. J. Fluor. Chem. 105, 285–293 (2000).
Dhungana, R. K., Kc, S., Basnet, P. & Giri, R. Transition metal-catalyzed dicarbofunctionalization of unactivated olefins. Chem. Rec. 18, 1314–1340 (2018).
Derosa, J., Apolinar, O., Kang, T., Tran, V. T. & Engle, K. M. Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem. Sci. 11, 4287–4296 (2020).
Luo, Y.-C., Xu, C. & Zhang, X. Nickel‐catalyzed dicarbofunctionalization of alkenes. Chin. J. Org. 38, 1371–1394 (2020).
Qi, X. & Diao, T. Nickel-catalyzed dicarbofunctionalization of alkenes. ACS Catal. 10, 8542–8556 (2020).
Kc, S. et al. Ni-catalyzed regioselective alkylarylation of vinylarenes via C(sp(3))-C(sp(3))/C(sp(3))-C(sp(2)) bond formation and mechanistic studies. J. Am. Chem. Soc. 140, 9801–9805 (2018).
Kc, S., Dhungana, R. K., Khanal, N. & Giri, R. Nickel-Catalyzed alpha-Carbonylalkylarylation of Vinylarenes: Expedient Access to gamma,gamma-Diarylcarbonyl and Aryltetralone Derivatives. Angew. Chem. Int. Ed. 59, 8047–8051 (2020).
Li, Y. et al. Oxidative Heck Reaction of Fluorinated Olefins with Arylboronic Acids by Palladium Catalysis. Eur. J. Org. Chem. 2015, 4340–4343 (2015).
Yang, J., Zhao, H.-W., He, J. & Zhang, C.-P. Pd-Catalyzed Mizoroki-Heck Reactions Using Fluorine-Containing Agents as the Cross-Coupling Partners. Catalysts 8, 23 (2018).
Cheng, R., Xu, C. & Zhang, X. Nickel-catalyzed regioselective coupling reaction of 3,3,3-trifluoropropene with arylzinc reagents. Chin. J. Org. Chem. 40, 3307 (2020).
Ichitsuka, T., Fujita, T. & Ichikawa, J. Nickel-Catalyzed Allylic C(sp3)–F Bond Activation of Trifluoromethyl Groups via β-Fluorine Elimination: Synthesis of Difluoro-1,4-dienes. ACS Catal. 5, 5947–5950 (2015).
Ding, D., Lan, Y., Lin, Z. & Wang, C. Synthesis of gem-Difluoroalkenes by Merging Ni-Catalyzed C-F and C-C bond activation in cross-electrophile coupling. Org. Lett. 21, 2723–2730 (2019).
Gu, J. W., Min, Q. Q., Yu, L. C. & Zhang, X. Tandem difluoroalkylation-arylation of enamides catalyzed by Nickel. Angew. Chem. Int. Ed. 55, 12270–12274 (2016).
Xu, C., Yang, Z. F., An, L. & Zhang, X. G. Nickel-catalyzed difluoroalkylation-alkylation of enamides. ACS Catal. 9, 8224–8229 (2019).
Xu, C., Cheng, R., Luo, Y.-C., Wang, M.-K. & Zhang, X. trans-selective aryldifluoroalkylation of endocyclic enecarbamates and enamides by Nickel Catalysis. Angew. Chem. Int. Ed. 59, 18741–18747 (2020).
Derosa, J., Tran, V. T., Boulous, M. N., Chen, J. S. & Engle, K. M. Nickel-Catalyzed beta,gamma-dicarbofunctionalization of alkenyl carbonyl compounds via conjunctive cross-coupling. J. Am. Chem. Soc. 139, 10657–10660 (2017).
Shrestha, B. et al. Ni-catalyzed regioselective 1,2-dicarbofunctionalization of Olefins by intercepting heck intermediates as imine-stabilized transient metallacycles. J. Am. Chem. Soc. 139, 10653–10656 (2017).
Kraft, B. M., Lachicotte, R. J. & Jones, W. D. Aliphatic carbon−fluorine bond activation using (C5Me5)2ZrH2. J. Am. Chem. Soc. 122, 8559–8560 (2000).
Kraft, B. M. & Jones, W. D. Mechanism of Vinylic and Allylic carbon−fluorine bond activation of non-perfluorinated Olefins using Cp*2ZrH2. J. Am. Chem. Soc. 124, 8681–8689 (2002).
Rieth, R. D., Brennessel, W. W. & Jones, W. D. Activation of Aromatic, Aliphatic, and Olefinic Carbon–Fluorine Bonds Using Cp*2HfH2. Eur. J. Inorg. Chem. 2007, 2839–2847 (2007).
Kühnel, M. F. & Lentz, D. Titanium-catalyzed C-F activation of fluoroalkenes. Angew. Chem. Int. Ed. 49, 2933–2936 (2010).
Bakewell, C., White, A. J. P. & Crimmin, M. R. Reactions of fluoroalkenes with an aluminium(I) complex. Angew. Chem. Int. Ed. 57, 6638–6642 (2018).
Sakaguchi, H., Ohashi, M. & Ogoshi, S. Fluorinated vinylsilanes from the copper-catalyzed defluorosilylation of fluoroalkene feedstocks. Angew. Chem. Int. Ed. 57, 328–332 (2018).
Hu, X. Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective. Chem. Sci. 2, 1867–1886 (2011).
Schley, N. D. & Fu, G. C. Nickel-catalyzed Negishi arylations of propargylic bromides: a mechanistic investigation. J. Am. Chem. Soc. 136, 16588–16593 (2014).
Xiao, Y. L., Guo, W. H., He, G. Z., Pan, Q. & Zhang, X. Nickel-catalyzed cross-coupling of functionalized difluoromethyl bromides and chlorides with aryl boronic acids: a general method for difluoroalkylated arenes. Angew. Chem. Int. Ed. 53, 9909–9913 (2014).
Standley, E. A. & Jamison, T. F. Simplifying nickel(0) catalysis: an air-stable nickel precatalyst for the internally selective benzylation of terminal alkenes. J. Am. Chem. Soc. 135, 1585–1592 (2013).
Xiao, Y. L., Min, Q. Q., Xu, C., Wang, R. W. & Zhang, X. Nickel-Catalyzed Difluoroalkylation of (Hetero)Arylborons with Unactivated 1-Bromo-1,1-difluoroalkanes. Angew. Chem. Int. Ed. 55, 5837–5841 (2016).
Xu, C. et al. Difluoromethylation of (hetero)aryl chlorides with chlorodifluoromethane catalyzed by nickel. Nat. Commun. 9, 1170 (2018).
Feng, Z., Min, Q.-Q., Xiao, Y.-L., Zhang, B. & Zhang, X. Palladium-catalyzed difluoroalkylation of aryl boronic acids: a new method for the synthesis of aryldifluoromethylated phosphonates and carboxylic acid derivatives. Angew. Chem. Int. Ed. 53, 1669–1673 (2014).
Chierchia, M., Xu, P., Lovinger, G. J. & Morken, J. P. Enantioselective radical addition/cross-coupling of organozinc reagents, alkyl iodides, and alkenyl boron reagents. Angew. Chem. Int. Ed. 58, 14245–14249 (2019).
Xu, C. et al. Difluoromethylation of (hetero)aryl chlorides with chlorodifluoromethane catalyzed by nickel. Nat. Commun. 9, 1170 (2018).
Lin, Q. & Diao, T. Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc. 141, 17937–17948 (2019).
Diccianni, J. B., Katigbak, J., Hu, C. & Diao, T. Mechanistic Characterization of (Xantphos)Ni(I)-Mediated Alkyl Bromide Activation: Oxidative Addition, Electron Transfer, or Halogen-Atom Abstraction. J. Am. Chem. Soc. 141, 1788–1796 (2019).
An, L., Xu, C. & Zhang, X. Highly selective nickel-catalyzed gem-difluoropropargylation of unactivated alkylzinc reagents. Nat. Commun. 8, 1460 (2017).
Shrestha, R., Dorn, S. C. M. & Weix, D. J. Nickel-catalyzed reductive conjugate addition to enones via allylnickel intermediates. J. Am. Chem. Soc. 135, 751–762 (2013).
Mohadjer Beromi, M., Banerjee, G., Brudvig, G. W., Hazari, N. & Mercado, B. Q. Nickel(I) aryl species: synthesis, properties, and catalytic activity. ACS Catal. 8, 2526–2533 (2018).
Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (21931013, 21991122), the National Key R&D Program of China (2021YFF0701700), the Science and Technology Committee of Shanghai Municipality (21XD1404400) and SIOC.
Author information
Authors and Affiliations
Contributions
X.Z. and C.X. conceived and designed the experiments. X.Z. directed the project. C.X. performed the experiments. M.-K.W. prepared some starting materials, purified some products, and conducted mechanism studies. X.Z. wrote the paper. S.Z. revised the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Norio Shibata and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, C., Wang, MK., Zhang, S. et al. Nickel-catalyzed alkyl-arylation of 3,3,3-trifluoropropene. Commun Chem 5, 41 (2022). https://doi.org/10.1038/s42004-022-00659-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00659-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
