
Abstract
Existing synthetic routes for accessing dibenzofuran core have intrinsic regioselectivity, limiting the substitution patterns available in heteropolycyclic arene products. Here we report a double 1,4-conjugate addition/intramolecular annulation cascade reaction between propargylamines and two equivalents of imidazolium methylides that allows efficient access of structurally versatile dibenzofurans. This transition metal-free protocol proceeds smoothly under bench-top air atmosphere and offers easy manipulation of substituents on the dibenzofuran core, and also provides good-to-excellent product yields with good functional group tolerance, particularly the –Br and –Cl groups which are often incompatible with existing metal-catalyzed C–C and/or C–O bond ring-forming processes. It is worth noting that ladder-type π-systems with all-arene quarternary carbon structure can be straightforwardly generated upon simple late-stage functionalization.
Similar content being viewed by others
Introduction
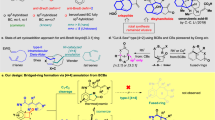
Dibenzofuran-containing heterocycle constitutes an important structural motif in many natural products, pharmaceutically intermediates, and functional materials1,2,3,4,5,6,7. Indeed, these flanked-arenes furans are shown to have particularly appealing characteristics, such as antibiotics, antimalarial, antiallergy, anti-inflammatory, and anticancer activities8,9,10,11. They also express their versatility in photoelectronic materials, especially in phosphorescent organic light-emitting diodes (PhOLEDs)12,13,14, and chemical probes in biological process investigations15,16,17. Given the significance of these unique benzofuran scaffolds, efforts have been made extensively toward the exploration of a new transition metal-catalyzed coupling approach allowing modern access to dibenzofuran with fascinating structural complexity. Representative synthetic strategies in this context are intramolecular ring closure pathways for achieving targeted dibenzofurans, either from a starting material of diaryl ether or ortho-arylphenol, via an aromatic carbon–carbon or carbon–oxygen bond-forming process, respectively (Fig. 1). Recent examples of intramolecular cyclization of non-substituted (X = H) or ortho-substituted aryloxybenzenes (X = halides, CO2H, OH, OTf, BF3K and etc.) were shown to be successful in constructing dibenzofuran framework under palladium18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35, silver36, rhodium37, or gold38 catalyst systems (Fig. 1a). In addition to C−C bond formation protocol, intramolecular O-arylation of ortho-arylphenols also exhibited as an alternative synthetic scheme for making dibenzofuran skeleton (Fig. 1b)39,40,41,42,43,44,45,46,47,48,49.

A brief glance of modern synthetic methods for preparing multiple arene-substituted dibenzofurans. a Recent examples of intramolecular cyclization of non-substituted or ortho-substituted aryloxybenzenes in constructing dibenzofuran framework under transition metals catalyst systems. b Intramolecular O-arylation of ortho-arylphenols for making dibenzofuran skeleton. c Direct electrophilic functionalization of dibenzofuran molecules. d A cascade reaction between propargylic amine and imidazolium methylide to access dibenzofuran skeletons (this work).
Nevertheless, employing expensive transition metal catalysts and associated ligands, and particularly limited direct availability of substrate SM-1 with various substituents, would hamper their generality for synthesizing unsymmetrical dibenzofuran with wanted substitution patterns. Despite the advancement of transition metal-catalyzed strategy, direct functionalization of dibenzofuran molecule is also adoptable (Fig. 1c)50. However, the availability of pattern-desirable starting material SM-2 may not be straightforward, and in principle, the regioselectivity of electrophilic substitution is not favorable at the meta-position of arene to the oxygen atom in dibenzofurans. Recently, impressive synthetic method establishments were disclosed in which they do not require transition metal catalysts, for instance, the diazotization-comprised cyclization of ortho-(aryloxy)aniline promoted by visible light51 or NaNO2/TFA;52,53 the substrate-dependent arylation of phenols with bromonitroarenes;54 the reaction of 2-aryl-3-nitrochromenes with pyridinium ylides;55 and the three-component reaction between 2-hydroxy-β-nitrostyrenes, sulfur ylides, and alkynes for the synthesis of dibenzofuran acrylates56,57.
ortho-Quinone methide (o-QM) is a versatile reagent for a variety of polycycle syntheses58. In particular, the alkyne-containing o-QM, namely alkynyl ortho-quinone methide (o-AQM) displays unique feature of possible exo- or endo-cyclization towards the alkynyl moiety59,60,61,62,63,64,65,66. In continuing our recent interest in investigating complex polycyclic structure assembly67,68 and o-AQM chemistry69,70,71, we are intrigued whether the acyl carbene motif can be doubly incorporated to o-AQM in achieving the densely-substituted arene on one side of dibenzofuran framework.
Herein, we report a cascade reaction between propargylic amines and imidazolium methylides (Fig. 1d). This base-promoted protocol allows modular assembly of dibenzofurans with a complementary substitution pattern, and exhibits good functional group tolerance, particularly tolerating -Br and -Cl groups which offer potential for late-stage product modification via cross-coupling technology. It is worth noting that this method is operationally simple, as the reaction can be conveniently performed under bench-top air atmosphere, and is complementary to frequently Br-group-incompatible transition metal-catalyzed annulative C(Ar)−C(Ar’) and C(Ar)−O bond construction reactions.
Results and discussion
Reaction optimization
Propargylic amine 1a and imidazolium salt 2a were chosen as the prototypical substrates for our initial reaction condition investigations (Table 1). Putting KOt-Bu in the reaction system allowed the formation of desired product 3aa (entries 1–3). Upon comparing imidazolium salt 2a to other similar reaction partners, e.g., 2-quin, 2-iso-quin, 2-p-NMe2py, and 2-py, imidazolium salt 2a gave the best result (entry 2 vs entries 4–7). To our delight, mixing the commonly used bases provided better outcomes, particularly the KOt-Bu/K2CO3 and KOt-Bu/KOH combinations (entries 8–13). Further investigation of the base stoichiometry offered better product yields (entries 14–16). Having the fruitful base combination, we next surveyed the regularly used solvents (entries 17–23). MeCN and DMF were found to be the best solvents of choice (entries 15 and 17). We intended to use MeCN for further screening owing to its operational simplicity. Room temperature conditions did not allow the reaction to proceed (entry 24). Further variation of reaction temperatures revealed that 80 °C is the optimal condition (entries 25–26). Extension of reaction time did not find beneficial to the product yield (entry 28). Thus, the optimal reaction conditions were found to be KOt-Bu/KOH in MeCN at 80 °C for 3 h. It is worthy to note that this cascade reaction proceed-well under bench-top air atmosphere.
Substrate scope
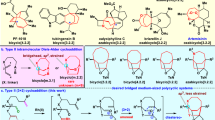
With the optimized reaction conditions in hand, we next investigated the scope of propargylic amine with various substituents (Fig. 2). In general, the desired dibenzofurans were delivered in good-to-excellent yields. No significant electronic effect of the alkynyl arene moiety at the propargylic scaffold was found (products 3da, 3ea, 3fa, and 3ga). Similarly, the electronically-varied substituents (e.g., –Me and –Cl) at the para-position of the phenolic moiety did not affect the product yields (products 3ea vs 3ha). Structure of 3ha was unambiguously characterized by single-crystal X-ray crystallography (see Supplementary Fig. 1 and Table 1, Supplementary Data 1 for CIF file). The –Br substituent at para- and ortho-position of the phenolic arene unit remained intact during the course of the reaction (products 3la, 3ma, 3na, and 3oa). This –Br group compatibility is of high attractiveness as this group can be further straightforwardly functionalized using established cross-coupling technology. In addition to halo-substituents, the steric bulky tert-butyl group at the ortho-phenolic-position was found to be tolerable towards the ring-forming process (product 3pa). Cyclohexenyl and thienyl units remained untouched in this cascade annulation and furnished the expected products 3ra and 3sa in 78% and 73% yields, respectively.

Reaction conditions: 1 (0.2 mmol), 2a (0.4 mmol), KOt-Bu (0.2 mmol), KOH (0.4 mmol) were stirred in acetonitrile (2 mL) at 80 °C for 3 h under bench-top air atmosphere. Isolated yields were reported. Isolated yield of 10-fold scale in parenthesis.
The scope of the reaction was further tested using various benzoyl imidazolium methylide derivatives (Fig. 3). Essentially no steric influence of the arene at 2 was found and the desired products were afforded in good yields (products 3ab, 3ac, 3ad, and 3ae). Substrates bearing halo groups, e.g., fluoro, chloro, and bromo were successfully employed in this reaction, yielding the resulting products 3ag-3al in 77% to 90% yields. Highly electron-withdrawing substituent –CF3 was compatible, affording the corresponding products 3am. With appropriate imidazolium methylide, the π-extended structure was able to be constructed in 92% yield (product 3an).

Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), KOt-Bu (0.2 mmol), KOH (0.4 mmol) were stirred in acetonitrile (2 mL) at 80 °C for 3 h under bench-top air atmosphere. Isolated yields were reported.
Mechanistic studies
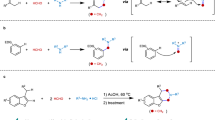
A competition-study between two electronically different imidazolium methylides 2f and 2m were performed (Fig. 4). Two main products 3afm and 3am were isolated in 32% and 36% yields, respectively. This experiment clearly reflected that the electronically poor methylide 2m underwent deprotonation faster and thus subsequent 1,4-conjugate addition to o-AQM. The product of 3af was not detected essentially that further added to this mechanistic proposal.

A competition-study between two electronically different imidazolium methylides 2f and 2m were performed.
A plausible reaction mechanism is proposed in Fig. 5. The alkynyl o-quinone methide (o-AQM) intermediate is generated upon releasing of piperidine via intramolecular 1,5-H shift under basic conditions58. Subsequent 1,4-conjugate addition of imidazolium methylide 2 furnishes adduct Int-A. Intramolecular 5-exo-dig annulation of Int-A gives intermediate Int-B, which subsequently forms Int-C via a facile β-H elimination. The second 1,4-conjugate addition of another molecule of 2 delivers Int-D, and then converts to Int-E under an elimination process. The Int-F is formed upon deprotonation and generates Int-G via a second annulation step. Rearomatization of Int-G finally furnishes product 3.

A hypothetical reaction mechanism for the formation of multiple arene-substituted dibenzofurans is presented.
Synthetic transformations and applications
Pentacene has been successful in advancing organic material applications72. The high charge carrier mobility of acenes is believed to be originated from the highly ordered π-π-interaction stacking between adjacent molecules73. Nevertheless, the stability of this pentacene towards practical application still has room for improvement. Recently, the ladder-type heteroacenes was reported to have both high charge mobility and stability under ambient conditions74. With regard to this impressive finding, we are interested to devise an investigation for assembling fluorene-containing spiro-ladder-type heteroacenes75. The all-arene quaternary carbon unit would be useful for easy management of distinctive spatial arrangement of the π-system for desirable mobility-tuning76,77. To our delight, the initial attempt of using superacid-promoted synthetic strategy78,79 was successful in delivering a spiro-ring-fused system (Fig. 6). In the presence of TfOH in toluene at room temperature, a series of spiro-ladder-type molecules 4aa, 4ca, and 4ia were able to be obtained in 91%, 94%, and 93% yields, respectively (Fig. 6). It is interesting to show that toluene served as both reagent and solvent for this transformation. Conversely, upon alternating the solvent to dichloromethane, the serendipitous products 5aa, 5af, 5ba, and 5oa were formed (the tertiary alcohol product 5aa was unambiguously confirmed by single-crystal X-ray crystallography, see Supplementary Fig. 2 and Table 2, Supplementary Data 2 for CIF file). These products indeed offer high opportunity for further functionalization via a simple –OH group transformation80,81, and thus allows rich entities for new material investigations.

Reaction conditions: dibenzofurans 3 (0.2 mmol), TfOH (0.6 mmol) were stirred in toluene (2 mL, synthesis of compound 4 for 1 h) or in DCM (2 mL, synthesis of compound 5 for 2 h) at room temperature. Isolated yields were reported.
In conclusion, we have succeeded in showing a doubly 1,4-conjugate addition/annulation cascade process for facile access of structurally versatile dibenzofurans. This transition metal-free protocol proceeds smoothly under bench-top air atmosphere and realizes easy manipulation of substituents on one of the flanked-arenes of dibenzofuran core. The present modular method exhibits good product framework diversity and complexity, decent product yields, and allows good functional group tolerance, particularly the –Br and –Cl groups where they are often found impermissible in existing Pd-catalyzed aromatic C–C or C–O bond ring-forming processes. It is worthy to note that materially attractive spiro-ladder-type π-system with all-arene quaternary carbon feature can also be simply attained via a one-step subsequent functionalization.
Methods
General procedures for the synthesis of propargylamines 1
To a 25 mL round-bottom flask equipped with a magnetic stir bar were added pyrrolidine (1.2 mmol), aldehyde (1.0 mmol), acetylene (1.2 mmol), copper (I) iodide (10 mol%), and toluene (3 mL). The mixture was degassed and backfilled with nitrogen, and then stirred in an oil bath preheated to 100 °C for 5 h (monitored by TLC). After the reaction completed (as determined using TLC), the reaction mixture was cooled to room temperature, diluted with CH2Cl2 (10 mL), and filtered through a thin pad of silica gel. The filter cake was washed with CH2Cl2, and the combined filtrate was concentrated in a vacuum. The crude product was purified by flash column chromatography on silica gel to afford the corresponding propargylamines.
General procedure for the synthesis of imidazolium methylides 2
1-Methyl-1H-imidazole (410 mg, 5.0 mmol) was added to 2-bromoethanones derivatives (5.0 mmol) in dry acetonitrile (5 mL). After stirring for 2 to 5 h at room temperature, the precipitate formed was filtered off and washed with acetonitrile to afford the desired imidazolium methylides 2, which can be used to next reaction without further purification. Other ylides used were synthesized according to these procedures.
General procedures for the synthesis of dibenzofurans 3
A mixture of propargylamines 1 (0.2 mmol), imidazolium methylides 2 (0.4 mmol), potassium t-butoxide (0.2 mmol), and potassium hydroxide (0.4 mmol) were added to a resealable screw-capped Schlenk tube under air atmosphere. Acetonitrile (2 mL) was then added. The tube sealed with a Teflon-coated cap and the resulting mixture was stirred in an oil bath preheated to 80 °C for 3 h (monitored by TLC). Upon completion of the reaction, the reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure. The residue was purified using flash column chromatography with a silica gel (200–300 mesh), using ethyl acetate and petroleum ether (1:20, v/v) as the elution solvent to give desired products 3. For NMR spectra, see Supplementary Figures 3–72).
General procedure for the competition experiment (synthesis of 3afm, and 3am)
A mixture of 2-(3-phenyl-1-(pyrrolidin-1-yl)prop-2-yn-1-yl)phenol (1a) (0.2 mmol, 0.56 g), 3-(2-(4-methoxyphenyl)-2-oxoethyl)-1-methyl-1H-imidazol-3-ium bromide (2 f) (0.4 mmol), 1-methyl-3-(2-oxo-2-(4-(trifluoromethyl)phenyl)ethyl)-1H-imidazol-3-ium bromide (2m) (0.4 mmol), potassium t-butoxide (0.2 mmol, 0.22 g), and potassium hydroxide (0.4 mmol, 0.22 g) were added to a resealable screw-capped Schlenk tube. Acetonitrile (3 mL) was then added. The tube sealed with a Teflon-coated cap and the resulting mixture was stirred in an oil bath preheated to 80 °C for 3 h (monitored by TLC). Upon completion of the reaction, the reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure. The residue was purified using flash column chromatography with a silica gel (200-300 mesh), using ethyl acetate and petroleum ether (1:20, v/v) as the elution solvent to give desired product 3fm and 3am in 32% and 36% yield, respectively. For NMR spectra, see Supplementary Figures 73, 74).
10-Fold scale synthesis of compound 3aa
A mixture of 2-(3-phenyl-1-(pyrrolidin-1-yl)prop-2-yn-1-yl)phenol (1a) (2.0 mmol, 0.56 g), 1-methyl-3-(2-oxo-2-phenylethyl)-1H-imidazol-3-ium bromide (2a) (4.0 mmol, 1.12 g), potassium t-butoxide (2.0 mmol, 0.22 g), and potassium hydroxide (4.0 mmol, 0.22 g) were added to a resealable screw-capped Schlenk tube. Acetonitrile (10 mL) was then added. The tube sealed with a Teflon-coated cap and the resulting mixture was stirred in an oil bath preheated to 80 °C for 3 h (monitored by TLC). Upon completion of the reaction, the reaction mixture was cooled to room temperature, and the solvent was removed under reduced pressure. The residue was purified using flash column chromatography with a silica gel (200–300 mesh), using ethyl acetate and petroleum ether (1:20, v/v) as the elution solvent to give desired product 3aa in 75% yield.
General procedures for the synthesis of compound 4
Dibenzofuran 3 (0.2 mmol) was mixed with TfOH (0.6 mmol) in a round bottom flask. Toluene (2 mL) was then added. The resulting mixture was stirred at room temperature (25 °C) for 1 h (monitored by TLC). Upon completion of the reaction, the solvent was removed under reduced pressure. The residue was purified using flash column chromatography with a silica gel (200–300 mesh), using ethyl acetate and petroleum ether as the elution solvent to give desired product 4. For NMR spectra, see Supplementary Figures 75–80).
General procedures for the synthesis of compound 5
Dibenzofuran 3 (0.2 mmol) was mixed with TfOH (0.6 mmol) in a round bottom flask. Dichloromethane (2 mL) was then added. The resulting mixture was stirred at room temperature (25 °C) for 2 h (monitored by TLC). Upon completion of the reaction, the solvent was removed under reduced pressure. The residue was purified using flash column chromatography with a silica gel (200–300 mesh), using ethyl acetate and petroleum ether as the elution solvent to give desired product 5. For NMR spectra, see Supplementary Figures 81–88).
Data availability
The data sets generated and analyzed during the current study are included in the Supplementary Information File and also available from the corresponding authors on request. For full methods, see Supplementary Methods. For 1H NMR, and 13C NMR spectra see Supplementary Figs. 3–88 and the GC-MS spectra for mechanistic investigation see Supplementary Figs. 89–95. For X-ray crystallographic figures, see Supplementary Fig. 1 for compound 3ha and Fig. 2 for compound 5aa. The X-ray crystallographic coordinates for structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2022149 (3ha – Supplementary Data 1) and 2022150 (5aa – Supplementary Data 2). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The coordinates for the corresponding structures are available in Supplementary Tables 1 and 2.
References
Millot, M., Dieu, A. & Tomasi, S. Dibenzofurans and derivatives from lichens and ascomycetes. Nat. Prod. Rep. 33, 801–811 (2016).
Luzina, O. A. & Salakhutdinov, N. F. Usnic acid and its derivatives for pharmaceutical use: a patent review (2000-2017). Expert. Opin. Ther. Pat. 28, 477–491 (2018).
Tsuji, H. & Nakamura, E. Design and functions of semiconducting fused polycyclic furans for optoelectronic applications. Acc. Chem. Rev. 50, 396–406 (2017).
Paul, K., Jalal, S., Kundal, S. & Jana, U. Synthesis of fused dibenzofuran derivatives via palladium-catalyzed domino C–C bond formation and iron-catalyzed cycloisomerization/aromatization. J. Org. Chem. 81, 1164–1174 (2016).
Andernach, L. et al. Terphenyl derivatives from allantophomopsis lycopodina. J. Nat. Prod. 79, 2718–2725 (2016).
Ebrahim, H. Y., Akl, M. R., Elsayed, H. E., Hill, R. A. & Sayed, K. A. E. Usnic acid benzylidene analogues as potent mechanistic target of rapamycin inhibitors for the control of breast malignancies. J. Nat. Prod. 80, 932–952 (2017).
Love, B. E. Isolation and synthesis of polyoxygenated dibenzofurans possessing biological activity. Eur. J. Med. Chem. 97, 377–387 (2015).
De Lombaert, S. et al. Potent and Selective Non-Peptidic Inhibitors of Endothelin-Converting Enzyme-1 with Sustained Duration of Action. J. Med. Chem. 43, 488–504 (2000).
Ye, Y. Q. et al. Structural elucidation and synthesis of vialinin C, a new inhibitor of TNF-α production. Bioorg. Med. Chem. 22, 2442–2446 (2014).
Beekman, A. M. & Barrow, R. A. Syntheses of the fungal metabolites Boletopsins 7, 11, and 12 from the Papua new guinea medicinal mushroom Boletopsis sp. J. Org. Chem. 79, 1017–1024 (2014).
Beekman, A. M., Wossa, S. W., Kevo, O., Ma, P. & Barrow, R. A. Discovery and synthesis of Boletopsins 13 and 14, brominated fungal metabolites of terrestrial origin. J. Nat. Prod. 78, 2133–2135 (2015).
Zhang, L. et al. Highly efficient blue phosphorescent organic light-emitting diodes employing a host material with small bandgap. ACS Appl. Mater. Interfaces 8, 16186–16191 (2016).
Deng, L., Li, J., Wang, G.-X. & Wu, L.-Z. Simple bipolar host materials incorporating CN group for highly efficient blue electrophosphorescence with slow efficiency roll-off. J. Meter Chem. 1, 8140–8145 (2013).
Van Dijken, A. et al. Carbazole compounds as host materials for triplet emitters in organic light-emitting diodes: polymer hosts for high-efficiency light-emitting diodes. J. Am. Chem. Soc. 126, 7718–7727 (2004).
Berndt, F. et al. 7-Amino-dibenzofuran-3-carboxylate: a new probe for femtosecond dynamic microsolvation studies of biomolecules. J. Photochem. Photobiol. A 234, 164–170 (2012).
Hagimori, M. et al. Synthesis of radioiodinated probes targeted toward matrix metalloproteinase-12. Bioorg. Med. Chem. Lett. 28, 193–195 (2018).
Butsch, V. et al. Design, (Radio)synthesis, and in vitro and in vivo evaluation of highly selective and potent matrix metalloproteinase 12 (MMP-12) inhibitors as radiotracers for positron emission tomography. J. Med. Chem. 61, 4115–4134 (2018).
Shiotani, A. & Itatani, H. Dibenzofurans by intramolecular ring closure reactions. Angew. Chem. Int. Ed. 13, 471–472 (1974).
Shiotani, A. & Itatani, H. Palladium-catalysed dibenzofuran synthesis by dehydrogenative ring closure. J. Chem. Soc. Perkin Trans. 1, 1236–1241 (1976).
Ames, D. E. & Opalko, A. Synthesis of dibenzofurans by palladium-catalysed intramolecular dehydrobromination of 2-Bromophenyl phenyl ethers. Synthesis 1983, 234–235 (1983).
Ames, D. E. & Opalko, A. Palladium-catalysed cyclisation of 2-substituted halogenoarenes by dehydrohalogenation. Tetrahedron 40, 1919–1925 (1984).
Liu, Z. & Larock, R. C. Synthesis of carbazoles and dibenzofurans via cross-coupling of o-Iodoanilines and o-Iodophenols with silylaryl triflates. Org. Lett. 6, 3739–3741 (2004).
Campeau, L.-C., Parisien, M., Jean, A. & Fagnou, K. Catalytic direct arylation with aryl chlorides, bromides, and iodides: intramolecular studies leading to new intermolecular reactions. J. Am. Chem. Soc. 128, 581–590 (2006).
Liu, Z. & Larock, R. C. Synthesis of carbazoles and dibenzofurans via cross-coupling of o-Iodoanilines and o-Iodophenols with silylaryl triflates and subsequent Pd-catalyzed cyclization. Tetrahedron 63, 347–355 (2007).
Liégault, B., Lee, D., Huestis, M. P., Stuart, D. R. & Fagnou, K. Intramolecular Pd(II)-catalyzed oxidative biaryl synthesis under air: reaction development and scope. J. Org. Chem. 73, 5022–5028 (2008).
Xu, H. & Fan, L.-L. Synthesis of dibenzofurans directly from aryl halides and ortho-Bromophenols via one-pot consecutive SNAr and intramolecular palladium-catalyzed aryl–aryl coupling reactions. Chem. Pharm. Bull. 56, 1496–1498 (2008).
Wang, C., Piel, I. & Glorius, F. Palladium-catalyzed intramolecular direct arylation of benzoic acids by tandem decarboxylation/C−H activation. J. Am. Chem. Soc. 131, 4194–4195 (2009).
Du, Z., Zhou, J., Si, C. & Ma, W. Synthesis of dibenzofurans by palladium-catalysed tandem denitrification/C-H activation. Synlett 20, 3023–3025 (2011).
Nervig, C. S., Waller, P. J. & Kalyani, D. Palladium-catalyzed intramolecular C–H arylation of arenes using tosylates and mesylates as electrophiles. Org. Lett. 14, 4838–4841 (2012).
Shen, Z., Ni, Z., Mo, S., Wang, J. & Zhu, Y. Palladium-catalyzed intramolecular decarboxylative coupling of arene carboxylic acids/esters with aryl bromides. Chem. –Eur. J. 18, 4859–4865 (2012).
Niu, L., Yang, H., Jiang, Y. & Fu, H. Efficient synthesis of dibenzoxaborininols from diaryl ethers and their application to dibenzofuran synthesis. Adv. Synth. Catal. 355, 3625–3632 (2013).
Ferguson, D. M., Rudolph, S. R. & Kalyani, D. Palladium-catalyzed intra- and intermolecular C–H arylation using mesylates: synthetic scope and mechanistic studies. ACS Catal. 4, 2395–2401 (2014).
Panda, N., Mattan, I. & Nayak, D. K. Synthesis of dibenzofurans via C–H activation of o-Iodo diaryl ethers. J. Org. Chem. 80, 6590–6597 (2015).
Okita, T. et al. Dibenzofuran synthesis: decarbonylative intramolecular C−H arylation of aromatic esters. Asian J. Org. Chem. 7, 1358–1361 (2018).
Asahara, K. K. et al. Pd-catalyzed denitrative intramolecular C–H arylation. Org. Lett. 21, 4721–4724 (2019).
Lockner, J. W., Dixon, D. D., Risgaard, R. & Baran, P. S. Practical radical cyclizations with arylboronic acids and trifluoroborates. Org. Lett. 13, 5628–5631 (2011).
Maetani, S., Fukuyama, T. & Ryu, I. Rhodium-catalyzed decarbonylative C–H arylation of 2-Aryloxybenzoic acids leading to dibenzofuran derivatives. Org. Lett. 15, 2754–2757 (2013).
Corrie, T. J. A., Ball, L. T., Russell, C. A. & Lloyd-Jones, G. C. Au-catalyzed biaryl coupling to generate 5- to 9-membered rings: turnover-limiting reductive elimination versus π-complexation. J. Am. Chem. Soc. 139, 245–254 (2017).
Kawaguchi, K., Nakano, K. & Nozaki, K. Synthesis of ladder-type π-conjugated heteroacenes via palladium-catalyzed double N-arylation and intramolecular O-arylation. J. Org. Chem. 72, 5119–5128 (2007).
Liu, J., Fitzgerald, A. E. & Mani, N. S. Facile assembly of fused Benzo[4,5]furo heterocycles. J. Org. Chem. 73, 2951–2954 (2008).
Xiao, B. et al. Synthesis of dibenzofurans via palladium-catalyzed phenol-directed C–H activation/C–O cyclization. J. Am. Chem. Soc. 133, 9250–9253 (2011).
Wei, Y. & Yoshikai, N. Oxidative cyclization of 2-arylphenols to dibenzofurans under Pd(II)/Peroxybenzoate catalysis. Org. Lett. 13, 5504–5507 (2011).
Zhao, J., Wang, Y., He, Y., Liu, L. & Zhu, Q. Cu-catalyzed oxidative C(sp2)–H cycloetherification of o-Arylphenols for the preparation of dibenzofurans. Org. Lett. 14, 1078–1081 (2012).
Zhao, J. et al. CuI-mediated sequential iodination/cycloetherification of o-arylphenols: synthesis of 2- or 4-Iododibenzofurans and mechanistic studies. Org. Lett. 14, 5362–5365 (2012).
Zhao, H. et al. A one-pot synthesis of dibenzofurans from 6-Diazo-2-cyclohexenones. Org. Lett. 17, 5744–5747 (2015).
Solórzano, P. C., Brigante, F., Pierini, A. B. & Jimenez, L. B. Photoinduced synthesis of dibenzofurans: intramolecular and intermolecular comparative methodologies. J. Org. Chem. 83, 7867–7877 (2018).
Li, J., Xu, Q.-N., Wang, Z.-B., Li, Y. & Liu, L. Synthesis of dibenzofurans from cyclic diaryliodonium triflates and water via oxygen–iodine exchange approach. ACS Omega 3, 12923–12929 (2018).
Janardhanan, J. C. et al. Synthesis of hybrid polycycles containing fused hydroxy benzofuran and 1H-indazoles via a domino cyclization reaction. New. J. Chem. 43, 10166–10175 (2019).
Sumida, Y. et al. Synthesis of Dibenzofurans by Cu-Catalyzed Deborylative Ring Contraction of Dibenzoxaborins. Org. Lett. 22, 6687–6691 (2020).
Smith, M. B. & March, J. Eds., March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structures, 6th Ed. John Wiley & Sons (2006).
Cho, J. Y., Roh, G.-B. & Cho, E. J. Visible-light-promoted synthesis of dibenzofuran derivatives. J. Org. Chem. 83, 805–811 (2018).
Kumar, A., Sattar, M., Verma, A., Dara, A. & Kumar, S. Double functionalization of 2-amino-2′-hydroxy-1,1′-biaryls: synthesis of 4-nitro-dibenzofurans and benzofuro-indoles. RSC Adv. 5, 44728–44741 (2015).
Sanz, R., Fernández, Y., Castroviejo, M. P., Pérez, A. & Fañanás, F. J. A route to regioselectively functionalized carbazoles, dibenzofurans, and dibenzothiophenes through anionic cyclization of Benzyne-tethered aryllithiums. J. Org. Chem. 71, 6291–6294 (2006).
Kumar, A. et al. Chemoselective arylation of phenols with bromo-nitroarenes: synthesis of nitro-biaryl-ols and their conversion into benzofurans and carbazoles. Chem. Commun. 50, 9481–9484 (2014).
Sun, J., Jiang, W. & Yan, C.-G. Convenient construction of dibenzo[b,d]furanes and 2,6-diaryl-4-(2-hydroxyphenyl)pyridines via domino reaction of pyridinium ylides with 2-aryl-3-nitrochromenes. Org. Chem. Front. 6, 1428–1432 (2019).
Shao, J. et al. Sulfur Ylide initiated [4 + 1]/[4 + 2] annulation reactions: a one-pot approach to dibenzofuran acrylate derivatives. Org. Lett. 21, 6370–6373 (2019).
Chen, W. et al. Efficient access to fluorescent benzofuro[3,2-b]carbazoles via TFA-promoted cascade annulations of sulfur ylides, 2-hydroxy-β-nitrostyrenes and indoles. Org. Chem. Front. 7, 873–878 (2020).
Bai, W.-J. et al. The domestication of ortho-Quinone methides. Acc. Chem. Res. 47, 3655–3664 (2014). and references therein.
Uyanik, M., Nishioka, K., Kondo, R. & Ishihara, K. Chemoselective oxidative generation of ortho-quinone methides and tandem transformations. Nat. Chem. 12, 353–362 (2020).
Zielke, K. & Waser, M. Formal (4 + 1)-addition of allenoates to o-Quinone methides. Org. Lett. 20, 768–771 (2018).
Du, J.-Y. et al. Metal-free one-pot synthesis of 3-phosphinoylbenzofurans via phospha-michael addition/cyclization of H-phosphine oxides and in situ generated ortho-Quinone methides. Org. Lett. 20, 477–480 (2018).
He, X. et al. FeCl3-promoted tandem 1,4-conjugate addition/6-endo-dig cyclization/oxidation of propargylamines and benzoylacetonitriles/malononitriles: direct access to functionalized 2-aryl-4H-chromenes. Org. Biomol. Chem. 16, 7191–7202 (2018).
Yu, X.-Y. et al. Catalytic asymmetric cycloaddition of in situ‐generated ortho‐Quinone methides and azlactones by a triple Brønsted acid activation strategy. Chem. –Eur. J. 22, 6774–6778 (2016).
Ma, J. et al. Dual catalysis: proton/metal-catalyzed tandem benzofuran annulation/carbene transfer reaction. Org. Lett. 18, 1322–1325 (2016).
Saha, S. & Schneider, C. Directing group assisted nucleophilic substitution of propargylic alcohols via o-Quinone Methide intermediates: Brønsted acid catalyzed, highly enantio- and diastereoselective synthesis of 7-Alkynyl-12a-acetamido-Substituted Benzoxanthenes. Org. Lett. 17, 648–651 (2015).
Li, Q. et al. Base‐mediated 1,4‐conjugate addition/intramolecular 5‐exo‐dig annulation of propargylamines with benzoylacetonitriles and β‐Keto esters for polysubstituted Furans and Furo[3,4‐c]coumarins formation. Adv. Synth. Catal. 361, 1874–1886 (2019).
Zhao, Q., Fu, W. C. & Kwong, F. Y. Palladium‐catalyzed regioselective aromatic extension of internal alkynes through a norbornene‐controlled reaction sequence. Angew. Chem., Int. Ed. 57, 3381–3385 (2018).
Li, M. & Kwong, F. Y. Cobalt‐catalyzed tandem C−H activation/C−C Cleavage/C−H cyclization of aromatic amides with alkylidenecyclopropanes. Angew. Chem., Int. Ed. 57, 6512–6516 (2018).
He, X. et al. Organocatalytic approach for assembling flavanones via a cascade 1,4-conjugate addition/oxa-Michael addition between propargylamines with water. Org. Lett. 22, 4306–4310 (2020).
He, X. et al. DMAP-catalyzed annulation approach for modular assembly of furan-fused chromenes. Org. Lett. 22, 9444–9449 (2020).
He, X. et al. A ZnI2-catalyzed regioselective cascade 1,4-conjugate addition/5-exo-dig annulation pathway for one-pot access to heterobiaryl frameworks. Chem. Commun. 55, 15069–15072 (2019).
Klauk, H. Organic Electronics: Materials, Manufacturing and Applications, Wiley-VCH: Weinheim, Germany (2006).
Cornil, J., Beljonne, D., Calbert, J.-P. & Brédas, J.-L. Interchain interactions in organic π‐conjugated materials: impact on electronic structure, optical response, and charge transport. Adv. Mater. 13, 1053–1067 (2001).
Saragi, T. P., Spehr, T., Siebert, A., Fuhrmann-Lieker, T. & Salbeck, J. Spiro compounds for organic optoelectronics. Chem. Rev. 107, 1011–1065 (2007).
Fu, W. C. & Kwong, F. Y. A denitrogenative palladium-catalyzed cascade for regioselective synthesis of fluorenes. Chem. Sci. 11, 1411–1417 (2020).
Ye, S. et al. Solution-processed solid solution of a novel carbazole derivative for high-performance blue phosphorescent organic light-emitting diodes. Adv. Mater. 22, 4167–4171 (2010).
Dong, Q. et al. Novel spirofluorene/indole/carbazole-based hole transport materials with high triplet energy for efficient green phosphorescent organic light-emitting diodes. Dyes Pigm. 137, 84–90 (2017).
Wong, K.-T., Wang, Z.-J., Chien, Y.-Y. & Wang, C.-L. Synthesis and properties of 9,9-diarylfluorene-based triaryldiamines. Org. Lett. 3, 2285–2288 (2001).
Ohta, T., Shudo, K. & Okamoto, T. Reaction of triphenylmethyl cation in trifluoromethanesulfonic acid. Reaction of carbodications. Tetrahedron Lett. 24, 71–74 (1983).
Zhao, W., Wang, Z., Chu, B. & Sun, J. Enantioselective formation of all‐carbon quaternary stereocenters from indoles and tertiary alcohols bearing a directing group. Angew. Chem. Int. Ed. 54, 1910–1913 (2015).
Zhou, C. et al. A Sc(OTf)3 catalyzed dehydrogenative reaction of electron-rich (hetero)aryl nucleophiles with 9-aryl-fluoren-9-ols. Org. Biomol. Chem. 17, 9615–9619 (2019).
Acknowledgements
We thank the Anhui Provincial Natural Science Foundation (No. 1808085MB41), the National Natural Science Foundation of China (No. 21772001), Cultivation Project for University Outstanding Talents of Anhui Province (2019), the Research Grants Council of Hong Kong (GRF: 14304519-19P), the CUHK Direct Fund (4442353) and Guangdong Basic and Applied Basic Research Foundation (No. 2019A1515011357) for financial support. We are thankful for financial support from the Innovation and Technology Commission (HKSAR, China) to the State Key Laboratory of Synthetic Chemistry.
Author information
Authors and Affiliations
Contributions
F.Y.K. and X.H. conceived and supervised the project. X.H., R.L., and M.X. designed and performed the experiments. R.L. and P.Y.C. carried out the data analysis. J.D. and Q.T. prepared the supporting information. Y.S. participated in the scientific discussions. P.Y.C. and F.Y.K. wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, X., Li, R., Choy, P.Y. et al. A cascade double 1,4-addition/intramolecular annulation strategy for expeditious assembly of unsymmetrical dibenzofurans. Commun Chem 4, 42 (2021). https://doi.org/10.1038/s42004-021-00478-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-021-00478-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
