
Abstract
Pyrrolidines and piperidines are important building blocks in organic synthesis. Numerous methods exist for constructing substituted pyrrolidines and piperidines. However, efficient syntheses of pyrrolidines and piperidines bearing chiral tertiary alcohols are limited. Here we report an efficient enantioselective nickel-catalyzed intramolecular reductive cyclization of N-alkynones. A P-chiral bisphosphorus ligand DI-BIDIME is designed and applied in the synthesis of tertiary allylic siloxanes bearing pyrrolidine and piperidine rings in high yields and excellent enantioselectivities, with triethylsilane as reducing reagent. The highest turn over number achieved is 1000 (0.1 mol% catalyst loading) with > 99:1 er. This reaction provides a practical way to synthesize pyrrolidine and piperidine derivatives with chiral tertiary alcohols from easily accessible starting materials under mild conditions. The products can be scaled up and transformed to various useful chiral intermediates. The P-chiral bisphosphorus ligand developed in this study represents one of the few ligands for highly enantioselective cyclization of alkynones.
Similar content being viewed by others

Introduction
Transition metal-catalyzed coupling of alkynals/alkynones has become a powerful method for efficient construction of allylic alcohol derivatives in current organic chemistry1,2,3. Recent development by employing various transition metal catalysts such as Ti4, Ni5,6,7,8,9,10,11,12,13,14,15,16,17,18, Rh19,20,21,22,23,24, Ir25, Ru26,27,28, and Pd29,30 in combination with a variety of coupling components and reducing/alkylative agents has greatly expanded its scope and applications. Among them, the nickel-catalyzed coupling of π systems pioneered by Mori5,8, Montgomery6,7,10, and Jamison9,15, is particularly attractive and offers a broad substrate scope and good functional group compatibility. However, the enantioselective cyclization of these substrates, especially for constructing chiral tertiary alcohols is limited16. The chiral tertiary alcohols construction is synthetically more difficult as the asymmetric addition to ketones (tetrasubstituted carbon synthesis) is generally more challenging than addition to aldehydes. Furthermore, the nickel-catalyzed reactions usually demonstrated in considerably high catalytic loadings (5 to 30 mol% of Ni catalyst), which is not “green” enough for the practicality of nickel-catalyzed synthesis. Thus, the development of efficient enantioselective nickel-catalyzed reactions remains a major challenge for synthetic chemists.
Herein, we report an efficient regiospecific and enantioselective nickel-catalyzed intramolecular reductive cyclization of N-alkynones with up to 1000 TON (0.1 mol% catalyst loading) and > 99:1 er under mild conditions.
Results
Ligand design
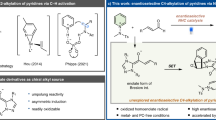
Despite the recent progress in nickel-catalyzed reactions, few reports are available on ligand engineering in seeking for a more robust and active nickel catalyst. Based on our previous experience on ligand design, we proposed this objective could be achieved by the development of our privileged ligand BIDIME31,32,33,34. We intuitively designed DI-BIDIME in hope to develop a more efficient enantioselective nickel-catalyzed reaction (Fig. 1a). Our design in DI-BIDIME are in two strategies: (1) DI-BIDIME is free of H7, not only avoiding the potential C–H functionalization at H7 position35,36,37 but also making the adjacent C–O bond more hindered and resistant to cleave in the presence of a nickel metal38,39; (2) both phosphorus atoms in DI-BIDIME should function independently with a reduced ligand entropy by half. This could be helpful for increasing the longevity of nickel catalyst. We herein report the development of DI-BIDIME (L4) by using this strategy.

Ligand design and reaction discovery of nickel-catalyzed regiospecific asymmetric reductive cyclization of N-alkynones. a Design of a more robust ligand for Ni-catalyzed reductive coupling. b Several therapeutic agents and bioactive molecules bearing a multi-substituted pyrrolidine/piperidine moiety. c–h Previous work of constructing functionalized multi-substituted pyrrolidines/piperidines via metal-catalyzed cyclizations. i This work: efficient nickel-catalyzed regiospecific asymmetric reductive cyclization of N-alkynones to synthesize pyrrolidines/piperidines with chiral tertiary alcohol sillyl ether
Reaction discovery
Multi-substituted pyrrolidines and piperidines are widely present in the structures of bioactive natural products and drugs40,41,42,43 (Fig. 1b). In general, their optical active version led to enhanced bio-activities44,45. However, methods for efficient construction of chiral substituted pyrrolidines and piperidines were limited46. In the past two decades, many metal-catalyzed cyclizations were developed for constructing functionalized pyrrolidines and piperidines (Fig. 1c–h). From 2000s, Montgomery group have first studied Ni-catalyzed regioselective cyclization of tethered N-alkynals without investigated their enantioselectivity6,7,11 (Fig. 1c). Krische and Tanaka then developed the rhodium-catalyzed enantioselective cyclizations of N-alkynals to form pyrrolidines with chiral secondary alcohols/ethers20,22,23 (Fig. 1d). In 2007, Zhou and colleagues21 first reported one enantioselective rhodium-catalyzed hydrosilylation/cyclization of 1,6-enynes (Fig. 1e) and followed by a enantioselective Ni-catalyzed intramolecular hydroalkenylation of N-1,6-dienes to synthesize chiral functionalized pyrrolidines and piperidines18 (Fig. 1f). In 2013, the first regioselective cyclization of N-alkynones was reported by Lu and colleagues30, which is catalyzed by a Pd precursor to construct a series of pyrrolidines with tertiary alcohols (Fig. 1g). Then Li and Xu24 reported an asymmetric rhodium-catalyzed cyclization for chiral tertiary allylic alcohols (Fig. 1h). To the best of our knowledge, there is no efficient Ni-catalyzed cyclization with high TONs (> 100), which can provide pyrrolidines and piperidines with chiral tertiary alcohols. We herein describe a highly efficient and practical nickel-catalyzed intramolecular reductive cyclization of N-alkynones for the first time with the nickel loading as low as 0.1 mol%, by employing the newly developed chiral bisphosphorus ligand DI-BIDIME through ligand engineering. A variety of pyrrolidine/piperidine derivatives bearing a chiral tertiary alcohol silyl ether moiety are prepared in high yields and excellent enantioselectivities (Fig. 1i). Further transformations of the silyl ether to alcohol and related derivatives were demonstrated. The applications of this method to the synthesis of key intermediates of prodine and (–)-peroxetine were also accomplished. We believed that this asymmetric Ni-catalyzed intramolecular reductive cyclization of N-alkynones would provide an efficient access to many useful building blocks of bioactive pyrrolidine and piperidine molecules.
Ligand effects and catalyst loading optimization
We began this study by choosing N-alkynone (1a) as the standard substrate to investigate the nickel-catalyzed intramolecular reductive cyclization of N-alkynones with triethylsilane (Et3SiH) as the reducing reagent (Fig. 2). The reactions were performed under nitrogen in dioxane at 25 °C for 12 h in the presence of a nickel catalyst prepared in situ from 10 mol % Ni(cod)2, 10 mol% monophosphorus ligand/5 mol% bisphosphorus ligand, and 3 equiv. Et3SiH. First, we evaluated various commercial available mono-, bis-phosphorus ligands and ligands developed in our laboratory47,48. As shown in Table 1, monophosphorus ligands: PPh3, PCy3, Ru-Phos, S-Phos, and X-Phos provided the desired product 2a in racemic form with high to excellent yields (PPh3: 98%; PCy3: 82%; Ru-Phos: 95%; S-Phos: 98%; X-Phos: 85%, entries 1–5). Chiral ligands (R)-MONOPHOS, (R)-Me-DuPhos, (R)-Binapine, and L1 showed no reactivity for this cyclization, whereas (R)-BINAP, (R)-(S)-JosiPhos, and (R)-DTBM-SEGPhos only give trace (< 5%) cyclized product (entries 6–12). Surpringly, our ligands (S)-Me-BIDIME (L2), (S)-BIDIME (L3), and (S, S)-DI-BIDIME [L4, see details in Supplementary Information (SI) for the synthesis] were applicable to the cyclization to obtain tertiary allylic alcohol silyl ethers 2a in good to excellent yields and enantioselectivities at 10 mol % nickel loading [L2: 68% yield, 89:11 er; L3: 98% yield, 97:3 er; L4: 98% yield, 99:1 er; entries 13–15]. We are excited to explore the potential of new ligand L4 in consideration of its excellent yield and enantioselectivity, especially for its catalyst loading with Ni(cod)2. To our delight, decreasing the nickel loading to 1 mol%, the highest yield and enantioselectivity (98% yield, 99:1 er) remained with L4 (entry 17), but only partial conversions were observed for L3 (40% yield), although the high er was maintained (97:3 er, entry 16) under standard conditions. We further reduced the catalyst loading to 0.1 mol% (TON = 1000); excellent enantioselectivity (99:1 er) was achevied for L4 with a full conversion at 60 °C (entry 19), but a partial conversion at 25 °C (45% yield, entry 18). Thus, fortunately we find that substrate 1a was completely converted to the product 2a with excellent yield and enantioselectivity at 60 °C for 12 h in the presence of 0.1 mol% Ni(cod)2 and 0.05 mol% (S,S)-DI-BI-DIME when scaled up to 2 mol substrate scale. The absolute configuration of 2a was assigned as R on the basis of the absolute configuration of desilylation product 3a determined by X-ray crystallography (see details in SI).

Reaction optimization. Nickel-catalyzed regiospecific asymmetric reductive cyclization of N-alkynones (1a) applying different ligands. Full screening conditions are reported in Table 1
Substrate scope
The high activity of L4 encouraged us to look into the substrate scope of the Ni-catalyzed intramolecular reductive coupling of N-alkynones. For this investigation, we adopted catalyst L4 and the conditions of entry 17 rather than those of entry 19 (Table 1) due to efficiency for convenience. As shown in Fig. 3, a series of aromatic N-alkynones with various substitution patterns and electronic properties were utilized to form products 2b-h in excellent yields (76–98%) and enantioselectivities (98:2 – > 99:1 er). Substrates with either electron-donating (2b, 2f, 2g) or electron-withdrawing (2c, 2d) substituents were all affordable to this asymmetric cyclization. Different substitution pattern with different electronic properties may have effects on the reactivity (especially for the yields), as both of 2e and 2f have better yields than 2b–2d even with lower catalyst loading [0.5 mol% compared with 1 mol% Ni(cod)2]. o-OMe substituted 2g was obtained under higher temperature (60 °C) with the full conversion of the substrate. A substrate containing an indole moiety 1h was also applicable for this transformation to obtain product 2h in an excellent yield and er (80% yield, 99:1 er) at 80 °C with 2 mol% Ni(cod)2 and 1 mol% L4. To our delight, under the same conditions as 2h (80 °C, 2 mol% Ni(cod)2, 1 mol% L4), aliphatic ketone 1i was also converted to the cyclization product 2i in 94% yield and > 99:1 er; an aliphatic alkyne was also transformed to a pyrrolidine product 2j in 84% yield and 92:8 er besides aromatic alkynes. The different N-protecting group also had a significant influence on its reactivity. Substrates with an N-Tf group and N-Bn group were less reactive, and products 2k and 2l were obtained in 19% yield and 10% yield, respectively, albeit with excellent er (98:2, > 99:1 separately). No formation of 2m was observed when an N-Boc group was employed for the substrate. The vastly different reactivity could be due to the conformational difference of the substrates exerted by various N-protecting groups. The bulky N-sulfonamides could have allowed the alkyne and ketone moieties to a close proximity, facilitating the cycloaddition mediated by a nickel catalyst to occur. We were curious about the size of the ring closure of this cyclization. Six-membered heterocycle were also synthesized from the accessible substrates 1n–1q. Piperidine products 2n–2q also gave in excellent yields and good enantioselectivities (95–98% yields, 90:10–89:11 er), albeit with the requirement of more Ni(cod)2 and ligand (5 mol% and 2.5 mol%, respectively). The diminished enantioselectivity of six-membered heterocycles in comparison with that of five-membered ones is possibly due to the conformational difference of the substrate. Six-membered cyclization could be more facile and efficient by development of a suitable new ligands. The absolute configuration of 2n–2q was confirmed as R on the basis of the X-ray crystallographic analysis of 3o, which is the desilylation product of 2o.

Substrate scope of asymmetric nickel-catalyzed reductive cyclization of N-alkynones. a Unless otherwise specified, the reactions were performed under nitrogen in dioxane (0.5 mL) at 25 °C for 12 h with 1 (0.2 mmol) in the presence of Ni(cod)2 (1 mol%), L4 (0.5 mol%), and triethylsilane (0.6 mmol); b 0.5 mol% Ni(cod)2, 0.25 mol% L4; c T = 60 °C. d 2 mol% Ni(cod)2, 1 mol% L4, T = 80 °C; e 4 mol% Ni(cod)2, 2 mol% L4; f 3o was the corresponding product after the desilylation of 2o, the absolute configuration of 2n–2q was assigned on the basis of the absolute configuration of 3o determined by X-ray crystallography, the absolute configuration of 2b–2l was assigned the same as 2a
Mechanistic considerations
The high enantioselectivities and yields prompted us to study the mechanism of this catalytic reaction and develop a stereochemical model. To understand whether the cycloaddition of 1a with the nickel complex takes place before the action of Et3SiH, a stoichiometric amount of [Ni(cod)2] was mixed with ligand L4 with stirring in dioxane at room temperature. The solution was turned dark brown quickly after the addition of substrate 1a. This process was monitored by React-IR; the three-dimensional spectra and trend chart of peak variation were shown in Fig. 4 and Fig. 5 separately. An absorption peak at 1701cm−1 of ketone functional group appeared after the addition of 1a and persisted before the addition of phosphorus ligand L4, indicating the cycloaddition of the carbonyl group did not proceed without the phosphorus ligand. However, the absorption peak slowly disappeared after the addition of a stoichiometric amount of L4, indicating that the cycloaddition of 1a with a nickel species bearing a bisphosphorus ligand L4 has occurred, which generate the key intermediate for the following reduction with Et3SiH.

Reaction monitoring. Three-dimensional spectra of stoichiometric reaction of [Ni(cod)2], 1a, and L4 monitored by React-IR

Kinetics of consumption of 1a. Trend chart of peak variation at 1701 cm−1 of stoichiometric reaction of [Ni(cod)2], 1a, and L4 monitored by React-IR
Based on these results and previous mechanistic study by Baxter and Montgomery49, and computational studies by Houk and colleagues47,48 on nickel-catalyzed intermolecular or intramolecular ynal reductive cyclizations, we proposed the catalytic cycle of this intramolecular asymmetric reductive cyclization of N-alkynones as dimeric metallacyclic model, which was depicted in Fig. 6. Bisphosphorus ligand L4 reacted with Ni(cod)2 to form the Ni0 species I, then addition of N-alkynone 1a generated the cyclization process through the stage II and provided dimer NiII metallacycle Ш. This is followed by coordination and σ-bond metathesis with triethylsilane to produce NiII hydride species IV. Final reductive elimination of IV provided the desired product 2a and regenerated the Ni0 catalyst, which completed the catalytic circulation of this trend. The enantioselectivity and stereoselectivity are apparently controlled by the cycloaddition stage Ш, two equivalent of Ni source elegantly coordinated to one equivalent of the unique bisphosphorus ligand L4 and two equivalent of N-alkynone 1a by the ketone and alkyne formed the cyclized intermediate Ш in high enantioselectivity and regioselectivity. Conformational analysis of metallacycle Ш with L4 as the ligand indicates that the steric interactions between the phenyl group on the metallacycle ring and the 2, 6-dimethoxyl phenyl moiety and P-chiral t-Bu group of L4 are rigid, which favored the R configuration of the following stages to the cyclization product 1a. The reason for the enantioselectivity enhancement of DI-BIDIME (L4) than BIDIME (L2) is possibly due to the greater steric hindrance of the stage Ш. The absolute configuration of product 3a determined by X-ray crystallography confirmed this hypothesis for the asymmetric control cycle.

Proposed mechanistic pathway. The proposed mechanism involves a dimeric metallacyclic model of asymmetric reductive cyclization of N-alkynones with nickel0 and bisphosphorus ligand L4
Synthetic utility of cyclization product
The highly enantioselective transformation allowed us to synthesize pyrrolidines with chiral tertiary alcohols and related derivatives. A gram-scale cyclization of 1a was carried out in dioxane at 60 °C in the presence of 0.1 mol% Ni(cod)2 and 0.05 mol% L4, and the product 2a (1.26 g) was obtained in 98% yield and 99:1 er (Fig. 7). Direct hydrogenation of the double bond in 2a over 10% Pd/C and H2 took place stereo specifically at the opposite side to the OSiEt3 group, forming compound 4a quantitatively with diasteroselectivity of 7:1 dr. After desilylation with TBAF (tetrabutylammonium fluoride), the two separable diastereoisomers with tertiary alcohol can be produced in 84% yield (5a) and 12% yield (5a’). To investigate the stereo effects of chirality and hindrance of OSiEt3 to the adjacent double bond reactions, we are curious to hydrogenate the free tertiary allylic alcohol 3a under the same condition as 4a, the isolated products 5a and 5a’ were provided in 39% yield and 60% yield separately. These results showed that the more bulky group at C3 position induced the major product as 5a with good diasteroselectivity (7:1 dr), but less diasteroselectivity (3:2 dr) with the other diastereoisomer 5a’ as major product. The results showed that the bulkiness of OH relating group has significant impact for the diasteroselectivity of hydrogenated product. This information may give suggestions for the synthesis of more complicated molecules with more than one chiral center. The double bond in 2a could be easily treated with ozone (O3) to give the ketone intermediate 6a in 90% yield, which is one versatile building block for many useful reactions, such as reductive aminations, Wittig reactions, and so on. For example, the olefin 8a can be obtained in 85% yield with CH3PPh3Br as the Wittig reagent from ketone 6a. By the way, deprotection of 6a could easily prepare chiral α-tertiary alcohol ketone 7a in 96% yield. The derivatives 6a, 7a, 8a, 9a, and 10a were anticipated to find wider applications in organic synthesis, which expanded the synthetic utility of this methodology.

Synthetic utility of cyclization products. Gram-scale reaction and further transformation of cyclization product 2a
Synthetic application to drug intermediates
To further demonstrate the synthetic utility of this methodology, we then focused on the synthesis of intermediates of the important drugs from available cyclized products. As showed in Fig. 8a, the chiral cyclization product 2o was converted to ketone compound 11 by ozonolysis reaction with 90% yield, followed by Wittig reaction utilizing CH3PPh3Br as the reagent to give the olefin 12 in 88% yield, then deprotection of silyl group with TBAF to afford the desired tertiary alcohol olefins 13 (96% yield), which could be transformed to the drug prodine (analgesic) by procedures16,18,44 reported in literature. The key intermediate of antidepressant (–)-paroxetine 16 was also achieved through similar steps (Fig. 8b). Ozonolysis of 2q provided ketone 14 in 90% yield and then delivered to olefin 15 with 86% yield, which underwent deprotection of trimethylsilyl group to give the desired tertiary alcohol olefins 16 in 96% yield. This intermediate could be converted to (–)-paroxetine through the similar procedures reported by Tang and colleagues16, and Zhou and colleagues18.

Synthetic application to drug intermediates. a Synthetic application to intermediates of prodine from 2o. b Synthetic application to intermediates of (–)-paroxetine from 2q
Discussion
In summary, we have developed a highly efficient regiospecific and enantioselective nickel-catalyzed intramolecular reductive cyclization of N-alkynones mediated by a nickel catalyst bearing P-chiral diphosphorus ligand with triethylsilane as the reducing reagent. A variety of pyrrolidine/piperidine derivatives bearing a chiral tertiary allylic alcohol silyl ether moiety were synthesized in high yields (up to 98% isolated yield) and excellent enantioselectivities (> 99:1 er). The highest TON of Ni-catalyzed reactions was reported as 1000 [0.1 mol% catalyst loading of Ni(cod)2] with developed unique ligand (S, S)-DI-BIDIME (L4), which is designed by ligand engineering through C7-bicoupling of privileged monophosphorus ligand (S)-BIDIME (L2). This method has proved to be a practical process for concise synthesis of pyrrolidines/piperidines with a chiral tertiary alcohol group. Mechanistic studies showed that the cycloaddition stage dimer NiII metallacycle Ш is the enantioselectivity-determining step, whereas the ligand L4 had an important role for providing a large π-conjugated system or steric interactions. A gram-scale preparation and a number of transformations of the cyclization product were conducted to show the capability and scalability of this methodology. The efficient synthesis of key intermediates of prodine and (–)-paroxetine has also been demonstrated for its applications in drug synthesis. Ligand design and engineering has proved to be an effective strategy for developing practical efficient asymmetric nickel-catalyzed transformations. The catalyst developed in this study has potential for application in other cyclization reactions. Further exploration on more efficient nickel catalyst, its detailed mechanistic study, and development of various efficient nickel-catalyzed asymmetric reactions are under investigation in our laboratory.
Methods
General procedure for asymmetric nickel-catalyzed intramolecular reductive cyclization of N-alkynones
Ni(cod)2 (2 μmol, 1 mol%), (S,S)-DI-BIDIME (L4, 1 μmol, 0.5 mol%), and dioxane (0.5 mL) were added to a 5 mL screw-cap vial equipped with a magnetic stirring bar in the glove box. Substrate 1 (0.2 mmol, 1.00 equiv.) was added to the solution in one portion, stirred for 5 min, followed by the addition of triethylsilane (Et3SiH, 0.6 mmol, 3.00 equiv.). The vial was closed with a screw cap and the resulting mixture was stirred at 25 °C for 12 h. Quenched with saturated sodium bicarbonate (NaHCO3), extracted with ethyl acetate (EtOAc), washed with saturated NaCl, dried over anhydrous sodium sulfate (Na2SO4), filtered, and concentrated under vacuum. The residue was purified by column chromatography (10% petroleum ether/EtOAc) to afford compound 2.
Synthesis and characterization
Full synthetic procedures and characterization for ligand DI-BIDIME and substrates 1a–1q are available in the Supplementary Methods and Supplementary Figures 1-9. High-performance liquid chromatography charts for racemic and chiral products 2a–2q are available in Supplementary Figures 10, 12-22, and 24-25. Full procedures for synthetic transformations to compounds 3a-16 are available in Supplementary Figures 26 and 27. 1H, 13C, 31P, and 19F NMR spectra of purified compounds are available in Supplementary Figures 28-83.
Crystallography
X-ray crystallographic CIF files, RFT files, and checkcif files for compounds 3a and 3o are available in Supplementary Data 1-6. X-ray-derived ORTEP representations of 3a and 3o are available in Supplementary Figures 11 and 23
Data availability
The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon reasonable request. The X-ray crystallographic coordinates for structures that support the findings of this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the accession code CCDC1532676 (3a) and CCDC1532677 (3o). These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: (+44)1223-336-033; or deposit@ccdc.cam.ac.uk).
References
Ojima, I., Tzamarioudaki, M., Li, Z. & Donovan, R. J. Transition metal-catalyzed carbocyclizations in organic synthesis. Chem. Rev. 96, 635–662 (1996).
Montgomery, J. Nickel-catalyzed cyclizations, couplings, and cycloadditions involving three reactive components. Acc. Chem. Res. 33, 467–473 (2000).
Skucas, E., Ngai, M.-Y., Komanduri, V. & Krische, M. J. Enantiomerically enriched allylic alcohols and allylic amines via C–C bond-forming hydrogenation: asymmetric carbonyl and imine vinylation. Acc. Chem. Res. 40, 1394–1401 (2007).
Kablaoui, N. M. & Buchwald, S. L. Development of a method for the reductive cyclization of enones by a titanium catalyst. J. Am. Chem. Soc. 118, 3182–3191 (1996).
Sato, Y. et al. Novel stereoselective cyclization via –allylnickel complex generated from 1,3-diene and hydride nickel complex. J. Am. Chem. Soc. 116, 9771–9772 (1994).
Oblinger, E. & Montgomery, J. A new stereoselective method for the preparation of allylic alcohols. J. Am. Chem. Soc. 119, 9065–9066 (1997).
Chevliakov, M. V. & Montgomery, J. Nickel and palladium catalysis in the stereoselective synthesis of functionalized pyrrolidines: enantioselective formal synthesis of (+)-α-allokainic acid. Angew. Chem. Int. Ed. 37, 3144–3146 (1998).
Sato, Y., Saito, N. & Mori, M. A novel asymmetric cyclization of ω-formyl-1,3-dienes catalyzed by a zerovalent nickel complex in the presence of silanes. J. Am. Chem. Soc. 122, 2371–2372 (2000).
Miller, K. M. & Jamison, T. F. Ligand-switchable directing effects of tethered alkenes in nickel-catalyzed additions to alkynes. J. Am. Chem. Soc. 126, 15342–15343 (2004).
Chaulagain, M. R., Sormunen, G. J. & Montgomery, J. New N-heterocyclic carbene ligand and its application in asymmetric nickel-catalyzed aldehyde/alkyne reductive couplings. J. Am. Chem. Soc. 129, 9568–9569 (2007).
Ni, Y., Kassab, R. M., Chevliakov, M. V. & Montgomery, J. Total syntheses of isodomoic acids G and H: an exercise in tetrasubstituted alkene synthesis. J. Am. Chem. Soc. 131, 17714–17718 (2009).
Ohashi, M., Taniguchi, T. & Ogoshi, S. Nickel-catalyzed formation of cyclopentenone derivatives via the unique cycloaddition of α,β-unsaturated phenyl esters with alkynes. J. Am. Chem. Soc. 133, 14900–14903 (2011).
Stolley, R. M., Duong, H. A., Thomas, D. R. & Louie, J. The discovery of [Ni(NHC)RCN]2 species and their role as cycloaddition catalysts for the formation of pyridines. J. Am. Chem. Soc. 134, 15154–15162 (2012).
Nakai, K., Yoshida, Y., Kurahashi, T. & Matsubara, S. Nickel-catalyzed redox-economical coupling of alcohols and alkynes to form allylic alcohols. J. Am. Chem. Soc. 136, 7797–7800 (2014).
Tasker, S. Z., Standley, E. A. & Jamison, T. F. Recent advances in homogeneous nickel catalysis. Nature 509, 299–309 (2014).
Fu, W., Nie, M., Wang, A., Cao, Z. & Tang, W. Highly enantioselective nickel-catalyzed intramolecular reductive cyclization of alkynones. Angew. Chem. Int. Ed. 54, 2520–2524 (2015).
Clarke, C., Incerti-Pradillos, C. A. & Lam, H. W. Enantioselective nickel-catalyzed anti-carbometallative cyclizations of alkynyl electrophiles enabled by reversible alkenylnickel E/Z isomerization. J. Am. Chem. Soc. 138, 8068–8071 (2016).
Li, K., Li, M.-L., Zhang, Q., Zhu, S.-F. & Zhou, Q.-L. Highly enantioselective nickel-catalyzed intramolecular hydroalkenylation of N- and O-tethered 1,6-dienes to form six-membered heterocycles. J. Am. Chem. Soc. 140, 7458–7461 (2018).
Shitani, R., Okamoto, K., Otomaru, Y., Ueyama, K. & Hayashi, T. Catalytic asymmetric arylative cyclization of alkynals: phosphine-free rhodium/diene complexes as efficient catalysts. J. Am. Chem. Soc. 127, 54–55 (2005).
Rhee, J. U. & Krische, M. J. Highly enantioselective reductive cyclization of acetylenic aldehydes via rhodium catalyzed asymmetric hydrogenation. J. Am. Chem. Soc. 128, 10674–10675 (2006).
Fan, B.-M., Xie, J.-H., Li, S., Wang, L.-X. & Zhou, Q.-L. Highly enantioselective hydrosilylation/cyclization of 1,6-enynes catalyzed by rhodium(I)complexes of spiro diphosphines. Angew. Chem. Int. Ed. 46, 1275–1277 (2007).
Tanaka, R., Noguchi, K. & Tanaka, K. Rhodium-catalyzed asymmetric reductive cyclization of heteroatom-linked 5-alkynals with heteroatom-substituted acetaldehydes. J. Am. Chem. Soc. 132, 1238–1239 (2010).
Masuda, K., Sakiyama, N., Tanaka, R., Noguchi, K. & Tanaka, K. Rhodium-catalyzed enantioselective cyclizations of γ-alkynylaldehydes with acyl phosphonates: ligand- and substituent-controlled C–P or C–H bond cleavage. J. Am. Chem. Soc. 133, 6918–6921 (2011).
Li, Y. & Xu, M.-H. Rhodium-catalyzed asymmetric tandem cyclization for efficient and rapid access to underexplored heterocyclic tertiary allylic alcohols containing a tetrasubstituted olefin. Org. Lett. 16, 2712–2715 (2014).
Ngai, M.-Y., Barchuk, A. & Krische, M. J. Iridium-catalyzed C−C bond forming hydrogenation: direct regioselective reductive coupling of alkyl-substituted alkynes to activated ketones. J. Am. Chem. Soc. 129, 280–281 (2007).
Patman, R. L., Chaulagain, M. R., Williams, V. M. & Krische, M. J. Direct vinylation of alcohols or aldehydes employing alkynes as vinyl donors: a ruthenium catalyzed C−C bond-forming transfer hydrogenation. J. Am. Chem. Soc. 131, 2066–2067 (2009).
Park, B. Y., Nguyen, K. D., Chaulagain, M. R., Komanduri, V. & Krische, M. J. Alkynes as allylmetal equivalents in redox-triggered C–C couplings to primary alcohols: (Z)-homoallylic alcohols via ruthenium-catalyzed propargyl C–H oxidative addition. J. Am. Chem. Soc. 136, 11902–11905 (2014).
Liang, T., Nguyen, K. D., Zhang, W. & Krische, M. J. Enantioselective ruthenium-catalyzed carbonyl allylation via alkyne–alcohol C–C bond-forming transfer hydrogenation: allene hydrometalation vs oxidative coupling. J. Am. Chem. Soc. 137, 3161–3164 (2015).
Zhao, L. & Lu, X. PdII-catalyzed cyclization of alkynes containing aldehyde, ketone, or nitrile groups initiated by the acetoxypalladation of alkynes. Angew. Chem. Int. Ed. 41, 4343–4345 (2002).
Shen, K., Han, X. & Lu, X. Cationic Pd(II)-catalyzed reductive cyclization of alkyne-tethered ketones or aldehydes using ethanol as hydrogen source. Org. Lett. 15, 1732–1735 (2013).
Hu, N., Li, K., Wang, Z. & Tang, W. Synthesis of chiral 1,4-benzodioxanes and chromans by enantioselective palladium-catalyzed alkene aryloxyarylation reactions. Angew. Chem. Int. Ed. 55, 5044 (2016).
Hu, N. et al. Synthesis of chiral α-amino tertiary boronic esters by enantioselective hydroboration of α-arylnamides. J. Am. Chem. Soc. 137, 6746–6749 (2015).
Xu, G., Fu, W., Liu, G., Senanayake, C. H. & Tang, W. Efficient syntheses of korupensamines A, B and michellamine B by asymmetric Suzuki-Miyaura coupling reactions. J. Am. Chem. Soc. 136, 570–573 (2014).
Du, K. et al. Enantioselective palladium-catalyzed dearomative cyclization for the efficient synthesis of terpenes and steroids. Angew. Chem. Int. Ed. 54, 3033–3037 (2015).
Nakao, Y., Kashihara, N., Kanyiva, K. S. & Hiyama, T. Nickel-catalyzed alkenylation and alkylation of fluoroarenes via activation of C−H bond over C−F bond. J. Am. Chem. Soc. 130, 16170–16171 (2008).
Johnson, S. A., Huff, C. W., Mustafa, F. & Saliba, M. Unexpected intermediates and products in the C−F bond activation of tetrafluorobenzenes with a bis(triethylphosphine)nickel synthon: direct evidence of a rapid and reversible C−H bond activation by Ni(0). J. Am. Chem. Soc. 130, 17278–17280 (2008).
Song, W., Lackner, S. & Ackermann, L. Nickel-catalyzed C-H alkylations: direct secondary alkylations and trifluoroethylations of arenes. Angew. Chem. Int. Ed. 53, 2477–2480 (2014).
Tobisu, M. & Chatani, N. Cross-couplings using aryl ethers via C−O bond activation enabled by nickel catalysts. Acc. Chem. Res. 48, 1717–1726 (2015).
Tobisu, M., Takahira, T., Morioka, T. & Chatani, N. Nickel-catalyzed alkylative cross-coupling of anisoles with grignard reagents via C–O bond activation. J. Am. Chem. Soc. 138, 6711–6714 (2016).
Moloney, M. G. Excitatory amino acids. Nat. Prod. Rep. 15, 205–219 (1998).
Wagstaff, A. J., Cheer, S. M., Matheson, A. J., Ormrod, D. & Goa, K. L. Paroxetine. Drugs 62, 655–703 (2002).
Ma, D. et al. Synthesis and biological evaluation of 1,3,3,4-tetrasubstituted pyrrolidine CCR5 receptor antagonists. Discovery of a potent and orally bioavailable anti-HIV agent. ChemMedChem 2, 187–193 (2007).
Hu, Y. et al. (±)-Homocrepidine A, a pair of anti-inflammatory enantiomeric octahydroindolizine alkaloid dimers from Dendrobium crepidatum. J. Nat. Prod. 79, 252–256 (2016).
Bell, K. H. & Portoghese, P. S. Stereochemical studies on medicinal agents. 14. relative stereochemistries and analgetic potencies of diastereomeric 3-allyl and 3-propyl derivatives of 1-methyl-4-phenyl-4-propionoxypiperidine. J. Med. Chem. 16, 203–205 (1973).
Bell, K. H. & Portoghese, P. S. Stereochemical studies on medicinal agents. 17. Synthesis, absolute configuration, and analgetic potency of enantiomeric diastereomers of 3-ethyl and 3-propyl derivatives of 1-methyl-4-phenyl-4-propionoxypiperidine. J. Med. Chem. 17, 129–131 (1974).
Yu, J., Shi, F. & Gong, L.-Z. Brønsted-acid-catalyzed asymmetric multicomponent reactions for the facile synthesis of highly enantioenriched structurally diverse nitrogenous heterocycles. Acc. Chem. Res. 44, 1156–1171 (2011).
McCarren, P. R., Liu, P., Cheong, P. H.-Y., Jamison, T. F. & Houk, K. N. Mechanism and transition-state structures for nickel-catalyzed reductive alkyne−aldehyde coupling reactions. J. Am. Chem. Soc. 131, 6654–6655 (2009).
Haynes, M. T. II et al. Dimer involvement and origin of crossover in nickel-catalyzed aldehyde–alkyne reductive couplings. J. Am. Chem. Soc. 136, 17495 (2014).
Baxter, R. D. & Montgomery, J. Mechanistic study of nickel-catalyzed ynal reductive cyclizations through kinetic analysis. J. Am. Chem. Soc. 133, 5728–5731 (2011).
Acknowledgements
Grants from NSFC (numbers 21432007 and 21572246), CAS (QYZDY-SSW-SLH029), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000) and ‘Jun-Ma’ Innovation Program of Inner Mongolia Province, and Inner Mongolia University are gratefully acknowledged. We also appreciate Miss Wen-min Wu for her generous help in the analysis of NMR and HRMS.
Author information
Authors and Affiliations
Contributions
W.T. and G.L. conceived and designed the experiments. G.L. and W.F. performed the experiments and prepared the Supplementary Information. X.M. and T.W. helped in substrates synthesis. M.N. helped in ligand synthesis. K.L. and X.X. helped in isolating and analyzing the data. W.T and G.L. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, G., Fu, W., Mu, X. et al. Pyrrolidines and piperidines bearing chiral tertiary alcohols by nickel-catalyzed enantioselective reductive cyclization of N-alkynones. Commun Chem 1, 90 (2018). https://doi.org/10.1038/s42004-018-0092-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-018-0092-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
