
Abstract
Nε-Methylation of lysine residues in histones plays an essential role in the regulation of eukaryotic transcription. The ‘highest’ methylation mark, Nε-trimethyllysine, is specifically recognised by Nε-trimethyllysine binding ‘reader’ domains, and undergoes demethylation, as catalysed by 2-oxoglutarate dependent JmjC oxygenases. We report studies on the recognition of the closest positively charged Nε-trimethyllysine analogue, i.e. its trimethylphosphonium derivative (KPme3), by Nε-trimethyllysine histone binding proteins and Nε-trimethyllysine demethylases. Calorimetric and computational studies with histone binding proteins reveal that H3KP4me3 binds more tightly than the natural H3K4me3 substrate, though the relative differences in binding affinity vary. Studies with JmjC demethylases show that some, but not all, of them can accept the phosphonium analogue of their natural substrates and that the methylation state selectivity can be changed by substitution of nitrogen for phosphorus. The combined results reveal that very subtle changes, e.g. substitution of nitrogen for phosphorus, can substantially affect interactions between ligand and reader domains / demethylases, knowledge that we hope will inspire the development of highly selective small molecules modulating their activity.
Similar content being viewed by others
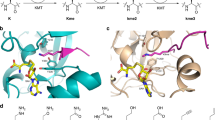


Introduction
Eukaryotic histones are subject to numerous posttranslational modifications (PTMs) that regulate expression in a context dependent manner. Histone lysine-residues are amongst the most frequently modified of all residues, including by acylation-type modifications, most commonly acetylation1. They are also iteratively Nε-methylated to give Nε-monomethylated (Kme1), Nε-dimethylated (Kme2), and Nε-trimethylated lysine (Kme3) residues (Fig. 1a). The roles of lysine Nε-methylation depend on factors including methylation state and location in nucleosomal complexes. Typically, histone H3 Nε-trimethyllysine 4 (H3K4me3), H3K36me3, and H3K79me3 are linked to transcriptional activation, while H3K9me3, H3K27me3 and H4K20me3 are linked to suppression2. Nε-Methyllysine groups are installed by histone Nε-lysine methyltransferases (KMTs, “writers”) and removed by histone Nε-lysine demethylases (KDMs, “erasers”) (Fig. 1b). Nε-Methyllysine chromatin binding proteins (“readers”) bind to specific Nε-methylated lysines to enable gene regulation3,4. KDMs have either an amine-oxidase or, more commonly, a JumonjiC (JmjC) catalytic domain4. The JmjC KDMs are Fe(II) and 2-oxoglutarate (2OG) dependent dioxygenases, which normally couple two electron substrate oxidation, e.g., Nε-methyllysine demethylation, to conversion of 2OG/O2 to succinate/CO2. JmjC KDMs catalyse removal of mono-, di-, or trimethyl groups via hydroxylation of an Nε-methyl group followed by decomposition of the hemiaminal intermediate producing formaldehyde and the demethylated product4.

a JmjC KDMs catalyse demethylation of Nε-trimethyllysine residues. Our work explored recognition of the simplest positively charged Nε-trimethyllysine analogue, i.e., the trimethylphosphonium derivative, by Nε-methyllysine binding proteins and Nε-methyllysine demethylases. n: number of methyl groups (3–1). b View from a structure of a JmjC KDM (KDM4AJmjC, light blue) complexed with H3K9me3 (yellow) and NOG (N-oxalylglycine, a 2OG analogue, white) (PDB: 2OQ6). c View from a structure of a reader (TAF3PHD, purple) complexed with H3K4me3 (yellow) (PDB: 2K17). Nitrogen: dark blue; oxygen: red; sulphur: yellow; zinc: grey; nickel: orange.
The interplay between KMTs and KDMs regulates lysine methylation status, which in turn regulates binding of methylation state-specific chromatin binding modules. Four identified non-catalytic domains interact with Nε-trimethyllysines: plant homeodomains (PHD) (Fig. 1c), tandem tudor domains (TTD), chromodomains (CHD), and malignant brain tumour (MBT) proteins3, all of which bind Nε-trimethyllysine in a cage comprised of typically hydrophobic and aromatic residues4. Experimental and computational studies have shown that binding of Nε-trimethyllysine by readers is driven by cation–π interactions between the positively charged quaternary ammonium group of Nε-trimethyllysine and electron-rich aromatic residues and by release of water molecules from the cage5,6,7,8,9.
Misregulation of histone modification is linked to human disease. For example, DNA encoding for the PHD3 finger of the KDM5A demethylase can fuse with that of nuclear pore protein 98 (NUP98) leading to the NUP98-KDM5A-PHD3 fusion protein, which is linked to acute myeloid leukaemia10. BPTF, a core subunit of the ATP-dependent nucleosome remodelling factor (NURF), can fuse with NUP98 to result in primary refractory acute megakaryoblastic leukaemia protein11. Other readers forming NUP98 fusion proteins include PHF23, NSD1, and NSD312,13.
Research on KMTs and KDMs4 has led to potent and partially selective inhibitors, of use in studying their roles and therapeutic potential. However, there remain challenges in achieving selectivity for particular KDM isoforms (there are >25 JmjC oxygenases and >60 2OG oxygenases in humans14,15). Development of selective inhibitors for chromatin binding modules, which are often present in epigenetic writers and erasers, has been particularly challenging, with reported inhibitors of the >100 human PHD16 and TTD17 domains being relatively weak and non-selective binders18,19,20. Knowledge of the selectivity of ligand binding by chromatin binding proteins and modifying enzymes is thus of both fundamental and medicinal interest21. To investigate the extent to which histone Nε-methyllysine readers and erasers can manifest selectivity, we synthesised a peptide containing the simplest possible positively charged Nε-trimethyllysine analogue, i.e., ε-trimethylphosphonium lysine (KPme3), and studied its interactions with histone Nε-methyllysine readers and erasers (Fig. 1). The results reveal that, at least some, readers and erasers can discriminate between KPme3 and Kme3 peptides, suggesting that identification of drug-like selective inhibitors should be possible.
Results
To study the effect of nitrogen substitution of Nε-trimethyllysine to phosphorus under in vitro conditions, the Fmoc-protected Pε-trimethylphosphonium analogue of Nε-trimethyllysine (Fmoc-KPme3-OH) was synthesised from L-lysine in nine steps (Supplementary Scheme 1). The Fmoc-KPme3-OH and the Fmoc-Kme3-OH control were incorporated into human histone H3-tail fragment peptides (histone H3 residues 1-10, ART(KPme3)QTARKS: H3KP4me3/ART(Kme3)QTARKS: H3K4me3; and histone H3 residues 1–15, ARTKQTAR(KPme3)STGGKA: H3KP9me3/ARTKQTAR(Kme3)STGGKA: H3K9me3) using Fmoc mediated solid-phase peptide synthesis (SPPS), followed by preparative HPLC (Supplementary Scheme 1).
ITC analysis of H3K4me3 and H3KP4me3 with reader domains
We investigated the thermodynamics of association of the H3KP4me3 peptide with five representative human reader domains containing either a PHD zinc finger or TTD, i.e. KDM5APHD3 (JARID1A-PHD3, residues M1489–V1641)10, TAF3PHD (R857–K924)22, BPTFPHD (L2583–N2751)23, SGF29TTD (R115–K293)24 and KDM4ATTD (JMJD2A, Q897-P1011)25, selected on the basis of their preference for binding Νε-trimethyllysine over other (non)methylation marks (Kme0 < Kme1 < Kme2 < Kme3), and their domain and aromatic cage diversity. The recombinant readers were produced in E. coli following reported procedures26. Isothermal titration calorimetric (ITC) analyses were used to determine the dissociation constant (Kd), the Gibbs free energy of binding (ΔG°), the enthalpy of binding (ΔH°), and the entropy of binding (ΔS°). Results with the H3K4me3 control peptide correlated with reported values10,23,24,25,27 (Table 1, Fig. 2, Supplementary Fig. 1).

Representative ITC results from the interaction of (a) KDM5APHD3 and (b) KDM4ATTD with H3K4me3 (black) or H3KP4me3 (red) substrates. Top panels show the raw ITC data and the bottom panels show the processed results.
Interestingly, for four of the readers, ITC experiments with H3KP4me3 indicated stronger complex formation than with H3K4me3 (Table 1). The largest increase was observed with KDM4ATTD, which manifested ~12-fold stronger binding with H3KP4me3 compared to H3K4me3. SGF29TTD exhibits comparable binding affinity for H3K4me3 and H3KP4me3; note that it is the only reader tested not possessing a Trp residue in its hydrophobic cage24. This result correlates with the observed unusually strong binding of the neutral Nε-trimethyllysine carbon-analogue to SGF29TTD compared with other reader proteins5. The increase in affinity for H3KP4me3 relative to H3K4me3 is generally a result of a more favourable ΔH°, with the values for the ΔS° remaining largely unchanged for four of the five readers, the exception being KDM4ATTD, (Table 1). Although the observed decreases in ΔH° are small (ΔΔH°: − 0.4 to −1.3 kcal mol−1) for KDM5APHD3, TAF3PHD, BPTFPHD and SGF29TTD, the decrease for H3KP4me3 relative to H3K4me3 is relatively large (ΔΔH°: −4.7 kcal mol−1) for KDM4ATTD. The more favourable ΔH° for binding for H3KP4me3 over H3K4me3 implies more favourable cation–π interactions between the trimethylphosphonium cation and the electron-rich aromatic cages, as found in related systems5,7,8. The longer C-P bond (1.87 Å) in H3KP4me3 compared to H3K4me3 (C-N bond in H3K4me3 is 1.47 Å) and increased volume (+Pme4: 115 Å3, +Nme4: 105 Å3)28 may help position the methyl hydrogens of the quaternary phosphonium cation closer to the aromatic cage residues. Note that, the limited added volume of H3KP4me3 compared to H3K4me3 means both are likely to release the same number of water molecules from the cages, suggesting equal contributions to affinity due to reader desolvation. Overall, the ITC results imply that the readers efficiently recognise the phosphonium analogue of Nε-trimethyllysine: importantly, despite the subtle nature of the difference between H3KP4me3 compared to H3K4me3, differences in the relative binding efficiencies of the readers for the peptides were observed.
Molecular dynamics simulations of histones with readers
We used molecular dynamics (MD) simulations to study how the readers bind to H3K4me3 and H3KP4me3. The Nε-trimethyllysine residue of H3K4me3 in structures of reader protein complexes was replaced with KPme3 with solvation in a 10 Å truncated octahedral box of TIP3P water29 and neutralised explicitly with sodium or chloride ions. AMBER1230 was used to simulate the systems for 10 ns (Supplementary Figs. 2–7, Supplementary Tables 1 and 2)31. Although this timescale is not long enough to observe events such ligand binding or substantial conformational changes, such simulations have been shown to be valuable in recent studies evaluating the stability of protein–ligand complexes and identifying potential favourable or unfavourable non-covalent interactions, including for reader–H3K4me3 complexes7,8,32,33. Over the simulation time, the SGF29TTD–H3KP4me3 complex manifests a similar pose to the SGF29TTD–H3K4me3 complex (Fig. 3), including in the hydrophobic cage (Y238, Y245, and F264) (Supplementary Figs. 2–7). The H3KP4me3 residue mimics the binding pose of H3K4me3 with respect to the cage residues, except for KDM5APHD3 (Supplementary Fig. 4). With KDM5APHD3, H3KP4me3 showed large fluctuations in the distance to W18 of the cage, an observation apparently reflected in previous MD studies where modifications to H3K4me3 yield less favourable interactions with the KDM5APHD3 W18 compared with W287,8.

Snapshots of simulations for SGF29TTD complexed with a histone H3 fragment (liquorice) containing KP4me3 (cyan) or K4me3 (white) at 0 ns and 10 ns.
Quantum chemical analyses in the gas and aqueous phase
We then analysed the energetics of binding for Kme3 and KPme3 (the side chains of H3K4me3 and H3K4Pme3, respectively) with TRP2, a model for two aromatic cage-comprising tryptophan residues, using quantum chemical methods. This model was chosen because KDM5APHD3 has only two aromatic residues present in its aromatic cage (W1625 and W1635, Supplementary Figs. 4 and 5). Such a simple model cannot respect the dynamics of complex protein-protein interactions; however, the results are informative with respect to the interactions of Kme3 and KPme3 side chains with KDM5APHD35,7,8. We used dispersion-corrected density functional theory (DFT) employing BLYP-D3BJ/TZ2P and COSMO for simulating aqueous solutions with ADF. The model complex TRP2–KPme3 presents a 1.2 kcal mol−1 stronger bonding interaction than TRP2–Kme3, with a ∆E(aq): −11.4 and −10.2 kcal mol−1 for the KPme3 and Kme3 complexes, respectively (Table 2). The Kme3 and KPme3 side chains in the modelled complexes have similar conformations, despite the larger size of P (Table 2 and Supplementary Fig. 8). The models imply that both the Kme3 and KPme3 side chains undergo only small deformations on TRP2 binding, as reflected in the strain energies: ∆E(aq)strain: 0.1 and 0.8 kcal mol−1 for the Kme3 and KPme3 complexes, respectively. The preference for KPme3 over Kme3 is also manifested in the absence of water, although to a lesser extent: ∆Eint: −27.6 and −28.0 kcal mol−1 for the Kme3 and KPme3 complexes, respectively. This result supports the above proposal that, energetically, desolvation effects (∆E(desolv)int) are similar for Kme3 and KPme3.
We investigated why the TRP2 aromatic cage interacts more favourably with KPme3 than Kme3, using quantitative Kohn-Sham molecular orbital (KS-MO) theory and energy decomposition analysis (EDA) of ∆Eint (Table 2). The results imply that the more stabilizing interaction ∆Eint for H3KP4me3 originates from more attractive electrostatic (by 0.8 kcal mol−1), orbital (by 0.6 kcal mol−1), and dispersion (by 1.6 kcal mol−1) interactions. The stronger electrostatic attraction of KPme3 is due to the somewhat more positively charged methyl H atoms of the phosphonium group (Fig. 4a). The more attractive ∆Eoi term in TRP2–KPme3 results from stronger, more stabilizing donor–acceptor orbital interactions from π orbitals to the σ*C-H type orbitals on the KPme3 side chain: the charge transfer is 0.09 electrons to KPme3 and only 0.04 electrons to Kme3 (Fig. 4b). The preference for KPme3 is caused by the lower energy of the σ*C-H type orbitals of KPme3 and their better overlap with π orbitals (Supplementary Table 3). Our bonding analyses show that these cation–π interactions can be viewed as cationic CH–π interactions. Note that the more favourable bonding terms in the TRP2–KPme3 complex leads to a shorter d(HMe-CTRP-5MR) distance between the phosphonium group and the cage, which slightly amplifies all interaction terms, including the steric (Pauli) repulsion (by 2.7 kcal mol−1, Table 2).

The TRP2 model employs the two tryptophan residues found in KDM5APHD3 (W1625, W1635). a Calculated VDD atomic charges (in mili-a.u.) for H3K4me3 and H3KP4me3 (red: negative, blue: positive). b Frontier orbitals (with orbital energies in eV) for Kme3, KPme3, and TRP2, (isosurface drawn at 0.03), computed at the BLYP-D3BJ/TZ2P level using an X-ray structure for TRP2 (PDB: 3GL6).
We carried out EDA analyses for the homolytic formation of the C-H bonds in Kme3 and KPme3 at the BLYP-D3BJ/TZ2P level (Supplementary Table 4 and Supplementary Fig. 9). The larger proton affinity for Kme3 compared to KPme3 is maintained both in solution and in the gas phase. The EDA results imply that this derives substantially from the more favourable electrostatic interactions for Kme3 compared to KPme3 (by 5.4 kcal mol−1), even though the orbital interactions are more favourable for KPme3, though only by 1.2 kcal mol−1. The VDD charge on CMe is −268 me and on N is +42 me, whereas for KPme3 the VDD charge for CMe is −348 me and +302 me on P. The difference in homolytic formation of C-H bonds for Kme3 or KPme3 thus seems to be a subtle interplay of a more favourable (i.e. more negative) charge on the methyl carbon plus a less favourable (more positive) charge on the P atom in KPme3.
MS and NMR studies show KDM4EJmjC can demethylate H3KP9me3
Having demonstrated that H3KP4me3 is a stronger binder than H3K4me3 with most of the readers, we investigated whether JmjC KDMs can catalyse demethylation of H3KP9me3, as occurs for H3K9me3 (Fig. 5a). We chose human KDM4EJmjC (M1–Q337), a histone H3K9me3/2 demethylase with relatively high demethylation activity as a model enzyme34. Reactions were monitored using MALDI-TOF mass spectrometry (Fig. 5b–d). KDM4EJmjC (0.5 µM) efficiently catalysed the di-demethylation of the positive control H3K9me3 (6.0 µM) as indicated by two -14 Da mass shifts, as anticipated (Fig. 5b)34. Michaelis-Menten kinetics yielded a KM of 6.1 µM and kcat of 5.3 min−1 (Vmax: 2.7 µM·min−1) (Supplementary Fig. 10), similar values to those reported using the shorter H37-14K9me3 substrate (KM: 21.3 µM and kcat: 4.6 min−1)35.

a KDM4EJmjC catalysed demethylation of H3KP9me3. b Mass spectra and time-course analysis of H3K9me3 (6.0 µM) and KDM4EJmjC at 0 min (black) and 12 min (red) showing the substrate H3K9me3 (orange) and demethylated products H3K9me2 (purple) and H3K9me1 (green). Mass spectra and time-course analysis of H3KP9me3 (5.0 µM) and KDM4EJmjC (c 0.5 µM, d 5.0 µM) at time points 0 (red), 5 (green) and 60 min (red) acquired using MALDI-TOF MS. Conditions: Asc (500 µM), Fe(II) (50 µM) and 2OG (100 µM). Errors represent standard deviations (n = 2 or 3).
When H3KP9me3 (5.0 µM) was treated with KDM4EJmjC (0.5, 5.0 µM) (Fig. 5b, c), H3KP9me3 was consistently observed to only undergo a single demethylation (−14 Da) to give H3KP9me2. Masses corresponding to potential subsequent demethylation to give H3KP9me1 or H3KP9 were not detected. Along with formation of H3KP9me2, time-dependent production of another product, assigned as the phosphine oxide (H3KP9me2O) (+16 Da), was observed (Fig. 5c, d). Reaction of stoichiometric amounts of KDM4EJmjC (5.0 µM) and H3KP9me3 (5.0 µM) showed faster H3KP9me2 and H3KP9me2O product formation, but no evidence for H3KP9me1 or H3KP9 formation. Controls demonstrated little or no H3KP9me2 or H3KP9me2O formation without KDM4EJmjC, ascorbate (Asc), Fe(II) or 2OG (Supplementary Fig. 11). By contrast, with H3K9me3 (which is a better substrate than H3KP9me3 – see below), without Asc and Fe(II) some demethylation was observed, likely reflecting co-purifying Fe(II) and 2OG (Supplementary Fig. 12). With Tris(2-carboxyethyl)phosphine (TCEP) as a reducing agent, rather than Asc, slightly increased yields of H3KP9me2 and H3KP9me2O were observed (Supplementary Fig. 13). Addition of catalase (to supress hydrogen peroxide formation36) with or without Asc or BSA did not substantially alter the amounts of H3KP9me2 or H3KP9me2O (Supplementary Fig. 13). To examine further whether reaction of H3KP9me2 to H3KP9me2O occurs enzymatically and / or non-enzymatically, reactions were quenched (H3K9me3: 5 min or H3KP9me3: 10 min) with formic acid, EDTA or 2,4-PDCA, incubated, then quenched again (H3K9me3: 30 min or H3KP9me3: 60 min) with formic acid (Supplementary Fig. 14). The H3K9me3 results show little variations in product profiles indicating that the reagents are experimentally effective. With H3KP9me3 where H3KP9me2 and H3KP9me2O are produced, on initial quenching with EDTA or 2,4-PDCA (which inhibits by chelating to Fe) we observed a slow increase in the peak corresponding to H3KP9me2O, but not H3KP9me2. The combined observations imply that slow production of H3KP9me2O from H3KP9me2 can occur via non-enzymatic as well as enzymatic oxidation. To verify that products detected using MALDI-TOF MS are not instrumental artefacts, time-course measurements were performed on H3K9me3 and H3KP9me3 with analysis by LC-MS. Similar demethylation and oxidation patterns, including production of H3KP9me2 and H3KP9me2O from H3KP9me3 were detected as observed with MALDI-TOF MS (Supplementary Fig. 15).
To directly compare the efficiency of demethylation of H3K9me3 (5.0 µM) and H3KP9me3 (5.0 µM), they were incubated with KDM4EJmjC (0.5 µM) in the same vessel (Supplementary Fig. 16). H3K9me3 was converted to H3K9me2/1 with the same efficiency as the control, but there was no evidence that H3KP9me3 was converted to H3KP9me2 or H3KP9me2O, although low levels of formation of H3KP9me2 cannot be ruled out as its peak overlaps with an isotope peak of H3K9me3. The combined results show H3K9me3 is a substantially better substrate than H3KP9me3.
KDM4EJmjC-catalysed demethylation of H3KP9me3 was analysed by 1H and 31P NMR; in both cases, the reaction proceeded to give signals corresponding to H3KP9me2 and H3KP9me2O (Fig. 6). In the 31P NMR, distinct resonances were observed for H3KP9me3 (25.9 ppm), H3KP9me2H (28.4 ppm), and H3KP9me2O (55.6 ppm). Notably, a different 31P resonance (δP: 28.4 ppm compared to 55.6 ppm) was observed when quenching the H3KP9me3 reaction with HCl (1 M); this was assigned as a protonated H3KP9me2H species, as supported by 1H-31P HMBC NMR, and comparison of chemical shifts with those for similar species, i.e. PMe3, PMe3H+, and P(O)Me3 (Supplementary Fig. 17), under identical conditions. 31P and 1H NMR time-course studies confirmed demethylation and conversion of 2OG to succinate (Supplementary Fig. 18).

a 31P NMR analyses of H3KP9me3 (◊) incubated with KDM4E, addition of acid (top) or quenched by heating (bottom). Evidence for formation of H3KP9me2H (□, δP = 28.4 ppm, top) and H3KP9me2O (○, δP = 55.6 ppm, bottom). b Overlay of 1H-31P HMBC analyses of the solutions after of H3KP9me3 incubated with KDM4EJmjC, quenched by heating (green cross peaks), or addition of acid (red cross peaks). Experimental conditions: H3KP9me3 (500 µM), Asc (1.00 mM), 2OG (1.00 mM), Fe(II) (100 µM), KDM4EJmjC (50 µM).
To test whether formaldehyde is generated by H3KP9me3 demethylation, a formaldehyde dehydrogenase (FDH) coupled assay was employed (Supplementary Fig. 19a)34,37. With H3K9me3, this assay gave KM: 5.1 µM and kcat: 6.1 min−1 (Vmax: 0.61 µM·min−1) (Supplementary Fig. 19b,c), values comparable to those obtained by MALDI-TOF MS. Measurements using H3KP9me3 also show an KDM4EJmjC and time-dependent increase in formaldehyde (Supplementary Fig. 20), but the low activity prohibited detailed kinetics analysis of H3KP9me3 using the FDH-assay.
Demethylation of H3KP9me3 by other KDM4s
To investigate whether H3KP9me3 can be demethylated by other human KDM4 subfamily members, recombinant KDM4AJmjC (M1–L359) and KDM4DJmjC (M1–L358) were produced in E. coli following an adaption of literature procedures38,39. At a relatively high enzyme concentration, KDM4AJmjC (2.4 µM) and KDM4DJmjC (2.4 µM) demonstrate clear demethylation activity on H3K9me3 (5.0 µM) (Supplementary Figs. 21 and 22). With H3KP9me3 (5.0 µM) substantial turnover to H3KP9me2 was observed with KDM4AJmjC and KDM4DJmjC, with little H3KP9me2O formation being observed. No evidence for further demethylation was accrued. Unlike the KDM4s, KDM3A/B (JMJD1A/B) and KDM7A/B (PHF8) do not catalyse demethylation of H3K9me3, but demethylate H3K9me2/1 to give the unmethylated lysine residue. To test the ability of KDM3 and KDM7 subfamily representatives to demethylate H3KP9me2 or H3KP9me3, KDM3AJmjC (T515–S1317) and KDM7BJmjC (M37–N483) were produced using baculovirus/sf9 and E. coli expression systems, respectively40,41. The synthesis of a histone H3 mimic peptide H3KP9me2 substrate is challenging as the tri-alkylated phosphine group is susceptible to oxidation during synthesis of the protected amino acid and during SPPS requiring oxygen-free conditions. Thus, to investigate if KDM3AJmjC or KDM7BJmjC can catalyse demethylation of H3KP9me2, an appropriate H3KP9me2 substrate was prepared in situ from H3KP9me3 using KDM4EJmjC (2.0 µM), in the presence of KDM3AJmjC (2.0 µM) or KDM7BJmjC (2.0 µM). [Note, KDM7BJmjC exhibits significantly higher H3K9me2 demethylation rates with trimethylated lysine 4 (H3K4me0 < H3K4me3), but is also active without the H3K4me3 modification40]. The results show that KDM3AJmjC and KDM7BJmjC demethylate their ‘natural’ H3K9me2 substrate40,42, but do not catalyse demethylation of H3K9me3 (as anticipated) or H3KP9me3 (Supplementary Fig. 23a–f). Unlike KDM4EJmjC alone, the combination of KDM4EJmjC with KDM3AJmjC or KDM7BJmjC and H3K9me3 manifests conversion to H3K9me1. The same combinations but with H3KP9me3, produced H3KP9me2 (due to KDM4EJmjC catalysis), but did not result in masses consistent with H3KP9me1 or H3KP9, implying that H3KP9me2 is not a substrate for KDM3AJmjC or KDM7BJmjC (Supplementary Fig. 23h, j, l).
Discussion
Methylation of carbon, nitrogen, oxygen and sulphur atoms in large and small biomolecules is of central biological importance; methyl groups linked via heteroatoms are common in drugs and agrochemicals. Alkylated phosphines are commonly used in organic synthesis, e.g., in Wittig reagents. It is thus perhaps surprising that methylphosphonium and related chemistry has, to our knowledge, not been more widely investigated in biochemistry43,44, in particular with respect to the possibility of demethylation.
Our studies on interactions between H3K4me3 and H3KP4me3 and readers demonstrate that H3KP4me3 can substitute for H3K4me345, in most cases with increased affinity, due to stronger cation–π interactions (bonding analyses reveal true cationic CH–π interactions). Notably, there are differences in the relative binding efficiencies of the readers with H3K4me3 compared to H3KP4me3, implying selective inhibition of readers by small drug-like molecules should be feasible. Similar observations have been made in relation to cation–π interactions between tetramethylammonium compounds and their tetramethylphosphonium analogues with respect to binding to aromatic cavities. Related studies with γ-butyrobetaine43 and the serine protease factor Xa44 suggest our observations may be of a general nature.
The results with H3KP4me3 contrast those for other H3K4me3 derivatives binding to readers, where typically comparable or lower affinities are observed (Supplementary Fig. 24, Supplementary Table 5) compared to H3K4me3. For example, studies comparing binding of H3K4me3 and H3KC4me3 to TAF3PHD and KDM4ATTD reveal impaired binding of H3KC4me3(ΔKd values of ~2-fold)46. By contrast, an increase in, or comparable, stability is observed for the H3KP4me3-reader complexes relative to the H3K4me3-reader complexes, with some showing much tighter binding (BPTFPHD, ΔKd: ~7-fold and KDM4ATTD, ΔKd: ~12-fold). For comparison, the difference in binding between H3K4me3 and unmodified-lysine is protein and condition dependent, but typically the ΔKd is >20-fold in favour of H3K4me310,23,24,25,47,48. Even more pronounced decreases in binding affinities are observed with KDM5APHD3 and TAF3PHD5 when the K4me3 in H3K4me3 is substituted for glycine, highlighting the importance of the lysine side chain in binding. Thus, the substitution of H3K4me3 for H3KP4me3 can have a positive effect on binding, knowledge that might be exploited in inhibitor design.
Previous studies revealed that some JmjC KDMs can catalyse oxidation of substrates other than the established Nε-methylated lysine substrates, e.g., H3K9me3/2 for KDM4E, as demonstrated with Nε-methyl-ethyl-lysine-9, a substrate that undergoes both demethylation and deethylation49. However, analysis with Nε-diethyllysine showed no evidence of reaction, demonstrating limitation of the plasticity of the KDM4E active site towards alkylated lysine substrates. Some JmjC KDMs can also catalyse N-methyl arginine demethylation and with appropriately sized Nε-substitutions some can catalyse formation of stable alcohol products49,50. We found that H3KP9me3 is a demethylation substrate for human KDM4A/D/E to give H3KP9me2; this observation is consistent with the relatively small increase in volume when H3KP9me3 is compared to H3K9me3 (Δ[+Pme4-+Nme4]: 10 Å3)28, though the demethylation rate is significantly slower for H3KP9me3 than for H3K9me3. Strikingly, although KDM4 enzymes (KDM4A/D/E) catalysed formation of H3KP9me2, they did not catalyse its further demethylation to give H3KP9me1, despite efficient conversion of H3K9me2 to H3K9me1. We propose that this, at least in part, is due to the decreased pKa of H3KP9me2 versus H3K9me2 – it seems that, at least for the KMD4 JmjC KDMs, the positively charged form of Nε-dimethyllysine H3K9me2 is the preferred substrate. Interestingly, we also observed conversion of H3KP9me2 to H3KP9me2O, possibly in part by non-enzymatic oxidation; we saw no evidence for formation of the analogous H3K9me2O N-oxide.
We also investigated whether the JmjC-domain of KDMs, which notably accept H3K9me2 can accept H3KP4me2 as a substrate. Since H3KP9me2 peptides are difficult to synthesise due to reactivity of the phosphine, we generated H3KP9me2 in situ from H3KP9me3 using KDM4EJmjC. The results with KDM3A and KDM7B, which naturally catalyse H3K9me2 demethylation, provide clear evidence they do not catalyse demethylation of H3KP9me2, revealing the ability of JmjC KDMs to accept P-analogues is subfamily dependent. As with the results for the readers, the results with JmjC KDMs show very small changes to the substrate, likely due to changes in size or charge, can make large differences in substrate selectivity. We hope that this knowledge will inspire medicinal chemistry efforts to identify JmjC KDM isoform specific inhibitors.
Phosphorous is essential for all life forms where it is principally found in its oxidised phosphate form, in nucleic acids, small molecules (e.g., ATP, NADPH), proteins and lipids, amongst other molecules. Alkylated phosphine compounds have, to our knowledge, not been identified in biology. In part this may be due to their tendency to be oxidized, as evident in our work where evidence for KDM4E-catalysed oxidation at H3KP9me2 to give H3KP9me2O, rather than H3K9me1 was accrued. However, phosphine (PH3) is present in the Earth’s atmosphere where it is proposed to be part of the phosphorus cycle51 and, may be present in the atmosphere of Venus52. Our results show that at least several related enzymes can act on reduced phosphine derivatives, highlighting the possibility that reduced phosphine derivatives might, at least in some specialised contexts, have a biological role, and/or that they may have played a role in the evolution of biology.
Methods
Protein production
The following purified reader domains: TAF3PHD (R857-K924), KDM5APHD3 (JARID1A, M1489-V1641), BPTFPHD (L2583-N2751), KDM4ATTD (JMJD2A, Q897-P1011) and SGF29TTD (R115-K293) were prepared as reported10,22,23,24,25,26. The histone lysine demethylase were produced and purified to high purity via reported procedures KDM3A (JMJD1A, T515-S1317)41, KDM4AJmjC39 (JMJD2A, M1-L359), KDM4EJmjC (JMJD2E, M1–Q337)34 and KDM7B40 (PHF8, M37-N483 [NP_001171]). KDM5DJmjC was produced using an N-terminal hexa-His tagged KDM4D (KDM4DM1-L358) DNA construct, transformed into BL21(DE3) competent cells for recombinant protein production. Colonies were used to inoculate of LB media (50 mL) containing kanamycin (50 µg·mL−1) and chloramphenicol (34 µg·mL−1), which was placed in a 37 °C shaker overnight. The starter culture (10 mL) was used to inoculate TB media (6 × 1 L) containing kanamycin (50 µg·mL−1) in 2 L baffled shaker flasks. After reaching an of OD600 of ~0.8, the temperature was reduced to 18 °C; at OD600 ~0.9 the cells were induced by the IPTG (0.5 mM) addition. After shaking overnight, the culture was centrifuged (5000 rpm, 10 mins), the media was decanted, and the cell pellet was suspended in Lysis Buffer [HEPES (50 mM), NaCl (500 mM), imidazole (20 mM), glycerol (5%) and TCEP (0.5 mM) in water (pH 7.4)]. The suspension was lysed by passaging through a high-pressure cell breaker (Avestin – EmulsiFlex-C5) for three rounds. The lysate was cleared by centrifugation (60 minutes, 36,000 × g, 4 °C), then loaded onto a Ni NTA gravity column. After extensive rinsing of the Ni-NTA gravity column with lysis buffer, the His-tagged protein was eluted in lysis buffer containing 300 mM imidazole. The eluted protein was concentrated and subjected to gel filtration chromatography using an AKTA Xpress system, an S200 16/600 gel filtration column, and GF buffer [HEPES (50 mM), NaCl (150 mM), glycerol (5%) and TCEP (0.5 mM) in water (pH 7.4)]. The protein identity was verified by LC-MS (ESI-TOF) observing a mass of 43759.7 Da, in accord with the predicted mass of 43751.6 Da.
Peptide synthesis
Histone H3 mimic-peptides were prepared (as C-terminal amides) using standard SPPS methodology with Nα-Fmoc protection. Reaction of the C-terminal amino acid with the Wang resin was done by suspending the Wang resin (1.0 g, 1.0 mmol·g−1, 1.0 equivalent) in CH2Cl2/DMF (9:1, 10 mL), followed by addition of diisopropylcarbodiimide (DIC, 252 mg, 2.0 mmol, 2 equivalents), HOBt (270 mg, 2.0 mmol, 2.0 equivalents), DMAP (12.0 mg, 0.10 mmol, 0.1 equivalents), and the Fmoc-Aa-OH (2.0 mmol, 2.0 equivalents). The solution was stirred slowly for (20 h, rt). Ac2O (200 μL, 2.10 mmol, 2.1 equivalents) and pyridine (200 μL, 2.40 mmol, 2.4 equivalents) were then added, and the suspension was stirred for 30 min at rt. The suspension was filtered and the resin washed with DMF, CH2Cl2 (40 mL), and MeOH (40 mL) (×3), and dried before using in coupling steps.
Manual approach
Each coupling reaction was performed in DMF with the appropriate Fmoc-protected amino acid (3.0 equivalents), diisopropylcarbodiimide (DIC, 3.3 equivalents) and hydroxybenzotriazole (HOBt, 3.6 equivalents). Note, coupling of Fmoc-KPme3-OH was done for an extended period (at least 16 hours). Subsequently, free N-terminal amines were capped (Ac2O, 2.0 equivalents, pyridine, 2.4 equivalents) before treatment with piperidine. Completion each coupling reactions was determined by the Kaiser test, followed by removal of the Fmoc group by treatment with piperidine (20% v/v in DMF) for 30 min, with completion being determined by the Kaiser test. Washing in between steps was done by treatment of the resin with DMF (3×). Before acidic deprotection and cleavage, the resin was treated with DMF (3×) and Et2O (3×), then dried under reduced pressure.
Peptide synthesizer approach
Peptides were synthesized using a Liberty Blue microwave assisted solid phase peptide synthesizer (CEM corporation). The coupling steps were carried out using DIC and Oxyma in DMF in a microwave vessel at 90 °C. Each coupling step was performed in DMF with an excess of Fmoc-protected amino acids (5.0 equivalents). Note that the coupling step of Fmoc-KPme3-OH (2.0 equivalents) was performed manually, using HATU (2.5 equivalents) in DMF for 16 h at rt. Subsequently, any free N-terminal amine was capped using Ac2O (2.0 equivalents), and pyridine (2.4 equivalents) before treatment with piperidine.
Cleavage of the peptides was achieved using a mixture of TFA 92.5%, H2O 2.5%, triisopropylsilane 2.5%, and ethane-1,2-dithiol 2.5%; the product was precipitated from Et2O after 3–4 h. The crude product was suspended in Et2O, then centrifuged (3500 rpm, 4 minutes); the supernatant was then decanted (3 times). Purification of the peptides was performed by preparative HPLC. Analysis of the peptides was done by LC-MS and analytical HPLC. Conditions for a typical HPLC purification run were starting conditions: MeCN (3%) in H2O (both supplemented with 0.1% (v/v) TFA), a gradient to 100% MeCN over 30 minutes. Sample fractions were pooled based on the results of LC-MS analysis, then lyophilised to yield the desired product as a fluffy powder.
Isothermal titration calorimetry
ITC studies followed a reported procedure26. The buffer used corresponded to that used in the final protein purification step. Briefly, TAF3PHD and KDM4ATTD: [Tris (50 mM) in water (pH 7.5)]; KDM5APHD3 and BTPFPHD: [Tris (50 mM), NaCl (20 mM) in water (pH 7.5)]; [Tris (25 mM), NaCl (50 mM), 1,4-dithiothreitol (1.0 mM) in water (pH 7.5)]. Experiments were conducted using ITC200 automated (GE Healthcare Life Sciences, USA) instrument at 25 °C. Histone peptide titrations were performed with the same reader batches. Solutions of the reader in buffer (25–40 µM) and of the histone H3 peptide (350–600 µM) in buffer were prepared. The prepared solutions were plated into a 96-well plate and inserted into the instrument for analysis. Experiments were performed according to manufacturer’s default settings: Plate pre-rinse syringe clean. A total of 19 injections were performed; each experiment was repeated 3–5 times. Heats of dilution for histone peptides determined in control experiments were subtracted from the titration binding data. Data were analysed with Origin 6.0 (Microcal Inc., Northampton, Massachusetts, USA) and curve fitting with one-site binding mode was applied.
MALDI-TOF demethylation experiments
A Bruker Daltonics MALDI-TOF/TOF AutoflexSpeed machine and Bruker MTP 384 target plates (polished steel BC, Part: 8280781) were used. The machine was controlled using Flex control (v. 3.4 build 135.10) and Compass for flex series (v. 1.4) software. Measurements were in the positive ion mode with the reflectron mode enabled. Incubations employed ProxiPlate-384™ (Perkin Elmer) plates into which was pipetted a solution of [H31-15K9me3 (10.0 µM), (+)-sodium L-ascorbate (1.0 mM), (NH4)2Fe(II)(SO4)2 (100 µM), di-sodium 2-oxoglutarate (200 µM) in buffer (HEPES (50 mM)) in MilliQ (pH 7.5)] (5.0 µL) using ClipTip™ (Thermo Scientific™) pipette tips and E1-ClipTip™ (Thermo Scientific™). The enzyme solution [KDM4EJmjC (1.0 µM) in HEPES (50 mM)] (5.0 µL) was added to initiate reaction at 37 °C. Reactions were quenched with formic acid in water (2%, 5.0 µL). Samples were then spotted onto a MALDI-TOF target plate (1.0 µL), MALDI matrix [sat. sol. α-cyano-4-hydroxy-cinnamic acid (10 mg·mL−1) in {trifluoro acetic acid, acetonitrile and MilliQ (0.1:50:50)}] (1.0 µL) was added, mixed, and dried in air. Samples were then analysed by MALDI-TOF. Specific experiments were supplemented with catalase from bovine liver (C3155-50MG, Merck) (5.0 µM), bovine albumin serum (BSA, Perkin Elmer, CR84-100, DTPA purified 7.5%) (5.0 µM), or Tris(carboxyethyl)phosphine hydrochloride salt (TCEP, M02624, Fluorochem) (500 µM).
Demethylation experiments using LC-MS
KDM4EJmjC demethylation studies of H31-15K9me3 or H31-15KP9me3 incubations using LC-MS were conducted as reported31, using Agilent RapidFire 365 and Agilent QTOF 6530 machines. In brief, samples were aspirated under vacuum (~50 µL), passed through a loop (10 µL, 400 ms) and wash on a SPE cartridge A (C4) using solvent A (1.5 mL·min−1, 4500 ms). Peptides were eluted using solvent B (1.25 mL·min−1, 4500 ms) and directed to the MS for measurements. The cartridge was equilibrated for the next sample (1.25 mL·min−1, 500 ms) and needle was cleaned with an organic wash solution. Between each sample, an alternating inorganic, organic and inorganic washes were performed to avoid any potential carry over on the SPE cartridge from previous sample. Solvent A: formic acid (0.1%) in water; Solvent B: formic acid (0.1%), acetonitrile (85%) in water; Inorganic wash: water; Organic wash: acetonitrile. Demethylation reactions were conducted in a temperature-controlled room (22 °C) and the MS machine Real-time monitoring mode. A solution [Peptide (6.0 µM), sodium L-ascorbate (600 µM), (NH4)2Fe(II)(SO4)2 (60 µM), disodium 2OG salt (120 µM) in buffer] (550 µL) was prepared. The first time point (~50 µL) was aspirated and acquired in the absence of KDM4EJmjC (t = 0 min). Subsequently, the enzyme solution [KDM4EJmjC (3.0 µM) in buffer] (100 µL) was added and samples from the solution were taken every 2–2.5 min and measured. Note that the time was recorded between the addition and the first aspiration of the enzymatic reaction mixture [Peptide and Enzyme solution mixture]. Each measurement with the corresponding mass profile was time-stamped and the data was processed using Agilent Masshunter (B.06.00), MicroSoft Excel™, GraphPad Prism© (v. 5.0) and Adobe illustrator (15.0.0) software.
Demethylation experiments studies using NMR
Incubations of H31-15KP9me3 with KDM4EJmjC were performed in Eppendorf tubes (1.5 mL). Conditions used for 1H and 31P NMR time-courses: H31-15KP9me3 (250 µM) was incubated with KDM4EJmjC (50 µM), sodium ascorbate (1.00 mM), 2OG (500 µM), and Fe(NH4)2(SO4)2 (100 µM) in HEPES-d18 buffer (50 mM, pH 7.5) in D2O ( > 95% 2H). Reactions (160 µL total volume) were quenched by addition of HCl (1 M, 10 equivalents) after the indicated time. The samples were centrifuged (1 min, 14,500 rpm) and the supernatant transferred to an NMR tube (3 mm, Norell). For characterisation of the products from the incubation of H31-15KP9me3 with KDM4EJmjC the following conditions were used: Peptide H31-15KP9me3 (500 µM) was incubated with sodium ascorbate (1.00 mM) 2-oxoglutarate (1.00 mM), Fe(NH4)2(SO4)2 (100 µM), and KDM4EJmjC (50.0 µM), in HEPES-d18 buffer (50 mM, pH 7.5) in D2O (>95% 2H) for 1 hour. Reactions were quenched by addition of HCl (1 M, 10 equivalents), or by heating (95 °C, 10 min). Precipitated proteins were removed by centrifugation (1 min, 14,500 rpm) and the supernatant transferred to an NMR tube (3 mm, Norell). Spectra were measured using a Bruker 600 MHz machine and analysed using MestReNova 14.1 (MestReLabs, Spain; www.mestrelab.com) and Topspin 3.6.1 (Bruker, Germany; www.bruker.com).
Quantum chemical analysis
Quantum chemical calculations were performed with the Amsterdam Density Functional software (ADF)53 using dispersion-corrected density functional theory at the BLYP-D3BJ/TZ2P level of theory54. Our BLYP-D3BJ/TZ2P approach provided results that are in excellent agreement with those of a recent high-level CCSD(T) benchmark study by Varma and coworkers (Supplementary Table 6)55. Solvation in water was simulated by means of the conductor like screening model (COSMO) of solvation implemented in ADF56,57,58,59. The cation–π interactions in TRP2–H3K4me3 and TRP2–H3KP4me3 complexes were analysed through quantitative Kohn–Sham molecular orbital theory combined with energy decomposition analysis (EDA)60,61. In this method the bond energy in water ∆E(aq) is a combination of the strain energy (∆Estrain(aq)) associated with deforming the cation and the reader from their equilibrium structures to the geometry they adopt in the complex, combined with the interaction energy (∆Eint(aq)) between these deformed fragments in the complex:
The role of desolvation in the complexation process can be analysed by splitting the solute–solute interaction (∆Eint(aq)) into the effect caused by the change in solvation (∆Eint(desolv)) and the remaining intrinsic interaction (∆Eint) between the unsolvated fragments in vacuum:
The interaction energy ΔEint can be further decomposed by:
where, ∆Velstat corresponds to the classical electrostatic interaction between the unperturbed charge distributions of the deformed fragments, which is usually attractive. The Pauli repulsion (∆EPauli) term comprises the destabilizing interactions between occupied orbitals and is responsible for steric repulsions. The orbital interaction (∆Eoi) accounts for charge transfer (donor–acceptor interactions between occupied orbitals on one moiety with unoccupied orbitals of the other, including the HOMO–LUMO interactions) and polarization (empty/occupied orbital mixing on one fragment due to the presence of another fragment). Finally, the ∆Edisp term accounts for the dispersion interactions based on Grimme′s DFT-D3BJ correction. The charge distribution was analysed using the Voronoi deformation density (VDD) method62.
Molecular dynamics simulations
MD simulations were carried out for 10 ns. Crystal structures for the models representing TAF3PHD (PDB: 2K17), KDM4ATTD (PDB: 2GFA), KDM5APHD3 (PDB: 2KGI), BPTFPHD (PDB: 2F6J), and SGF29TTD (PDB: 3ME9) readers were used as starting structures for the protein-ligand modelling. Starting structures were built by manually replacing the Kme3 residue of H3K4me3 with KP9me3 residue in the reader protein crystal structures complexes. AMBER1230 was used with the Amberff12SB force field to define protein partial charges. Hydrogen atom addition was performed with LEaP. Systems were solvated in a 10 Å truncated octahedral box of TIP3P29 water and neutralised explicitly with either sodium or chloride counterions. Non-bonding parameters of Zn(II), previously established from studies of KDM4A63, were employed. Atomic partial charges for H3Kp9me3 correspond to the Restrained Electrostatic Potential (RESP)64 charges, as shown in Supplementary Table 2. Parameters for Kme3 were taken from previous work31. The final systems were minimised for 1,000 cycles of steepest-descent minimization followed by 1,000 cycles of conjugate-gradient minimization to remove close van der Waals contacts using the sander program in AMBER12. Equilibration was achieved using PMEMD to heat the systems to 310 K followed by independent MD simulations performed with a periodic boundary condition at a constant pressure of 1 atm with isotropic molecule-based scaling at a time step of 2.0 fs. All simulations used a dielectric constant of 1.0, Particle Mesh Ewald summation65 to calculate long-range electrostatic interactions and bond-length constraints applied to all bonds to H atoms. Trajectories were saved at 20 ps intervals and visualised using VMD66.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that the main data supporting the findings of this study are available within the paper and its Supplementary Information file. Other relevant data are available from the corresponding authors upon reasonable request.
References
Shahbazian, M. D. & Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem 76, 75–100 (2007).
Lee, B. M. & Mahadevan, L. C. Stability of histone modifications across mammalian genomes: implications for ‘epigenetic’ marking. J. Cell. Biochem. 108, 22–34 (2009).
Taverna, S. D., Li, H., Ruthenburg, A. J., Allis, C. D. & Patel, D. J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040 (2007).
Kooistra, S. M. & Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 13, 297–311 (2012).
Kamps, J. J. A. G. et al. Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat. Commun. 6, 8911 (2015).
Hughes, R. M., Wiggins, K. R., Khorasanizadeh, S. & Waters, M. L. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc. Natl. Acad. Sci. USA. 104, 11184–11188 (2007).
Al Temimi, A. H. K. et al. Recognition of shorter and longer trimethyllysine analogues by epigenetic reader proteins. Chem. Commun. 54, 2409–2412 (2018).
Pieters, B. J. G. E. et al. Mechanism of biomolecular recognition of trimethyllysine by the fluorinated aromatic cage of KDM5A PHD3 finger. Commun. Chem. 3, 69 (2020).
Rahman, S., Wineman-Fisher, V., Al-Hamdani, Y., Tkatchenko, A. & Varma, S. Predictive QM/MM Modeling of Modulations in Protein–Protein Binding by Lysine Methylation. J. Mol. Biol. 433, 166745 (2021).
Wang, G. G. et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459, 847–851 (2009).
Roussy, M. et al. NUP98-BPTF gene fusion identified in primary refractory acute megakaryoblastic leukemia of infancy. Genes Chromosom. Cancer 57, 311–319 (2018).
Gough, S. M. et al. NUP98-PHF23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD histone reader function. Cancer Discov. 4, 564–577 (2014).
Wang, G. G., Cai, L., Pasillas, M. P. & Kamps, M. P. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat. Cell Biol. 9, 804–812 (2007).
Islam, M. S., Leissing, T. M., Chowdhury, R., Hopkinson, R. J. & Schofield, C. J. 2-Oxoglutarate-Dependent Oxygenases. Annu. Rev. Biochem. 87, 585–620 (2018).
Markolovic, S. et al. Structure-function relationships of human JmjC oxygenases - demethylases versus hydroxylases. Curr. Opin. Struct. Biol. 41, 62–72 (2016).
Musselman, C. A. & Kutateladze, T. G. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 39, 9061–9071 (2011).
Lu, R. & Wang, G. G. Tudor: A versatile family of histone methylation ‘readers’. Trends Biochem. Sci. 38, 546–555 (2013).
Bhushan, B. et al. Investigations on small molecule inhibitors targeting the histone H3K4 tri-methyllysine binding PHD-finger of JmjC histone demethylases. Bioorg. Med. Chem. 26, 2984–2991 (2018).
Wagner, E. K., Nath, N., Flemming, R., Feltenberger, J. B. & Denu, J. M. Identification and characterization of small molecule inhibitors of a plant homeodomain finger. Biochemistry 51, 8293–8306 (2012).
Miller, T. C. R. et al. Competitive binding of a benzimidazole to the histone-binding pocket of the pygo PHD finger. ACS Chem. Biol. 9, 2864–2874 (2014).
Maas, M. N., Hintzen, J. C. J., Porzberg, M. R. B. & Mecinović, J. Trimethyllysine: From carnitine biosynthesis to epigenetics. Int. J. Mol. Sci. 21, 9451 (2020).
van Ingen, H. et al. Structural insight into the recognition of the H3K4me3 mark by the TFIID subunit TAF3. Structure 16, 1245–1256 (2008).
Li, H. et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442, 91–95 (2006).
Bian, C. et al. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. 30, 2829–2842 (2011).
Lee, J., Thompson, J. R., Botuyan, M. V. & Mer, G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 15, 109–111 (2008).
Pieters, B., Belle, R. & Mecinović, J. The effect of the length of histone H3K4me3 on recognition by reader proteins. ChemBioChem 14, 2408–2412 (2013).
Zhao, S. et al. Kinetic and high-throughput profiling of epigenetic interactions by 3D-carbene chip-based surface plasmon resonance imaging technology. Proc. Natl Acad. Sci. USA 114, E7245–E7254 (2017).
Beyeh, N. K., Valkonen, A. & Rissanen, K. Encapsulation of tetramethylphosphonium cations. Supramol. Chem. 21, 142–148 (2009).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Case, D. A. et al. AMBER 12, University of California, San Francisco. (2012).
Belle, R. et al. Investigating D-lysine stereochemistry for epigenetic methylation, demethylation and recognition. Chem. Commun. 53, 13264–13267 (2017).
Kar, T. et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 10, 10895 (2020).
Xu, Y. & Meng, X. Molecular simulation elaborating the mechanism of 1β-hydroxy alantolactone inhibiting ubiquitin-conjugating enzyme UbcH5s. Sci. Rep. 10, 141 (2020).
Rose, N. R. et al. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J. Med. Chem. 51, 7053–7056 (2008).
Sakurai, M. et al. A miniaturized screen for inhibitors of Jumonji histone demethylases. Mol. Biosyst. 6, 357–364 (2010).
Khan, A., Schofield, C. J. & Claridge, T. D. W. Reducing Agent-Mediated Nonenzymatic Conversion of 2-Oxoglutarate to Succinate: Implications for Oxygenase Assays. ChemBioChem 21, 2898–2902 (2020).
Couture, J.-F., Collazo, E., Ortiz-Tello, P. A., Brunzelle, J. S. & Trievel, R. C. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat. Struct. Mol. Biol. 14, 689–695 (2007).
Whetstine, J. R. et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125, 467–481 (2006).
Ng, S. S. et al. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature 448, 87–91 (2007).
Horton, J. R. et al. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 17, 38–43 (2010).
Rose, N. R. et al. Plant growth regulator daminozide is a selective inhibitor of human KDM2/7 histone demethylases. J. Med. Chem. 55, 6639–6643 (2012).
Goda, S., Isagawa, T., Chikaoka, Y., Kawamura, T. & Aburatani, H. Control of histone H3 lysine 9 (H3K9) methylation state via cooperative two-step demethylation by jumonji domain containing 1A (JMJD1A) homodimer. J. Biol. Chem. 288, 36948–36956 (2013).
Kamps, J. J. A. G. et al. Cation-π interactions contribute to substrate recognition in γ-butyrobetaine hydroxylase catalysis. Chem. Eur. J. 22, 1270–1276 (2016).
Salonen, L. M. et al. Molecular recognition at the active site of factor Xa: Cation-π Interactions, stacking on planar peptide surfaces, and replacement of structural water. Chem. Eur. J. 18, 213–222 (2012).
Pieters, B. J. G. E. et al. Installation of Trimethyllysine Analogs on Intact Histones via Cysteine Alkylation. Bioconjug. Chem. 30, 952–958 (2019).
Hintzen, J. C. J. et al. Comparison of molecular recognition of trimethyllysine and trimethylthialysine by epigenetic reader proteins. Molecules 25, 1918 (2020).
Vermeulen, M. et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 (2007).
Huang, Y., Fang, J., Bedford, M. T., Zhang, Y. & Xu, R. M. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 312, 748–751 (2006).
Hopkinson, R. J. et al. Is JmjC oxygenase catalysis limited to demethylation? Angew. Chem. Int. Ed. Engl. 52, 7709–7713 (2013).
Walport, L. J. et al. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat. Commun. 7, 11974 (2016).
Roels, J. & Verstraete, W. Biological formation of volatile phosphorus compounds. Bioresour. Technol. 79, 243–250 (2001).
Greaves, J. S. et al. Phosphine gas in the cloud decks of Venus. Nat. Astron 5, 655–664 (2021).
te Velde, G. et al. Chemistry with ADF. J. Comput. Chem. 22, 931–967 (2001).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988).
Rahman, S. et al. Methyl-Induced Polarization Destabilizes the Noncovalent Interactions of N-Methylated Lysines. Chem. Eur. J. 27, 11005–11014 (2021).
Fonseca Guerra, C., van der Wijst, T., Poater, J., Swart, M. & Bickelhaupt, F. M. Adenine versus guanine quartets in aqueous solution: Dispersion-corrected DFT study on the differences in π-stacking and hydrogen-bonding behavior. Theor. Chem. Acc. 125, 245–252 (2010).
Padial, J. S., de Gelder, R., Fonseca Guerra, C., Bickelhaupt, F. M. & Mecinović, J. Stabilisation of 2,6-diarylpyridinium cation by through-space polar-π interactions. Chem. Eur. J. 20, 6268–6271 (2014).
Van Der Wijst, T., Fonseca Guerra, C., Swart, M., Bickelhaupt, F. M. & Lippert, B. A ditopic ion-pair receptor based on stacked nucleobase quartes. Angew. Chem. Int. Ed. 48, 3285–3287 (2009).
Simó Padial, J. et al. Stabilization of 2,6-diarylanilinum cation by through-space cation-π interactions. J. Org. Chem. 82, 9418–9424 (2017).
Baerends, E. J., Gritsenko, O. V. & Van Meer, R. The Kohn-Sham gap, the fundamental gap and the optical gap: The physical meaning of occupied and virtual Kohn-Sham orbital energies. Phys. Chem. Chem. Phys. 15, 16408–16425 (2013).
Bickelhaupt, F. M. & Baerends, E. J. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry. in 1–86 (John Wiley & Sons, Ltd, 2000).
Fonseca Guerra, C., Handgraaf, J. W., Baerends, E. J. & Bickelhaupt, F. M. Voronoi Deformation Density (VDD) Charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD Methods for Charge Analysis. J. Comput. Chem. 25, 189–210 (2004).
Cortopassi, W. A. et al. Cation–π interactions in CREBBP bromodomain inhibition: an electrostatic model for small-molecule binding affinity and selectivity. Org. Biomol. Chem. 14, 10926–10938 (2016).
Bayly, C. I., Cieplak, P., Cornell, W. & Kollman, P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280 (1993).
Wang, H., Fang, J. & Gao, X. The optimal particle-mesh interpolation basis. J. Chem. Phys. 147, 124107 (2017).
Phillips, J. C. et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005).
Acknowledgements
This work was supported by the European Research Council (ERC Starting Grant, ChemEpigen-715691, J.M.), Cancer Research UK Programme Grant (C8717/A18245; C8717/A28285 C.J.S., A.K.), EPSRC (EP/M50659X/1 and EP/L003376/1 R.B., A.K. and EP/L015838/1 J.J.A.G.K.), Clarendon Scholarship (J.J.A.G.K.), the Netherlands Organization for Scientific Research (NWO-ALW, NWO-CW, and NWO-EW, F.M.B.), the Spanish MINECO (PID2019-106830GB-I00 and MDM-2017-0767, J.P.), and a World Bank Education Grant (K.K).
Author information
Authors and Affiliations
Contributions
J.M. and C.J.S conceived and supervised the project. J.J.A.G.K. synthesized Fmoc-KPme3-OH. J.J.A.G.K. and R.B. prepared histone peptides. J.J.A.G.K. and J.M. carried out thermodynamic studies. J.J.A.G.K. and T.D.W.C. carried out NMR experiments. R.B., A.K. and C.J.S. performed KDM-catalysed demethylation assays. J.P. and F.M.B. carried out quantum-chemical analyses. K.K. and R.S.P. performed MD simulations. B.J.G.E.P. and E.S. produced proteins. R.B., J.J.A.G.K., C.J.S. and J.M. wrote the manuscript with contributions from J.P., R.S.P., F.M.B. and A.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Adegboyega Oyelere, Takayoshi Suzuki and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Belle, R., Kamps, J.J.A.G., Poater, J. et al. Reading and erasing of the phosphonium analogue of trimethyllysine by epigenetic proteins. Commun Chem 5, 27 (2022). https://doi.org/10.1038/s42004-022-00640-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00640-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
