
Abstract
A prevailing scenario of the origin of life postulates thioesters as key intermediates in protometabolism, but there is no experimental support for the prebiotic CO2 fixation routes to thioesters. Here we demonstrate that, under a simulated geoelectrochemical condition in primordial ocean hydrothermal systems (–0.6 to –1.0 V versus the standard hydrogen electrode), nickel sulfide (NiS) gradually reduces to Ni0, while accumulating surface-bound carbon monoxide (CO) due to CO2 electroreduction. The resultant partially reduced NiS realizes thioester (S-methyl thioacetate) formation from CO and methanethiol even at room temperature and neutral pH with the yield up to 35% based on CO. This thioester formation is not inhibited, or even improved, by 50:50 coprecipitation of NiS with FeS or CoS (the maximum yields; 27 or 56%, respectively). Such a simple thioester synthesis likely occurred in Hadean deep-sea vent environments, setting a stage for the autotrophic origin of life.
Similar content being viewed by others


Introduction
Thioesters occupy the central position in cellular metabolism as exemplified by acetyl-CoA. This fact has suggested thioesters to be the crucial components of prebiotic precursor of metabolism since as early as 1960’s1,2,3. A key enzyme in thioester biosynthesis is carbon monoxide dehydrogenase/acetyl-CoA synthase (CODH/ACS), in which the Ni-based active centers reduce CO2 to CO, react CO with a methyl group to form acetyl, and acetylate CoA to form acetyl-CoA. This apparently simple process with pivotal Ni has evoked the idea that a prebiotic CO2 fixation on a (Fe,Ni)S mineral preceded the CODH/ACS-based reaction4,5.
However, experimental support of this scenario remains limited. The CO2-to-CO conversion is endergonic even with H2 as the reductant (Supplementary Fig. 1), so biological CO production uses either electron bifurcation or a chemiosmotic pH gradient across the cell membrane to overcome the energy shortage. In the ACS catalytic cycle, the active Ni site serves as both the electron donor and accepter through changing its oxidation states between +1 and +3, facilitating both the reduction and oxidation intermediate steps6. Such redox bifunctionality of Ni has never been observed in natural Ni-bearing minerals. Although Huber and Wächtershäuser demonstrated S-methyl thioacetate (MTA) synthesis from CO and methanethiol in the presence of NiS as a potential prebiotic precursor of the ACS reaction7, the yield was very low (0.2% based on the initial amount of CO) even under their optimum condition (pH 1.6 and 100 °C), which is not so common in nature. A recent work suggested a one-pot pyruvate synthesis from CO2 and H2 on magnetite (Fe3O4) and awaruite (Ni3Fe) as a protometabolic acetyl-CoA pathway8. The proposed reaction, however, provides no explanation for the origin and antiquity of thioester-dependent metabolism. Thioesters might have played central roles in protometabolism not only as carbon sources but also as energy currencies in a manner analogous to ATP1,2,9, serving as an entry point of phosphate into metabolism5,10.
How were CO2-to-CO reduction and CO conversion realized under primordially realistic conditions? A clue lies in an on-site observation by Yamamoto et al.11 of electricity generation in deep-sea hydrothermal vent chimneys and mineral deposits. The geoelectricity arises from the redox coupling between hydrothermal fluid chemicals and seawater-dissolved species via electrically conducting yet thermally insulating sulfide rocks (Supplementary Fig. 1)12,13. Considering cool (0–50 °C) and slightly acidic (pH 6–7) character of the ancient seawater14 together with pH and temperature dependences of the H+/H2 and CO2/CO redox potentials, H2-rich alkaline hydrothermal systems must have readily provided negative electric potentials favorable for the CO2-to-CO reduction at the chimney-ancient seawater interface. We previously demonstrated efficient CO2 electroreduction to CO on certain metal sulfide catalysts (for example, cadmium sulfide) under a simulated early ocean geoelectrochemical condition15. It was later found that FeS undergoes day-scale electroreduction to Fe0 16. The resultant FeS-Fe0 assemblage, named FeS_PERM (FeS partially electroreduced to metal), showed the exceptional capability of promoting various prebiotically important reactions owing to the synergy between surface Fe sites with different oxidation states (Fe2+ and Fe0)17,18.
In our previous works15,16, NiS exhibited neither CO evolution nor Ni0 formation detectable by X-ray diffraction (XRD) analysis. However, our further investigation presented below found non-crystalline growth of Ni0 under geochemically feasible potential conditions, with a substantial amount of CO bound on the resultant surface Ni0 sites. We will also report here that the partially reduced NiS, that is, NiS_PERM realizes efficient conversion of CO and methanethiol to MTA even at room temperature and neutral pH. Notice that the accumulation of surface-bound CO during CO2 electroreduction is generally recognized as poisoning of surface (electro)catalytic activity19,20, but our results suggest that the CO accumulation process on NiS_PERM was rather a crucial step for subsequent primordial thioester formation in early ocean hydrothermal systems. Thioesters are versatile compounds in organic chemical synthesis as well as in biosynthesis, enabling diverse coupling reactions, including C–C bond formation, esterification, and amide bond formation owing to the activated acyl unit21,22,23. To the best of our knowledge, our study provides the first experimental demonstration of thioester synthesis from CO and thiol under mild aqueous conditions. Thus, besides rendering a realistic support for autotrophic scenarios of the origin of life1,2,3,4,5, our demonstrated CO-thiol reaction on NiS_PERM could be a practical approach to CO utilization24. In the following and in Supplementary Information, we also present the results for FeS and CoS and their influences on the NiS’s capabilities for CO production and MTA synthesis.
Results
Metal sulfide electroreduction
We prepared metal sulfides, including NiS by simply mixing an aqueous solution of the corresponding metal chloride and an aqueous solution of sodium sulfide. The obtained sulfides were exposed to a constant electric potential (–0.5 to –1.0 VSHE (volt versus the standard hydrogen electrode)) for 7 days in 100 mM NaCl at room temperature (25 ± 2 °C) (Supplementary Figs. 2 and 3). The solution pH was maintained at 6 ± 0.25 by continuous CO2 bubbling. The electrolyzed sulfides were then separated from the supernatant solution, dried under vacuum, and used for the subsequent experiments.
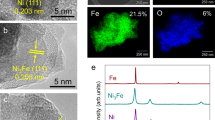
The NiS consists of aggregated nanoparticles with an average particle diameter of 15 ± 11 nm (Supplementary Fig. 6). Although no significant morphological change was discerned (Supplementary Fig. 6), exposure at –0.5 VSHE led to the desulfurization of NiS to heazlewoodite (Ni3S2) (Fig. 1a), consistent with thermodynamic calculation (Fig. 1b). Further reduction of Ni3S2 at lower electric potentials was indicated by energy-dispersive X-ray spectroscopy (EDS) mapping on the NiS particles electrolyzed at –1.0 VSHE (Fig. 1c), where a clear decrease in the sulfur signal intensity relative to nickel was observed in comparison with pure NiS, and with the NiS electrolyzed at –0.5 VSHE (Fig. 1d; all EDS data are scaled to have the same S signal intensities at 2.3 keV). In agreement with the EDS result, enhancement of the reducing potential from –0.5 to –1.0 VSHE led spectral changes in the nickel K-edge X-ray absorption near-edge structure (XANES) of NiS samples (Fig. 1e) attributable to the occurrence and growth of Ni0 in Ni3S2 up to the Ni0 percentage of 12% (Fig. 1f and Supplementary Fig. 11). Similarly, XANES analysis showed 18% conversion of CoS to Co0 at –1.0 VSHE (Supplementary Fig. 12). Thus, NiS_PERM and CoS_PERM are formed at –1.0 VSHE and at even less negative potentials (i.e., closer to 0 VSHE) near their sulfide/metal equilibria (Fig. 1b, f, Supplementary Figs. 11 and 12) just as in the FeS case. However, in contrast to FeS_PERM that exhibited broad but clear XRD signals for Fe0 even at –0.7 VSHE (Supplementary Fig. 8)16, NiS_PERM showed no XRD signal for Ni0 in the examined potential range (≤ –1.0 VSHE) (Fig. 1a). The Ni0 percentage up to 12% (Fig. 1f) should be well above the detection limit of XRD (Supplementary Fig. 10) if Ni0 formed a localized crystalline domain in the Ni3S2 structure. Thus, NiS_PERM would be with more finely dispersed zerovalent metal than FeS_PERM.

a XRD patterns showing NiS electroreduction to Ni3S2 at –0.5 VSHE or below. b EH-pH diagram for the sulfide/metal system of Ni. c EDX mapping results of the NiS electrolyzed at –1.0 VSHE. d Normalized EDX spectra of NiS before (blue) and after the electrolysis at –0.5 VSHE (green) or –1.0 VSHE (red). e First derivative nickel K-edge XANES spectra. f The percentages of Ni0 in the electrolyzed NiS quantified by the XANES analysis (see “Methods”).
CO production and accumulation on the metal-sulfide PERMs
To explore the possibility of CO production and accumulation on the metal-sulfide PERMs formed under CO2 atmosphere, we dissolved the resultant metal-sulfide PERMs completely in 35% hydrogen chloride, and quantified the released CO by gas chromatography (Supplementary Fig. 18 and Supplementary Movie 1). Except for FeS, all the sulfides exposed to ≤ –0.6 VSHE were found to retain CO (Fig. 2a and Supplementary Table 2). The threshold potential is close to the sulfide/metal equilibrium potentials for Ni and Co (–0.57 VSHE for both; Fig. 1b and Supplementary Fig. 4), and is also near the thermodynamic CO2/CO redox potential at the condition for sulfide electrolysis (–0.50 VSHE; Supplementary Fig. 1). The amount of CO increased with decreasing the potential up to 180 ± 40 μmol g−1 for the case of NiS. Because sulfurized Ni binds CO considerably more weakly than pure Ni0,25 the observed CO should be mostly due to the CO bound on the surface Ni0 sites. In fact, the maximum adsorption (180 ± 40 μmol g−1) corresponds to the surface coverage of one CO molecule per 8 ± 3 surface Ni atoms (Supplementary Fig. 13), consistent with the percentage of Ni0 grown at –1.0 VSHE (Fig. 1f). The similar interpretation should be applicable to the adsorption behavior of CO on the CoS_PERM26. Coprecipitation with FeS led to decline of the amounts of CO on NiS and CoS (Fig. 2a) probably because the resultant surface Fe sites were not involved in the CO accumulation owing to the stable Ni0–CO and Co0–CO bindings, as indicated by no CO evolution during the NiS and CoS electrolysis15.

a The amounts of surface-bound CO on the electrolyzed sulfides (μmol g−1). b The yields of MTA from the surface-bound CO (a) in the presence of methanethiol (75 μmol per 50 mg of sulfide). c The percentages of CO converted to MTA in the reactions shown in (b). d The yields of MTA from externally added CO and methanethiol (75 μmol for each). The MTA yield on (Co,Ni)S declined at the lowest potential (–1.0 VSHE) because of the depletion of methanethiol (see Supplementary Table 4). e Possible intermediate steps in the MTA synthesis via the formation of NiS_PERM with the surface-bound CO (red arrow) and the CO-methanethiol reaction on NiS_PERM (green arrows). The error bars in a–d were determined by multiple independent runs under the same reaction conditions (see “Methods”).
Nonenzymatic thioester synthesis
We mixed the metal-sulfide PERMs retaining the surface-bound CO (Fig. 2a) (50 mg for each) with an aqueous solution of sodium methanethiolate (75 μmol) in a serum bottle (13.8 ml), and agitated it at room temperature (25 ± 2 °C) for up to 7 days without externally imposing electric potential. pH was buffered at neutral (7.0 ± 0.5) by filling the gas space with CO2 (1 atm). Despite this very mild condition, gas chromatography–mass spectrometry analysis revealed efficient MTA formation in the presence of specific electrolyzed sulfides (Fig. 2b, c). Organosulfur compounds detected with the amount higher than 0.01 μmol were methanethiol, MTA, dimethyl sulfide, and dimethyl disulfide (Supplementary Figs. 19 and 20), among which MTA was the sole product built from CO and methanethiol:
Experiments with the NiS_PERM prepared at –1.0 VSHE exhibited the following product characteristics. The yield of MTA increased over one week, while the amounts of CO and methanethiol decreased from their maxima at the beginning (Supplementary Fig. 24). CO appeared in the gas-phase through competitive adsorption with methanethiol: in the absence of methanethiol under otherwise identical condition, only a small amount of CO gas was detected (0.03 μmol) and no MTA was observed (Supplementary Fig. 21a). As long as methanethiol was available, replacement of CO2 in the gas space with helium (He), and dissolution of 1 M phosphate (pH 7.0) into the sample solution had no significant influence on the yield of MTA (3.2 ± 0.6 μmol; Fig. 2b → 3.6 ± 0.7 μmol). It was also confirmed with 13C-labeled CO2 that the CO2 filled in the serum bottle did not serve as a component of MTA. The MTA formed under 13CO2 atmosphere showed the mass spectrum identical to that of the standard MTA with normal carbon isotopic composition.
In aqueous solutions, acetate (0.19 ± 0.04 μmol = 0.25 ± 0.05 mM in 0.75 ml H2O) and formate (0.03 ± 0.01 μmol = 0.035 ± 0.01 mM in 0.75 ml H2O) were observed (Supplementary Fig. 25). The acetate concentration increased to 2.0 ± 0.4 mM when the product solution was basified by 1 M sodium hydroxide due to MTA hydrolysis. Because MTA hydrolysis is accelerated at elevated temperatures27, a one-day experiment at 80 °C resulted in a higher yield of acetate (2.4 ± 0.5 μmol = 3.2 ± 0.6 mM in 0.75 ml H2O) and a lower yield of MTA (0.68 ± 0.14 μmol) (Supplementary Fig. 25). Other biologically relevant organic acids, such as pyruvate and lactate, were not detected in any product solutions with concentrations higher than 0.01 mM. We also treated solid samples with 8 M potassium hydroxide after the reaction, but neither pyruvate nor lactate was observed.
When CO and methanethiol were initially introduced with equal amounts (75 μmol, or 0.14 bar in the gas space) in the presence of NiS_PERM formed at –1.0 VSHE, the yield of MTA more than doubled, compared with the case with methanethiol alone in the initial gas-phase (3.2 ± 0.6 → 7.4 ± 1.5 μmol; Fig. 2d and Supplementary Fig. 21a). In contrast, under the same initial condition with CO, few or no detectable MTA was formed in the presence of pure NiS, the Ni3S2 prepared at –0.5 VSHE, or pure Ni0 (Supplementary Fig. 21 and Supplementary Table 4), indicating the necessity of the coexistence of zerovalent and non-zerovalent Ni surface sites for efficient MTA production. It is also notable that the MTA yield decreased steeply as the NiS electroreduction potential was changed from –1.0 to –0.8 VSHE (Fig. 2b, d) although –0.8 VSHE is still sufficient for the Ni0 generation (Fig. 1f). In light of reported chemical functions of Ni and other metals in organometallic analogues of ACS28,29, one possible interpretation is that the MTA synthesis involves at least two adjacent surface Ni0 sites (Fig. 2e): one Ni0 serves as an electron donor for the reductive cleavage of methanethiol C–S bond. Close location of another Ni0 (the closest Ni–Ni distance = 2.48 Å; Supplementary Fig. 13) allows transfer of the generated electrophilic methyl onto the nucleophilic carbon of adsorbed CO. After the formation of C–C bond to make the acetyl-Ni0, its oxidative thiolysis to form MTA occurs through coupling with the reduction of neighboring non-zelovalent Ni atoms. The cleavage of methanethiol C–S bond also accounts for the formations of CH4 and dimethyl sulfide (Supplementary Tables 3 and 4), where the resultant methyl converts to CH4 via protonation, while thiolysis of the methyl generates dimethyl sulfide. If a random Ni0 distribution is assumed, a 12% conversion of Ni2+ to Ni0 achieved at –1.0 VSHE (Fig. 1f) results in 1.4% occurrence of the adjacent Ni0–Ni0 surface pair, while this percentage drops to 0.16% with a 4% conversion achieved at –0.8 VSHE. Thus, our proposed process taking multiple Ni sites into account (Fig. 2e) explains the large potential dependence in MTA productivity of NiS_PERM (Fig. 2b, d) as well as the few or no MTA formation on Ni3S2 and pure Ni0. This nonenzymatic process is expected to be eventually deactivated due to cumulative surface sulfurization (Fig. 2e), but the surface activity may be restored by additional solid electrolysis.
Interestingly, even greater CO-to-MTA conversion ratios were obtained with the NiS coprecipitating with FeS or CoS (Fig. 2c). Up to 56 ± 10% of the surface-bound CO, produced by CO2 electroreduction, was converted to MTA on the electrolyzed (Co,Ni)S. Although the electrolyzed (Fe,Ni)S produced MTA with low yields (Fig. 2b) due to the low surface accumulation of CO during the electroreduction, the percentages of CO to form MTA were kept at high levels (~20%) even at ≥ –0.8 VSHE (Fig. 2c). In fact, (Fe,Ni)S produced a considerable amount of MTA (1.44 ± 0.29 μmol) from externally introduced CO even when –0.6 VSHE was applied to the electrolysis (Fig. 2d). Since the electrochemical reducibility of FeS is higher than that of NiS (Supplementary Figs. 7 and 8), it may well be the case that Fe in the electrolyzed (Fe,Ni)S assists the performance of Ni as an electron donor at an intermediate reduction step just as the supportive role of Fe in ferredoxin serving as the redox mediator in the ACS-reaction6. Co is also involved in the enzymatic process as a transporter of the methyl group6.
Discussion
Our experiments revealed that partial electroreduction product of NiS, that is, NiS_PERM, promotes thioester synthesis from CO and methanethiol. NiS_PERM accumulates surface-bound CO during the formation under CO2 atmosphere, so NiS offers a two-step CO2 fixation route to thioester with the aid of electric energy. The electric potential required for these reactions (≤ –0.6 VSHE; Figs. 1 and 2) is more reducing than that observed in a present-day black smoker system (≥ –0.022 VSHE)11. However, the value of –0.6 VSHE and even lower potentials are available in H2-rich alkaline hydrothermal fields (Supplementary Fig. 1)30,31. Deep-sea systems are more advantageous than terrestrial ones because the elevated pressure increases the solubility of H2, which indeed serves as an electron source for the reduction of seawater CO2 in the presence of (Fe,Ni)S precipitates32. On the primordial ocean floor, widespread occurrence of elevated hydrothermal H2 fluxes has been suggested from the presence of awaruite in many fossilized serpentinization systems33,34.
In the Archean to Hadean eons, submarine hydrothermal environments may also have favored NiS precipitation owing to the huge supply of mantle-derived Ni into the ocean. The resultant massive Ni sulfide deposits are seen today in association with Archean komatiite with the Ni/Fe weight ratios occasionally exceeding one35. Given the estimated Ni2+ and Fe2+ concentrations in the early Archean seawater with their sulfide solubilities, selective NiS precipitation relative to FeS is expected in micro- to semimicro-molal level presence of hydrogen sulfide (H2S and HS–) (Supplementary Fig. 26), which is a likely concentration range at the outer surface of ancient alkaline hydrothermal chimneys36.
Within deep-sea vent chimneys, dynamic mixing of hydrothermal fluids with seawater through the pore systems is envisioned (Fig. 3)37. Occasional increase of hydrogen sulfide concentration would remove the surface-bound CO (see “Methods”, Supplementary Figs. 27 and 28). On the other hand, such H2S-rich condition is advantageous to thiol synthesis from CO or CO2.38,39 Once H2S recedes, methanethiol may react with restored CO to form MTA on Ni sulfide_PERMs (Fig. 2b–d). Thus, fluid-mediated material transport should have favored the occurrence and combination of multiple reactions through interlinked mineral pores (Fig. 3). It is conceivable, for example, that the CO formed on NiS_PERM and CoS_PERM is trapped on (Fe,Ni)S_PERM with thiols, thereby realizing the thioester formation with mild electric potentials (Fig. 2d). Thus, the thioester synthesis via the formation of Ni sulfide_PERMs should have been possible in primordial ocean alkaline hydrothermal systems. Just as Huber and Wächtershäuser7, however, we have not yet discussed seriously the source of methanethiol. To make a realistic estimate for the activity and robustness of abiotic thioester synthesis, further investigation for the geochemical availabilities of thiols and the other necessary reaction parameters (the electric potential and the Ni-sulfide composition) is desirable.

Nowadays, growing consensus for CO2 as the dominant carbon species on the Hadean Earth has stimulated exploration of geo- and astro-chemical events that could have transiently realized highly reducing early atmosphere40. Subsequent photochemical and aqueous-phase processes with reactive carbon sources, powerful oxidizing/reducing agents, and/or UV light might have realized abiotic thioester synthesis41,42. Although these reactions might have played a role in enriching prebiotic soup, their relevance to biosynthesis is ambiguous. Consequently, numerous questions remain unanswered by the transient scenario as to the gap between prebiotic chemistry and early biochemistry conserved in the metabolic and phylogenetic architectures of life.
The two key facts: CO2 fixation utilizing various states of Ni due to our experiments (Fig. 2) and the well-known central role of Ni at the active sites of CODH/ACS6 may lead to a plausible scenario for the emergence of protometabolism prior to the origin of life. Sustained occurrence of electrochemical energy adequate for the Ni sulfide_PERM formation, ≤ –0.6 VSHE in the ancient seawater condition (Figs. 1 and 2), is limited in nature except for deep-sea hydrothermal settings (Supplementary Fig. 1). This environmental specificity with the known necessity of Ni in both biological and chemical CO2 fixation suggest that the Ni sulfide_PERM-promoted thioester synthesis in early ocean hydrothermal systems (Fig. 3) is a primordial precursor of the CODH/ACS reaction. The CO production on metal-sulfide_PERMs (Fig. 2a) may also have served as crucial carbon and energy sources for the early chemolithotrophic ecosystem, thereby supporting, or even directing, the origin and early evolution of autotrophs as suggested from the autotrophic metabolism core43.
Methods
Preparation of metal sulfides
All metal sulfides were prepared by adding 100 mM sodium sulfide (Na2S) dropwisely into the corresponding 100 mM metal chlorides (NiCl2, FeCl2, or CoCl2) or their binary mixtures (for example, 50 mM NiCl2 plus 50 mM FeCl2) under vigorous stirring to a final volume ratio of 1:1. Solid precipitates were then separated from the supernatant solutions by centrifugation (8000 rpm, 10 min) and were dried under vacuum. To prevent oxidation by atmospheric O2, the sample preparation was conducted in a glove box filled with N2 gas (>99.99995% purity), with 4% H2 (>99.99999% purity) being added (the Coy system). All chemicals were purchased from FUJIFILM Wako Pure Chemical Corporation as reagent grade. Deaerated Milli-Q water (18.2 megohms) was used as the solvent.
Pure metals (Fe0, Co0, and Ni0) were obtained from EM Japan. Their reported characteristics and XRD patterns measured in this study are presented in Supplementary Table 1 and Supplementary Fig. 9, respectively.
Sulfide electrolysis
Metal sulfide electrolysis was conducted under a simulated early ocean condition in accordance with the procedure reported previously16. Briefly, 400 mg of sulfide samples was deposited on a carbon working electrode (5.7 cm2) in a H-type cell (Supplementary Fig. 2), immersed in a deaerated 100 mM NaCl, and exposed to a flow (20 ml min−1) of CO2 (>99.995% purity) containing 4 ppm of H2S. The CO2 gas buffered the solution pH at slightly acidic (6.0 ± 0.25). The H2S partial pressure was determined by thermodynamic calculation to supply H2S and HS– into solution with the equilibrium total concentration of 0.5 μmol kg−1. Although the ionic strength and pressure conditions adopted in this experiment are different from the ancient deep-sea hydrothermal settings36,44, a thermodynamic calculation indicates that these differences have no significant influence on the redox potentials for the sulfide/metal and the CO2/CO systems (Supplementary Fig. 5)16.
While keeping the CO2 gas flow that was started at least 1 h before each experiment, a constant potential was applied on the carbon electrode for 7 days by using a multi-potentiostat (PS-08; Toho Technical Research). All potentials were measured against an Ag/AgCl reference electrode in saturated KCl and were converted to the SHE scale by the following equation:
After the electrolysis, the electrochemical cell was immediately transferred into a glove box filled with N2 and H2 gases (volume ratio, 96:4). The solid sample was then separated from the supernatant solution, dried under vacuum for 1 h, and stored inside the glove box. A dry vacuum pump (RDA-281H, ULVAC) with the reported ultimate pressure of 0.08 Pa was used for the dehydration. The surface-bound CO was expected to be intact at this vacuum condition25,45,46.
The prepared sulfide_PERMs were stable in the anaerobic glove box over weeks, but were oxidized rapidly in the air. Typically, a minute exposure to the air completely degraded their capabilities for promoting MTA synthesis. Their high susceptibilities to oxidation are not a severe problem in the geoelectrochemical scenario (Supplementary Fig. 1) because of the absence of O2 and other reactive oxidants (for example, H2O2) in primordial deep-sea environments.
Solid characterization
Scanning electron microscopy (SEM) imaging was performed on a Helios G4 UX (Thermo Fisher Scientific) equipped with a PP-3010 cryo preparation system (Quorum) and an Octane Super C5 EDS detector (AMTEC). An acceleration voltage of 2 kV was applied for secondary electron imaging and 20 kV for EDS analysis under a reduced pressure of <1 × 10−4 Pa at room temperature (23 ± 2 °C). Samples were prepared as follows. First, sulfide sample sealed in a serum bottle was placed in a vacuum chamber of the cryo-preparation system, whose internal atmosphere was replaced with N2 gas from liquid N2 vaporization by three times of evacuation-purge cycles. Under flow of N2 gas, the serum bottle was opened, and the sulfide was rubbed onto a carbon tape (Nisshin EM) attached on a transfer shuttle by a plastic spatula. The shuttle was transferred into a transfer device under ~102 Pa, and then to the SEM chamber via a cryo-preparation chamber (<10−4 Pa).
Particle size distribution (Supplementary Fig. 6) was determined by manual segmentation on Affinity Photo for iPad 1.6.8.77 (Serif (Europe) Ltd.) followed by image analysis with Image-Pro 3D v9.3 (Media Cybernetics). Particle diameters were calculated from the segmented areas assuming spherical morphology.
X-ray diffraction (XRD) patterns of sulfide samples were measured by using an X-ray diffractometer with Cu Kα radiation (MiniFlex 600, Rigaku). All runs were conducted with 2θ ranging from 10° to 90° using 0.02° 2θ step with a scan rate of 0.1 or 1° min−1. To prevent oxidation by atmospheric O2 during the measurement, the solid samples were shielded in an air-sensitive sample holder (Rigaku). Peak identifications were made on the basis of the reference patterns reported in the Powder Diffraction File published by the International Centre for Diffraction Data. The reference patterns are presented in Fig. 1a and Supplementary Figs. 8–11 with the measured XRD data.
X-ray absorption near-edge structure (XANES) spectra at Ni and Co K-edges were measured at BL-12C (bending magnet beamline) in a synchrotron radiation facility (Photon Factory) in High Energy Accelerator Research Organization (KEK), Tsukuba, Japan. In the beamline, X-ray from a synchrotron operated at 2.5 GeV (current: 450 mA) was monochromatized with a Si(111) double-crystal monochromator, and focused to an area of 0.5 × 0.5 mm2 with a bent cylindrical mirror, which also reduced the higher order. XANES spectra were obtained in transmission mode using two ion chambers to measure intensities of incident (I0) and transmitted (I) X-rays. Energy step within the XANES region was 0.25 eV, and the absorbance by the sample (μt) was obtained as μt = ln(I/I0). Sulfide samples were diluted with boron nitride (BN, < 150 nm, 99% purity) to yield the metal/BN molar ratio of 1:25, and shielded by an O2-impermeable polyethylene film. Measurement was conducted for a part of the sample with uniform thickness within the area of the X-ray beam.
The percentages of zerovalent metal in the electrolyzed NiS and CoS were estimated by a least-squares fitting of the sample spectra after background-subtraction and normalization with an X-ray absorption spectroscopy data processing software ATHENA47. For the NiS samples, the linear combination of pure Ni0 and the NiS electrolyzed at –0.5 VSHE were used for the fitting between 8300 and 8440 eV. The NiS electrolyzed at –0.5 VSHE was used as a reference of Ni3S2 based on the thermodynamic calculation (Fig. 1b), XRD pattern (Fig. 1e), and the XANES spectral profile (Supplementary Fig. 11a) identical to that of pure Ni3S2.48 The energy region higher than 8440 eV was not analyzed because of lower signal-to-noise ratio and of greater influence from the local environment of Ni atom. The best fit was determined by calculating the lowest R factor, which was defined as:
In this equation, μexp and μcal are the experimental and the calculated absorbances at a given energy E, respectively. For the CoS samples, the energy range 7690–7830 eV were fitted with the linear combination of pure Co0 and the CoS electrolyzed at –0.5 VSHE in a manner similar as Ni described above.
Search for the surface-bound CO on electrolyzed sulfides
Each sulfide sample (50 mg for each) was sealed in a serum bottle (124.7 ml) with a Teflon-laminated butyl rubber cap and an aluminum stopper in a glove box filled with N2 and H2 gases (volume ratio, 96:4). The bottle was then filled with pure He gas (>99.99995%) by flowing the He through a stainless needle at a rate of 100 ml min−1 over 10 min. This was followed by the addition of 5 ml of 35% HCl (super special grade; FUJIFILM) under vigorous agitation. After the sulfide sample dissolved completely under 60 rpm rotation of the bottle at room temperature (25 ± 2 °C) (typically, within 30 min), the headspace gas was analyzed by gas chromatography (GC) (Fig. 2a, Supplementary Fig. 18 and Supplementary Table 2). To ensure reproducibility, we carried out multiple independent runs, using several sulfide samples for each metal. Differences among the data obtained at the identical condition were less than 20%. The same was true for the following two experiments “Nonenzymatic thioester synthesis” and “Competitive adsorption of CO with H2S”.
To verify the CO production and accumulation on the NiS_PERM, we conducted the following experiment with isotopically labeled sodium bicarbonate (NaH13CO3, 99% purity) provided by Cambridge Isotope Laboratories, Inc. First, 400 mg of NiS was electrolyzed at –1.0 VSHE for 7 days in 60 ml of 0.5 M phosphate buffer solution (pH 6.0) under pure N2 gas flow (>99.9998%). Then, the gas headspace of the electrochemical cell (~40 ml) was purged with 13CO2 gas that was provided by a dropwise addition of 0.1 M phosphoric acid (H3PO4) onto NaH13CO3 powder under vigorous stirring in a closed vial connected with the electrochemical cell with a polyvinyl chloride tube and stainless needles. After additional two-hour electrolysis with the gas inlet and outlet closed, the NiS_PERM on the carbon paper was collected, dried under vacuum, and dissolved completely in 35% HCl. GC-MS analysis of the released gas revealed the formation of 13CO with the m/z = 29 (Supplementary Fig. 18c). No 13CO was detected in the 13CO2 formed by the H3PO4 addition to NaH13CO3 (Supplementary Fig. 18).
In the normal CO2 gas bubbled during the sulfide electrolysis, we detected no CO with our GC’s maximum performance (detection limit; ~1 ppm). Even if a very slight amount of CO impurity is present in the CO2, its contribution to the surface-bound CO (Fig. 2a and Supplementary Table 2) must be negligible because the electrolyzed FeS released almost no CO despite the fact that Fe0 has an even stronger CO binding energy than Ni0 and Co0.49
Supplementary Movie 1 presents a demonstration of the surface-bound CO on the NiS electrolyzed at –1.0 VSHE. A weak acid (1 M H3PO4) was used here to make sure that the gas bubble formation is not due to NiS dissolution but from the purging of the electrolyzed surface.
Nonenzymatic thioester synthesis
Each sulfide sample (50 mg for each) was sealed in a serum bottle (13.8 ml) with a Teflon-laminated butyl rubber cap and an aluminum stopper in a glove box filled with N2 and H2 gases (volume ratio, 96:4). The bottle was then filled with pure CO2 gas (>99.995%) by flowing the CO2 through a stainless needle at a rate of 100 ml min−1 over 5 min. This was followed by the addition of 0.75 ml of 100 mM sodium methanethiolate (CH3SNa) with or without 1.86 ml of CO gas (1 atm, 99.9% purity). After rotating the bottle at 60 rpm for 7 days at room temperature (25 ± 2 °C), the headspace gas was analyzed by GC (Fig. 2b–d, Supplementary Figs. 19–23 and Supplementary Tables 3 and 4). The aqueous suspension was then centrifuged (10,000 rpm, 2 min) and measured for pH by a portable pH meter (Seven2Go Pro, Mettler Toledo).
We did not carry out “one-pot” organic synthesis in an electrochemical cell because H2 evolution during sulfide electrolysis15 prevented us from keeping methanethiol in the cell in the reaction period due to pressurization. As discussed in the main text, however, occurrence and combination of multiple reactions are likely to be more realistic than a single-pot reaction in natural hydrothermal vent environments.
Competitive adsorption of CO with H2S
Sulfide sample (50 mg for each) was sealed in a serum bottle (19.5 ml) with a Teflon-laminated butyl rubber cap and an aluminum stopper in a globe box filled with N2 and H2 gases (volume ratio, 96:4). The bottle was then filled with pure He gas (>99.99995%) by flowing the He through a stainless needle at a rate of 100 ml min−1 over 5 min. This was followed by the addition of 1 ml of 300 mM Na2S and the subsequent addition of 1 ml of 450 mM HCl. After rotating the bottle at 60 rpm for 1 day at room temperature (25 ± 2 °C), the headspace gas was analyzed by GC (Supplementary Figs. 26 and 27 and Supplementary Table 5). No carbon-sulfur compounds (for example, methanethiol, carbon disulfide, carbonyl sulfide) were detected.
From the pH values of aqueous suspensions (7.5–8.0; Supplementary Table 5), the initial dissolved H2S and HS– concentrations are predicted to be 90–120 mM in total, equilibrium H2S distribution being assumed in the gas and liquid phases without the surface adsorption.
Sample analysis
Inorganic gases, including H2, CO, and CH4, were quantified by a Shimadzu GC system equipped with a BID-2010 Plus detector (Tracera). A MICROPACKED-ST column (Shinwa) was attached. He (>99.99995%) was used as the carrier gas at a column flow rate of 7 ml min−1. The column temperature was initially kept at 35 °C for 2.5 min, raised to 250 °C at a rate of 20 °C min−1, and then raised to 265 °C at a rate of 4 °C min−1. A chromatogram for a standard gas sample is shown in Supplementary Fig. 14 with obtained calibration curves.
Gas-phase sulfur compounds were analyzed by using a Shimadzu gas chromatograph-mass spectrometer (GCMS-QP2010 Ultra) equipped with a DB–SULFUR SCD column (60 m, 0.32 mm I.D, Agilent). He (>99.99995%) was used as the carrier gas at a column flow rate of 33.2 cm s−1. The column temperature was initially kept at 35 °C for 5 min, raised to 155 °C at a rate of 10 °C min−1, and then raised to 235 °C at a rate of 20 °C min−1. The scan range was set to m/z 5–200 Da. Compounds were identified by comparing the observed retention times and mass spectra with those of respective standards prepared from commercial reagents, and were quantified based on calibration curves (Supplementary Fig. 15). Several gas samples were also analyzed by the Tracera GC system with a DB–SULFUR SCD column at the measurement condition described in this paragraph (Supplementary Fig. 16).
Aqueous-phase products were characterized, after filtration with a polytetrafluoroethylene membrane filter (pore size, 0.2 μm), by using a Shimadzu HPLC system equipped with an electric conductivity detector and an anion exchange column (Shim-pack SCR-102H, Shimadzu) set at 40 °C. The p-toluenesulfonic acid aqueous solution (5 mM) was used as the eluent at a rate of 1.6 ml min−1. A chromatogram for several organic acids is shown in Supplementary Fig. 17 with the obtained calibration curves.
Note that basified aqueous samples stored in non-airtight containers over several days occasionally showed 1–10 μM of acetate and pyruvate signals (data not shown) because basification facilitates absorption of organic acids from ambient air. To avoid crucial contaminations such as pyruvate, we set our detection cutoff at 0.01 mM and analyzed all aqueous samples as soon as they were prepared.
Quantification of organosulfur products
The four organosulfur compounds observed in the present study (methanethiol, MTA, dimethyl sulfide, and dimethyl disulfide) distribute both the gas- and aqueous-phases with significant fractions. Thus, aqueous-phase concentrations of methanethiol, dimethyl sulfide, and dimethyl disulfide were calculated from the partial pressures of respective compounds quantified by GC with the Henry’s law constants reported in the literature (0.38, 0.56, and 0.58 mol l−1 atm−1, respectively)50. The constant for MTA was determined experimentally to be 5.8 ± 1.0 mol l−1 atm−1 from three independent measurements of the water-gas partitioning of MTA in a closed glass vial containing a certain volume of 100 mM phosphate buffer solution (pH 6.7). The constants for methanethiol, dimethyl sulfide, and dimethyl disulfide determined by this method were in agreement with the literature values with the percentage errors less than 15%.
The organosulfur compounds may also be present on the sulfide surfaces. Actually, when 1 M phosphoric acid (H3PO4) was added after the 7-day interaction of methanethiol with the NiS_PERM formed at –1.0 VSHE, the gas-phase concentrations of MTA and methanethiol increased by 30 and 70%, respectively, while that of dimethyl sulfide decreased by 50%. Nevertheless, because such chemical processes may cause undesirable reactions that consume MTA, we did not take account of the surface adsorption in the yield determination of organosulfur compounds. For the same reason, in the nonenzymatic thioester synthesis experiment, we report the amounts of inorganic gases (H2, CO, and CH4) quantified from the gas-phase analysis alone (Supplementary Figs. 19b, 22, and 23, Supplementary Tables 3 and 4).
Dimethyl disulfide was always observed in the presence of methanethiol with the amount typically less than a few percent of the initial amount of methanethiol (Supplementary Figs. 19a and 20). A probable cause of the dimethyl disulfide formation is methanethiol oxidation by atmospheric O2 (2 CH3SH + 0.5 O2 → CH3SSCH3 + H2O) at the timing of sample injections into the GC system owing to air intrusion (Supplementary Fig. 19b). Except for dimethyl disulfide, no oxidative organosulfur compound was detected (Supplementary Fig. 20). It was also confirmed that MTA, dimethyl sulfide, and dimethyl disulfide were stable in air.
Thermodynamic calculation
Redox potentials (Eh) of the H+/H2 and the CO2/CO couples as functions of temperature and pH (Supplementary Fig. 1) were calculated with the following equations:
In these equations, T, R, and F stand for temperature in kelvins, the gas constant (8.3145 J mol‒1 K‒1), and the Faraday constant (96,485 J mol‒1 V‒1), respectively. αi represents the activity of the species i that was calculated either using the extended Debye-Hückel equation51 for aqueous ionic species (H+) or setting the activity coefficient to unity for aqueous neutral species (H2, CO2, and CO). ∆fG°(i) signifies the standard Gibbs energy of formation of the species i at the temperature and pressure of interest, which were calculated according to the revised HKF equations of state52 using the relevant thermodynamic data and the revised HKF parameters reported in the literature53,54. ∆fG° of H2O was taken from Helgeson and Kirkham55.
The Eh–pH diagrams of the sulfide–metal systems (Fig. 1b and Supplementary Fig. 4) were computed with the Act2 program in Geochemist’s Workbench version 10.0.5 by using the thermodynamic dataset for aqueous species calculated by the above procedures, those for sulfide minerals listed in Supplementary Table 3 presented by Kitadai et al.16, and those for pure metals compiled in Robie and Hemingway56.
Data availability
All data needed to evaluate the conclusions of this study are presented in this article, the Supplementary Information and/or the Supplementary Movie. Additional data related to this study are available from the corresponding author on reasonable request.
References
de Duve, C. Blueprint For a Cell: The Nature and Origin of Life (Neil Patterson Publishers, 1991).
Hartman, H. Speculations on the origin and evolution of metabolism. J. Mol. Evol. 4, 359–370 (1975).
Wächtershäuser, G. Groundworks for an evolutionary biochemistry: The iron–sulphur world. Prog. Biophys. Molec. Biol. 58, 85–201 (1992).
Russell, M. J. & Martin, W. The rocky roots of the acetyl-CoA pathway. Trends Biochem. Sci. 29, 358–363 (2004).
Sousa, F. L. et al. Early bioenegetic evolution. Philos. Trans. R. Soc. B 368, 20130088 (2013).
Can, M., Giles, L. J., Ragsdale, S. W. & Sarangi, R. X-ray absorption spectroscopy reveals an organometallic Ni–C bond in the CO-treated form of acetyl-CoA synthase. Biochem. 56, 1248–1260 (2017).
Huber, C. & Wächtershäuser, G. Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 276, 245–247 (1997).
Preiner, M. et al. A hydrogen-dependent geochemical analogue of primordial carbon and energy metabolism. Nat. Eco. Evol. 4, 534–542 (2020).
Goldford, J. E., Hartman, H., Smith, T. F. & Segre, D. Remnants of an ancient metabolism without phosphate. Cell 168, 1126–1134 (2017).
Whicher, A., Camprubi, E., Pinna, S., Herschy, B. & Lane, N. Acetyl phosphate as a primordial energy currency at the origin of life. Orig. Life Evol. Biosph. 48, 159–179 (2018).
Yamamoto, M. et al. Spontaneous and widespread electricity generation in natural deep-sea hydrothermal fields. Angew. Chem. Int. Ed. 56, 5725–5728 (2017).
Nakamura, R. et al. Electrochemical current generation across a black smoker chimney. Angew. Chem. Int. Ed. 49, 7692–7694 (2010).
Ang, R. et al. Thermoelectricity generation and electron–magnon scattering in a natural chalcopyrite mineral from a deep-sea hydrothermal vent. Angew. Chem. Int. Ed. 54, 12909–12913 (2015).
Krissansen-Totton, J., Arney, G. N. & Catling, D. C. Constraining the climate and ocean pH of the early Earth with a geological carbon cycle model. Proc. Natl Acad. Sci. USA 115, 4105–4110 (2018).
Kitadai, N. et al. Geoelectrochemical CO production: Implications of the autotrophic origin of life. Sci. Adv. 4, eeao7265 (2018).
Kitadai, N. et al. Metals likely promoted protometabolism in early ocean alkaline hydrothermal systems. Sci. Adv. 5, eeav7848 (2019).
Zhu, W. et al. Nickel sulfide microsphere film on Ni foam as an efficient bifunctional electrocatalyst for overall water splitting. Chem. Commun. 52, 1486–1489 (2016).
Xiao, H., Goddard, W. A. III, Cheng, T. & Liu, Y. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl Acad. Sci. USA 114, 6685–6688 (2017).
Hori, Y. & Murata, A. Electrochemical evidence of intermediate formation of adsorbed CO in cathodic reduction of CO2 at a nickel electrode. Electrochim. Acta 35, 1777–1780 (1990).
Hansen, H. A., Varley, J. B., Peterson, A. A. & Nørskov, J. K. Understanding trends in the electrocatalytic activity of metals and enzymes for CO2 reduction to CO. J. Phys. Chem. Lett. 4, 388–392 (2013).
Kondo, T. & Mitsudo, T. Metal-catalyzed carbon–sulfur bond formation. Chem. Rev. 100, 3205–3220 (2000).
Hirschbeck, V., Gehrtz, P. H. & Fleisher, I. Metal-catalyzed synthesis and use of thioesters: recent developments. Chem. Eur. J. 24, 7092–7107 (2018).
Pietrocola, F., Galluzzi, L., Pedro, J. M. B., Madeo, F. & Kroemer, G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821 (2015).
Satanowski, A. & Bar-Even, A. A one-carbon path for fixing CO2. EMBO Rep. 21, e50273 (2020).
Erley, W. & Wagner, H. Sulfur poisoning of carbon monoxide adsorption on Ni(111). J. Catal. 53, 287–294 (1978).
Ma, S. H., Jiao, Z. Y., Wang, T. X. & Zu, X. T. Effect of preadsorbed S on the adsorption of CO on Co(0001). J. Phys. Chem. 113, 16210–16215 (2009).
Chandru, K., Gilbert, A., Butch, C., Aono, M. & Cleaves, H. J. II The abiotic chemistry of tholated acetate derivatives and the origin of life. Sci. Rep. 6, 29883 (2016).
Lindahl, P. A. Acetyl-coenzyme A synthase: the case for a Ni0p-based mechanism of catalysis. J. Biol. Inorg. Chem. 9, 516–524 (2004).
Horn, B., Limberg, C., Herwig, C. & Mebs, S. (2011) The conversion of nickel-bound CO into an acetyl thioester: organometallic chemistry relevant to the acetyl coenzyme A synthase active site. Angew. Chem. Int. Ed. 50, 12621–12625 (2011).
Morrill, P. L. et al. Geochemistry and geobiology of a present-day serpentinization site in California: the Cedars. Geochim. Cosmochim. Acta 109, 222–240 (2013).
Boyd, E. S., Amenabar, M. J., Poudel, S. & Templeton, A. S. Bioenergetic constraints on the origin of autotrophic metabolism. Philos. Trans. R. Soc. A 378, 20190151 (2019).
Hudson, R. et al. CO2 reduction driven by a pH gradient. Proc. Natl Acad. Sci. USA 117, 22873–22879 (2020).
Rajendran, S. & Nasir, S. Hydrothermal altered serpentinized zone and a study of Ni-magnesioferrite-magnetite-awaruite occurrences in Wadi Hibi, Northrn Oman Mountain: Discrimination through ASTER mapping. Ore Geol. Rev. 62, 211–226 (2014).
McCollom, T. M. Abiotic methane formation during experimental serpentinization of olivine. Proc. Natl Acad. Sci. USA 113, 13956–13970 (2016).
Bekker, A. et al. Atmospheric sulfur in Archean komatiite-hosted nickel deposits. Science 326, 1086–1089 (2009).
Shibuya, T., Russell, M. J. & Takai, K. Free energy distribution and hydrothermal mineral precipitation in Hadean submarine alkaline vent systems: Importance of iron redox reactions under anoxic conditions. Geochim. Cosmochim. Acta 175, 1–19 (2016).
Tivey, M. K. Generation of seafloor hydrothermal vent fluids and associated mineral deposits. Oceanography 20, 50–65 (2007).
Heinen, W. & Lauwers, A. M. Organic sulfur compounds resulting from the interaction of iron sulfide, hydrogen sulfide and carbon dioxide in an anaerobic aqueous environment. Orig. Life Evol. Biosph. 26, 131–150 (1996).
Schulte, M. D. & Rogers, K. L. Thiols in hydrothermal solution: standard partial molal properties and their role in the organic geochemistry of hydrothermal environments. Geochim. Cosmochim. Acta 68, 1087–1097 (2004).
Benner, S. A. et al. When did life likely emerge on Earth in an RNA-first process? ChemSystemsChem 1, e1900035 (2019).
Weber, A. L. Nonenzymatic formation of “energy-rich” lactoyl and glyceroyl thioesters from glyceraldehyde and a thiol. J. Mol. Evol. 20, 157–166 (1984).
Chevallot-Beroux, E., Gorges, J. & Moran, J. Energy conservation via thioesters in a non-enzymatic metabolism-like reaction network. ChemRxiv. Preprint at https://doi.org/10.26434/chemrxiv.8832425 (2019).
Ragsdale, S. W. Life with carbon monoxide. Crit. Rev. Biochem. Mol. Biol. 39, 165–195 (2004).
Amend, J. P. & McCollom, T. M. in Chemical Evolution II: From the Origins of Life to Modern Society, (eds. Zaikowski, L., Fredrich, J. M. & Seidel, S. R.) 63–94 (American Chemical Society, 2009).
Bridge, M. E., Comrie, C. M. & Lambert, R. M. Chemisorption studies on cobalt single crystal surfaces I. carbon monoxide on Co(0001). Surf. Sci. 67, 393–404 (1977).
Gonzalez, L., Miranda, R. & Ferrer, S. A thermal desorption study of the adsorption of CO on Fe(110); enhancement of dissociation by surface defects. Sur. Sci. 119, 61–70 (1982).
Ravel, B. & Newville, M. (2005) ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12, 537–541 (2005).
Van Loon, L. L., Throssell, C. & Dutton, M. D. Comparison of nickel speciation in workplace aerosol samples using sequential extraction analysis and X-ray absorption near-edge structure spectroscopy. Environ. Sci. Process. Impacts 17, 922–931 (2015).
Abild-Pedersen, F. & Andersson, M. P. CO adsorption energies on metals with correction for high coordination adsorption sites—a density functional study. Surf. Sci. 601, 1747–1753 (2007).
Sander, R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 15, 4399–4981 (2015).
Helgeson, H. C., Kirkham, D. H. & Flowers, G. C. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures: IV. Calculation of activity coefficients, osmotic coefficients, and apparent molal and standard and relative partial molal properties to 600oC and 5 KB. Am. J. Sci. 281, 1249–1516 (1981).
Shock, E. L., Oelkers, E. H., Johnson, J. W., Sverjensky, D. A. & Helgeson, H. C. Calculation of the thermodynamic properties of aqueous species at high pressures and temperatures. J. Chem. Soc. Faraday Trans. 88, 803–826 (1992).
Shock, E. L., Helgeson, H. C. & Sverjensky, D. A. Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: Standard partial molal properties of inorganic neutral species. Geochim. Cosmochim. Acta 53, 2157–2183 (1989).
Shock, E. L. & McKinnon, W. B. Hydrothermal processing of cometary volatiles–applications to triton. Icarus 106, 464–477 (1993).
Helgeson, H. C. & Kirkham, D. H. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures: I. summary of the thermodynamic/electrostatic properties of the solvent. Am. J. Sci. 274, 1089–1198 (1974).
Robie, R. A. & Hemingway, B. S. “Thermodynamic Properties of Minerals and Related Substances at 298.15 K and 1 Bar (105 Pascals) Pressure and at Higher Temperatures” (U.S. Geological Survey Bulletin 2131, U.S. Geological Survey, 1995).
Aono, M., Kitadai, N. & Oono, Y. A principled approach to the origin problem. Orig. Life. Evol. Biopsh 45, 327–338 (2015).
Reeves, E. P., McDermott, J. M. & Seewald, J. S. The origin of methanethiol in midocean ridge hydrothermal fluids. Proc. Natl Acad. Sci. USA 111, 5474–5479 (2014).
Acknowledgements
We thank Reiko Nagano and Atsuko Fujishima for their help in laboratory experiments. This study was supported by JSPS KAKENHI (Grant Nos. 18H04456 and 20H00209) and the Astrobiology Center Program of NINS (Grant No. AB292004).
Author information
Authors and Affiliations
Contributions
N.K and Y.O. conceived the whole project based on the principled approach57 and N.K. realized nonenzymatic thioester synthesis. Y.T. measured XANES spectra of the sulfide samples. S.O. performed SEM-EDX analysis, and W.T. prepared figures for the SEM-EDX data. N.K. performed all experiments except for the XANES and SEM-EDX measurements with technical support of Y.N., and prepared all figures and tables for the obtained results. S.O. considered possible intermediate steps in the MTA formation. N.K and Y.O. wrote the manuscript with input from R.N., M.Y., S.O., W.T., Y.N., Y.T., and K.T.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kitadai, N., Nakamura, R., Yamamoto, M. et al. Thioester synthesis through geoelectrochemical CO2 fixation on Ni sulfides. Commun Chem 4, 37 (2021). https://doi.org/10.1038/s42004-021-00475-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-021-00475-5
This article is cited by
-
Nickel-organo compounds as potential enzyme precursors under simulated early Earth conditions
Communications Chemistry (2024)
-
Electrosynthesis of amino acids from NO and α-keto acids using two decoupled flow reactors
Nature Catalysis (2023)
-
Liquid and supercritical CO2 as an organic solvent in Hadean seafloor hydrothermal systems: implications for prebiotic chemical evolution
Progress in Earth and Planetary Science (2022)
-
Geoelectrochemistry-driven alteration of amino acids to derivative organics in carbonaceous chondrite parent bodies
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
