
Abstract
Sarcopenia is a common skeletal muscle disease in older people. Lower limb muscle strength is a good predictive value for sarcopenia; however, little is known about its genetic components. Here, we conducted a genome-wide association study (GWAS) for knee extension strength in a total of 3452 Japanese aged 60 years or older from two independent cohorts. We identified a significant locus, rs10749438 which is an intronic variant in TACC2 (transforming acidic coiled-coil-containing 2) (P = 4.2 × 10−8). TACC2, encoding a cytoskeleton-related protein, is highly expressed in skeletal muscle, and is reported as a target of myotonic dystrophy 1-associated splicing alterations. These suggest that changes in TACC2 expression are associated with variations in muscle strength in older people. The association was consistently observed in young and middle-aged subjects. Our findings would shed light on genetic components of lower limb muscle strength and indicate TACC2 as a potential therapeutic target for sarcopenia.
Similar content being viewed by others
Introduction
Sarcopenia is a common skeletal muscle disease in older people, which leads to unfavorable outcomes such as falls, fractures and death1,2,3. Sarcopenia can be defined by using muscle mass, muscle strength and physical performance4,5,6. The loss of muscle mass has mainly been used as an indicator of sarcopenia; however, recent studies suggest that muscle strength is a better indicator that reflects adverse health outcomes of sarcopenia2,7,8,9. For example, Schaap et al. described that low handgrip strength was associated with incidence of falling, independent of a muscle mass2. Therefore, muscle strength, rather than a muscle mass, is adopted as the primary indicator for sarcopenia in the revised European Working Group on Sarcopenia in Older People (EWGSOP) algorithm5.
As an indicator for muscle strength, handgrip strength is commonly used; however, it only reflects the strength of the upper extremities, not that of lower extremities. A recent study reported that knee extension strength, a proxy of lower limb strength, is more strongly associated with performance-based sarcopenia compared to handgrip strength9,10. Correspondingly, Yeung et al. reported stronger association of knee extension strength with health characteristics than handgrip strength11. Thus, knee extension strength would be a more appropriate indicator of sarcopenia than handgrip strength.
Muscle strength has been known to be heritable12,13,14. Family studies showed the heritability of handgrip strength was 56%12 and genome-wide association studies (GWASes) of handgrip strength suggested the heritability was 13–24%15,16. Previous GWASes have discovered 170 variants associated with muscle strength15,16,17,18,19. One GWAS on maximum handgrip strength divided by weight in UK Biobank participants identified 101 loci and showed a shared genetic etiology of handgrip with cardiometabolic and cognitive health15. Another GWAS of muscle weakness based on handgrip strength in Europeans aged 60 years or older identified 15 loci17. However, there is only one GWAS of lower limb muscle strength, which did not identify any significant loci18 due to a lack of statistical power. GWASes of lower limb muscle strength with adequate sample size would add good information for understanding of the genetic architecture of sarcopenia.
In the present study, we performed a GWAS of knee extension strength using 3452 participants aged 60 years or older from two independent cohorts. We identified a locus with genome-wide significance, which has not been identified in previous GWASes of muscle strength. In the locus, we identified a candidate susceptibility gene, TACC2 (transforming acidic coiled-coil-containing 2) which is highly expressed in skeletal muscle. We also identified several suggestive loci, which include promising candidate susceptibility genes. The variants associated with handgrip strength in a previous GWAS15 showed an association in our dataset.
Results
Sample source, genotyping and imputation
The study design was illustrated in Supplementary Fig. 1. Three sets of samples from two independent cohorts were enrolled in this study, which consisted of a total of 3478 participants aged 60 years or older (Table 1). Set 1 and Set 2 consisted of 1014 and 841 participants. Both were from Shimane CoHRE Study20,21, but different arrays were used for genotyping. Set 3 consisted of 1623 participants from Bunkyo Health Study22. While the methods to measure knee extension strength were slightly different between the two study cohorts, the patterns of distributions of knee extension strength were very consistent (Supplementary Figs. 2, 3).
We used Illumina HumanOmniExpressExome BeadChip for genotyping 1014 participants in Set 1. After quality controls of samples and variants, data on a total of 1007 samples remained for the further analysis (Supplementary Fig. 1). We conducted a whole-genome imputation using an in-house reference panel containing a total of 3256 Japanese whole-genome sequence data and 2504 individuals in the 1000 Genomes Project (1KG phase 3v5). We set a threshold of Rsq more than 0.3 for variants to be included in the following analyses. An association analysis was conducted by the linear mixed model using fastGWA23. The top three principal components (PCs) were used as covariates. Association results in this data set are shown in Supplementary Fig. 4.
We used Illumina Asian Screening Array for genotyping 841 and 1623 participants in Set 2 and 3, respectively. After quality controls, 838 and 1607 samples remained in Set 2 and 3, respectively (Supplementary Fig. 1). We conducted the whole-genome imputation as described above. Results in each data set were shown in Supplementary Figs. 5 and 6.
Genome-wide association studies and a meta-analysis
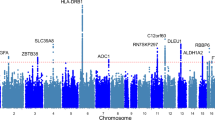
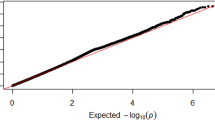
Then, we conducted a meta-analysis of three GWASes using the fixed-effect inverse-variance weighted method with the use of the METAL software24. We took an intersection of imputed variants across the three data sets, resulting in 9,146,474 autosomal variants and 197,639 chromosome X variants. The Manhattan plot and Q-Q plot of the meta-analysis are shown in Fig. 1. We did not find an inflation of statistics (inflation factor (λGC) of 1.02) and linkage disequilibrium score regression (LDSC) revealed an intercept of 1.00 (SE, 0.0071), indicating that the current results were not confounded by any bias and no apparent strong polygenic effects on the muscle strength in lower limbs presumably due to the limited sample size in the current study. A heritability calculated by LDSC was 8.9%, indicating a substantial contribution of genetic components on the lower limb muscle strength.

a Manhattan plot. X-axis: chromosomal location. Y-axis: −log10 p-value for each genetic variant. Horizontal red line: genome-wide significance (P < 5 × 10−8). Horizontal blue line: suggestive genome-wide significance (P < 1 × 10−5). A locus with genome-wide significance and a locus with suggestive genome-wide significance were identified on chromosomes 10 and 11, respectively. b Q-Q plot for the analysis. The genomic inflation factor (λGC) was 1.02.
A novel locus associated with knee extension strength
We identified a novel locus significantly associated with knee extension strength (Fig. 1 and Table 2). The lead variant is rs10749438, an intronic variant in TACC2 (transforming acidic coiled-coil-containing 2) located at 10q26 (Beta = −0.15, P = 4.2 × 10−8) (Fig. 2a and Table 2). The risk allele for muscle weakness is allele A. We observed consistent associations of this variant across the three data sets and no heterogeneity of association results was observed (I2 = 0, Fig. 3). rs10749438 was positioned at enhancer-like histone marks, H3K27ac in skeletal muscle according to the ENCODE database25 and HaploReg (v4.1)26. A statistical fine-mapping analysis revealed rs10749438 with the highest posterior probability (Supplementary Table 1). TACC2 encodes a cytoskeleton-related protein27 that concentrates at centrosomes throughout the cell cycle and is reported as a target of myotonic dystrophy 1-associated splicing alterations28. Skeletal muscle showed high TACC2 expression according to Genotype-Tissue Expression project version 8 (GTEx v8)29 (Supplementary Fig. 7).

a The significant locus with the lead variant (rs10749438) and (b) the suggestive locus with the lead variant (rs6483495) associated with knee extension strength.

Consistent associations were observed across the three datasets. META, the effect size of meta-analysis. Error bar, 95% confidence interval. Beta are shown in Table 2.
Since TACC2 was reported to be susceptible to transcriptional regulation effect of androgen receptor30 and our three data sets were female-dominant, we conducted sex-specific analysis and observed the effect size and direction of rs10749438 are consistent regardless of sex (Table 3 and Supplementary Tables 2, 3). The age-stratified analysis also showed that effect directions were consistent (Table 3 and Supplementary Table 4). We further conducted analyses using a total of 173 subjects aged under 60 years to evaluate whether this association was specific to older people or not (Supplementary Fig. 8 and Supplementary Table 5). The analysis of the participants aged under 60 years also showed a consistent trend of the association (Table 3 and Supplementary Table 6). While the effect size tended to be strong in the participants aged 75 years or older in comparison with that aged 60 years or older, the trend was not held in subjects under 60 years. The meta-analysis revealed a further increased association of this variant (P = 1.2 × 10−8, I2 = 0; Table 4 and Supplementary Fig. 9), suggesting that the association might be observed in a general population.
Suggestive loci associated with knee extension strength
We also identified 17 suggestive loci (P < 1.0 × 10−5, Table 2). Among these loci, rs1718074 in an intron of the dystrophin gene (DMD) located at Xp21.2-p21.1 is the most noteworthy (Beta = 0.14, P = 2.9 × 10−6) (Table 2). DMD is the disease gene for Duchenne muscular dystrophy and Becker muscular dystrophy, both of which show progressive deterioration of muscle tissue and resultant weakness31. The effect size and the effect direction of rs1718074 were consistent between males and females (Supplementary Tables 2 and 3).
rs6483495, an intronic variant of MAML2 located at 11q21, showed the borderline significant p-value (Beta = −0.14, P = 5.4 × 10−8) (Fig. 2b and Table 2). The locus has not been reported in muscle-related GWASes. Skeletal muscle does not highly express MAML2. While we observed consistent associations of this locus across the three data sets (I2 = 0, Table 2), when we expanded the participants to those aged under 60 years, we observed the opposite direction compared to the result of older participants (Supplementary Table 6). These findings suggest that further studies are necessary to confirm the association between MAML2 and knee extension strength.
Evaluation of the variants identified in a GWAS of handgrip strength
We further tested whether the variants associated with handgrip strength15,16,17 showed associations in our dataset. Out of 170 variants associated with three GWASes of handgrip strength, 132 were included in our dataset, and 18 proxy variants were selected for the test based on the linkage disequilibrium (LD) of Europeans. Among the 150 variants, 87 showed the same direction of effect and a binomial test p-value was 0.03. Among the 87 variants, five showed an association of nominal statistical significance (expected number: 4.35). These findings suggest that muscle strength of upper and lower limbs may share a small part of genetic architecture.
Discussion
We conducted the GWAS of knee extension strength. There are a few limitations to our study. First, the sample size is not large enough to detect many genome-wide significant loci. Future studies with large sample sizes would be necessary. Second, we used different devices to measure the knee extension strength in the two cohorts; that is, one used isometric testing and the other isokinetic testing. We believe that the impact of the difference of the testings on the association is expected to be small since these testings are reported to be highly correlated32. Their distributions were very consistent (Supplementary Figs. 2, 3).
Between knee extension strength and handgrip strength, a poor to moderate correlation has been reported9,33,34,35, which could explain their substantial but relatively weak shared directions of effects. Accordingly, our results suggest that only small fraction of genetic architecture is shared between muscle strength of upper and lower limbs. While lower limb muscle strength is reported to be more strongly associated with sarcopenia than handgrip strength9,10,11, most GWASes for muscle strength were based on handgrip strength15,16. Thus, our identification of the genetic variants associated with lower limb strength would shed light on the etiology of sarcopenia.
We successfully identified the locus with the genome-wide significance, which contained a candidate gene, TACC2. The association was the same as a meta-analysis with the random effect model (Beta = −0.148, P = 4.2 × 10−8). We additionally investigated if there is a possible confounding effect of knee osteoarthritis on knee extension muscle strength by using knee osteoarthritis as an additional covariate, but we found no confounding effect of knee osteoarthritis (Beta = −0.15, P = 3.7 × 10−8). To the best of our knowledge, the present study is the first GWAS for lower limb muscle strength that identified a significant locus. In line with our results, a previous GWAS of muscle weakness based on handgrip strength in European elderly showed the consistent association between rs10749438 and muscle weakness with nominal statistical significance (P = 0.037)17. We also investigated if rs10749438 is associated with other sarcopenia-related traits such as lean body mass36, frailty37, walking pace38, fatigue39, testosterone40 and IGF141 in the UK Biobank, which did not show any nominal significant association. rs10749438 is located at enhancer-like histone marks, H3K27ac in skeletal muscle and TACC2 is highly expressed in skeletal muscle. While the top posterior probability and overlapping with the enhancer region suggest rs10749438 as a promising candidate of a causal variant, functional follow-up is necessary to conclude this point. Regarding a responsible gene in this association, using cell cultures from human embryonic muscle, myotonic dystrophy 1-associated splicing alterations were significantly enriched in TACC2 which is one of cytoskeleton-related gene27,28. Although the variants are not an expression quantitative trait locus (eQTL) for TACC2 according to GTEx v829, there is a possibility that the variant’s functional effect is more context-dependent. In fact, sampling site of muscle in GTEx is not quadriceps femoris muscle but gastrocnemius muscle. These findings suggest that TACC2 is a good candidate gene for muscle strength and further experimental validation using animal models will be needed. Another possibility is that the variant regulates other distant genes, ATE1, NSMCE4A and BTBD16. These genes are also candidate of causal genes for knee extension strength. Additionally, there is a possibility that the variant’s functional effect on TACC2 or another gene is more context-dependent and existing eQTL studies may not have detected such effects yet.
TACC2 belongs to the TACC protein family which involves in the complex process of regulating microtubule dynamics during cell division42. TACC genes lie within a chromosomal region associated with tumorigenesis. Mammalian TACC proteins, namely TACC1, TACC2 and TACC3 interact with microtubules, and control cell growth and differentiation during cell division43. Several studies indicated high expression of TACC2 was involved in tumorigenesis of a variety of cancers30,44,45,46. These findings suggest TACC2 may function in muscle via modulating cell division. Further studies would be necessary to clarify the role of TACC2 in muscle strength and sarcopenia.
We also identified 17 suggestive loci including candidate causal variants. While these variants are good candidates for further replication analyses, we should be cautious of variants with low minor allele frequencies due to possible inaccurate imputation compared with common variants. That is another limitation of the study and future studies with larger dataset will be needed to confirm the associations. We investigated if those suggestive variants are associated with sarcopenia-related traits such as lean body mass, frailty, walking pace fatigue, testosterone and IGF1 in the UK Biobank. We did not find very consistent patterns of associations (Supplementary Note), suggesting that ancestry matching for GWAS and further expansion of sample size for muscle strength is necessary.
In summary, we identified a novel locus associated with knee extension strength. This finding provides insights into the genetic architectures underlying muscle strength in lower limbs. It would be interesting to integrate the current results with studies of sarcopenia in the future.
Methods
Shimane CoHRE study
This cross-sectional study is a part of the cohort study conducted by the Center for Community-based Healthcare Research and Education in Shimane University (Shimane CoHRE Study)19,20. Shimane CoHRE Study is an ongoing health examination for the community-dwelling people in Shimane prefecture, Japan. It complied with all relevant ethical regulations. The study protocol was approved by the Ethics Committee of Shimane University School of Medicine. Written informed consent was obtained from all participants. Based on different recruitment and genotyping terms, two data sets, Shimane 1st cohort (Set 1) and Shimane 2nd cohort (Set 2) were obtained. The participants of Set 1 and Set 2 were analyzed separately for those aged 60 years or older and those aged under 60 years. The characteristics of the participants were shown in Table 1 and Supplementary Table 5. The participants were all Japanese. We did not exclude any participants with knee osteoarthritis in the study.
Bunkyo Health Study
Bunkyo Health Study is a prospective cohort study of over 10 years21, which recruited older subjects aged 65–84 years living in Bunkyo-Ku, an urban area in Tokyo, Japan. Among the 68 communities in Bunkyo-Ku, we selected 13 communities based on probability proportionate to size sampling. We obtained the name and address of all residents aged 65–84 years in the selected communities from residential registries. The exclusion criteria were to have a pacemaker or defibrillator placement and diabetes mellitus requiring insulin therapy. All participants provided written informed consent. The details of the characteristics were shown in Table 1. The participants were all Japanese. We did not exclude any participants with knee osteoarthritis in the study.
Phenotype
In Shimane CoHRE Study, knee extension strength was measured by using the Quadriceps Training Machine (QTM) (QTM-05F, Alcare, Tokyo, Japan). The device has a knee holding part corresponding to the knee joint with approximately 30° flexion. Participants were asked to put some muscle as hard as possible for three seconds, and the maximum value that was reached during that time period was recorded. Both legs were measured in turn. We calculated relative knee extension strength as an average of measurements of the right and left legs divided by weight. The average of measurements was regressed and residualized by age and sex, and the residuals were inverse-rank normalized and used as quantitative phenotypes.
In Bunkyo Health Study (Set 3), knee extension muscle strength was measured by using the BIODEX system 4 (Biodex Medical Systems, Upton, New York, USA), which measures isokinetic knee muscle strength. To measure a value close to the maximum extension muscle force, we adopted the maximum torque at an angular velocity of 60°. As in Set 1 and Set 2, we calculated relative knee extension strength as an average of measurements of the right and left legs divided by weight. The average of measurements was regressed and residualized by age and sex, and the residuals were inverse-rank normalized and used as quantitative phenotypes.
Genotyping and quality control
We genotyped samples of Set 1 with the Illumina HumanOmniExpressExome BeadChip and those of Set 2 and Set 3 with the Illumina Asian Screening Array.
For quality control of samples, we excluded those with: (1) sex inconsistency between genotype and clinical data, (2) genetically identical to others (PI_HAT > 0.9, PI_HAT was based on identity by decent (IBD), i.e., P(IBD = 2) + 0.5*(IBD = 1)), (3) sample call rate < 0.98, and (4) outliers from East Asian clusters identified by PC analysis using genotypes in the HAPMAP project. For quality control of genotypes, we excluded variants meeting any of the following criteria: (1) call rate < 0. 99, (2) Hardy–Weinberg equilibrium p < 1.0 × 10−6, (3) the allele frequency show difference between the reference > = 6% compared with the reference panel.
Imputation
We utilized the 1000 Genomes Project Phase 3 [1KGP3v5; (May 2013, n = 2504)] and 3256 in-house Japanese whole-genome sequence data obtained from the Biobank Japan47 (JEWEL_3K) for imputation to achieve better imputation accuracy for the Japanese population as previously described48. In brief, samples were sequenced at high depth (15x, 30x) on various platforms. The whole-genome sequencing data was processed, following the standardized best practice method in Genome Analysis Toolkit (GATK). In addition to the process of the best practice, we put additional filters of approximate read depth and genotype quality before variant quality score recalibration (VQSR). The variants at multi-allelic sites were removed from the combined reference panel by vcftools (version 0.1.14). We estimated the haplotypes by SHAPE IT (version 2.778) and combined the data of the 1KG phase 3v5 and the BBJ by using IMPUTE249,50. Quality control was performed with bcftools (version 1.3.1) and vcftools (version 0.1.14). Variants at multi-allelic sites, monomorphic sites and singletons were excluded. We performed pre-phasing using EAGLE2.4.1 (https://alkesgroup.broadinstitute.org/Eagle/) to determine the haplotypes. We imputed the genotype dosages with minimac4 (v1.0.0)51. After imputation, we excluded variants with an imputation quality of Rsq < 0.3 and minor allele frequency <0.005. Imputation quality of Rsq is the estimated value of the squared correlation between imputed genotypes and true, unobserved genotypes.
GWAS and meta-analysis
We conducted GWAS using the fastGWA23 linear mixed model package and used the top three PCs as covariates. We excluded variants with minor allele frequencies <0.005. We conducted GWASes separately for three sets and performed an inverse variance fixed-effects meta-analysis by using METAL24. For the X chromosome, we performed GWAS in males and females separately and meta-analyzed using METAL24. METAL also calculates I2 which describes the percentage of variation across studies that is due to heterogeneity rather than chance. We annotated the variants which exceeded the significant threshold (P < 5 × 10−8) and the suggestive threshold (P < 1 × 10−5) in the GWAS by using ANNOVAR52, HaploReg26 and ENCODE database25.
LDSC
We estimated the heritability of the knee extension strength GWAS result using LDSC (version 1.0.0). We excluded variants in the human leukocyte antigen region (chromosome 6: 26–34 Mb). We further calculated heritability z-scores and standard errors (SEs) to assess the reliability of heritability estimation. The heritability is based on the variants in additive model53.
Age- and sex-stratified analyses
To investigate the effect of age in the association of significant and suggestive variants in the participants aged 60 years or older, we conducted age-stratified analyses, including participants aged under 60 years and those aged 75 years or older. Since the linear mixed model did not converge due to the small sample size in these analyses, we performed a linear regression by using PLINK 2.0 after excluding related individuals (PI_HAT > 0.25, Supplementary Fig. 8). We also conducted sex-stratified analyses by using PLINK 2.0 in the same manner. We showed the results of statistical power analyses in Supplementary Fig. 10.
Bayesian statistical fine-mapping analysis
We performed statistical fine-mapping analysis using FINEMAP software (version 1.3.1)54 to prioritize causal variants in susceptible loci. The FINEMAP computes a posterior probability of causality for each variant. We ranked candidate putative causal variants in a descending order of their posterior probabilities and created a 95% credible set of causal variants by adding the posterior probabilities of the ordered variants until their cumulative posterior probabilities reached 0.95. We used the default priors and parameters in FINEMAP.
Evaluation of the variants identified in a GWAS of handgrip strength
We investigated if 140 variants identified in the GWAS of handgrip strength15 had the same direction of effect in our GWAS. Since our dataset includes only 108 variants out of those variants, we used 12 high LD variants (r2 > 0.8) with other variants. We calculated LD based on 1KG European ancestry data. We conducted a binomial test; 120 variants were tested and 0.5 was the expected proportion of variants with the same direction of effect.
Statistics and reproducibility
We did not perform any statistical method to predetermine sample size because we used all available samples we have to maximize statistical power. GWAS were performed by using fastGWA23 linear mixed model package and used the top three PCs as covariates. A meta-analysis was performed by using METAL24. Significant threshold of the GWAS meta-analysis is p ≤ 5 × 10−8 accounting for multiple testing. For the evaluation of the variants identified in a GWAS of handgrip strength, we conducted binomial test by using R (version 4.0.2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The GWAS summary statistics generated in this study is available in the JENGER database and GWAS catalog (http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90319001-GCST90320000/GCST90319502). The remaining data are available with in the article, Supplementary Information and Source Data file. The source data behind the graphs in the paper can be found in Supplementary Data 1.
Code availability
The code of statistical analyses is available on GitHub (URL: https://github.com/Shuji2022/Code) and is also archived in Zenodo (URL: https://doi.org/10.5281/zenodo.10675274)55.
References
Bischoff-Ferrari, H. A. et al. Comparative performance of current definitions of sarcopenia against the prospective incidence of falls among community-dwelling seniors age 65 and older. Osteoporos. Int. 26, 2793–2802 (2015).
Schaap, L. A., van Schoor, N. M., Lips, P. & Visser, M. Associations of sarcopenia definitions, and their components, with the incidence of recurrent falling and fractures: the longitudinal aging study Amsterdam. J. Gerontol. A Biol. Sci. Med Sci. 73, 1199–1204 (2018).
De Buyser, S. L. et al. Validation of the FNIH sarcopenia criteria and SOF frailty index as predictors of long-term mortality in ambulatory older men. Age Ageing 45, 602–608 (2016).
Cruz-Jentoft, A. J. et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on sarcopenia in older people. Age Ageing 39, 412–423 (2010).
Cruz-Jentoft, A. J. et al. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing 48, 16–31 (2019).
Cruz-Jentoft, A. J. & Sayer, A. A. Sarcopenia. Lancet 393, 2636–2646 (2019).
Schaap, L. A., Koster, A. & Visser, M. Adiposity, muscle mass, and muscle strength in relation to functional decline in older persons. Epidemiol. Rev. 35, 51–65 (2013).
Menant, J. C. et al. Strength measures are better than muscle mass measures in predicting health-related outcomes in older people: time to abandon the term sarcopenia? Osteoporos. Int. 28, 59–70 (2017).
Harris-Love, M., Benson, K., Leasure, E., Adams, B. & McIntosh, V. The influence of upper and lower extremity strength on performance-based sarcopenia assessment tests. J. Funct. Morphol. Kinesiol 3, 53 (2018).
Kristensen, M. T., Hulsbæk, S., Faber, L. L. & Kronborg, L. Knee extension strength measures indicating probable sarcopenia is associated with health-related outcomes and a strong predictor of 1-year mortality in patients following hip fracture surgery. Geriatrics 6, 8 (2021).
Yeung, S. S. Y. et al. Knee extension strength measurements should be considered as part of the comprehensive geriatric assessment. BMC Geriatr. 18, 130 (2018).
Zempo, H. et al. Heritability estimates of muscle strength-related phenotypes: a systematic review and meta-analysis. Scand. J. Med Sci. Sports 27, 1537–1546 (2017).
Silventoinen, K., Magnusson, P. K. E., Tynelius, P., Kaprio, J. & Rasmussen, F. Heritability of body size and muscle strength in young adulthood: a study of one million Swedish men. Genet Epidemiol. 32, 341–349 (2008).
Matteini, A. M. et al. Heritability estimates of endophenotypes of long and health life: the long life family study. J. Gerontol. A Biol. Sci. Med Sci. 65A, 1375–1379 (2010).
Tikkanen, E. et al. Biological insights into muscular strength: genetic findings in the UK biobank. Sci. Rep. 8, 6451 (2018).
Willems, S. M. et al. Large-scale GWAS identifies multiple loci for hand grip strength providing biological insights into muscular fitness. Nat. Commun. 8, 16015 (2017).
Jones, G. et al. Genome-wide meta-analysis of muscle weakness identifies 15 susceptibility loci in older men and women. Nat. Commun. 12, 654 (2021).
Matteini, A. M. et al. GWAS analysis of handgrip and lower body strength in older adults in the CHARGE consortium. Aging Cell 15, 792–800 (2016).
Semenova, E. A., Pranckevičienė, E., Bondareva, E. A., Gabdrakhmanova, L. J. & Ahmetov, I. I. Identification and characterization of genomic predictors of sarcopenia and sarcopenic obesity using UK biobank data. Nutrients 15, 758 (2023).
Hamano, T. et al. Effect of environmental and lifestyle factors on hypertension: Shimane COHRE study. PLoS One 7, e49122 (2012).
Garu, A. et al. Effect of multimorbidity on fragility fractures in community-dwelling older adults: Shimane CoHRE Study. J. Clin. Med 10, 3225 (2021).
Someya, Y. et al. Skeletal muscle function and need for long-term care of urban elderly people in Japan (the Bunkyo Health Study): a prospective cohort study. BMJ Open 9, e031584 (2019).
Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet 51, 1749–1755 (2019).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Davis, C. A. et al. The Encyclopedia of DNA Elements (ENCODE): data portal update. Nucleic Acids Res. 46, D794–D801 (2018).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012).
Eden, E., Navon, R., Steinfeld, I., Lipson, D. & Yakhini, Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48 (2009).
Klinck, R. et al. RBFOX1 cooperates with MBNL1 to control splicing in muscle, including events altered in myotonic dystrophy type 1. PloS One 9, e107324 (2014).
The GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Takayama, K. et al. TACC2 is an androgen-responsive cell cycle regulator promoting androgen-mediated and castration-resistant growth of prostate cancer. Mol. Endocrinol. Balt. Md 26, 748–761 (2012).
Hoffman, E. P., Brown, R. H. & Kunkel, L. M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 (1987).
Lord, J. P., Aitkens, S. G., McCrory, M. A. & Bernauer, E. M. Isometric and isokinetic measurement of hamstring and quadriceps strength. Arch. Phys. Med Rehabil. 73, 324–330 (1992).
Bohannon, R. W. Dynamometer measurements of grip and knee extension strength: are they indicative of overall limb and trunk muscle strength? Percept. Mot. Skills 108, 339–342 (2009).
Felicio, D. C. et al. Poor correlation between handgrip strength and isokinetic performance of knee flexor and extensor muscles in community-dwelling elderly women: handgrip strength and isokinetic performance. Geriatr. Gerontol. Int 14, 185–189 (2014).
Singhal, S. et al. Low one‐repetition‐maximum knee extension is significantly associated with poor grip strength, female sex, and various aging‐related syndromes. AGING Med. 3, 125–131 (2020).
Pei, Y.-F. et al. The genetic architecture of appendicular lean mass characterized by association analysis in the UK Biobank study. Commun. Biol. 3, 608 (2020).
Atkins, J. L. et al. A genome‐wide association study of the frailty index highlights brain pathways in ageing. Aging Cell 20, e13459 (2021).
Timmins, I. R. et al. Genome-wide association study of self-reported walking pace suggests beneficial effects of brisk walking on health and survival. Commun. Biol. 3, 634 (2020).
International Consortium for Blood Pressure GWAS, CHARGE Consortium Aging and Longevity Group, CHARGE Consortium Inflammation Group. et al. Genetic contributions to self-reported tiredness. Mol. Psychiatry 23, 609–620 (2018).
Ruth, K. S. et al. Using human genetics to understand the disease impacts of testosterone in men and women. Nat. Med. 26, 252–258 (2020).
Sinnott-Armstrong, N. et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet 53, 185–194 (2021).
Peset, I. & Vernos, I. The TACC proteins: TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol. 18, 379–388 (2008).
Gergely, F. et al. The TACC domain identifies a family of centrosomal proteins that can interact with microtubules. Proc. Natl. Acad. Sci. 97, 14352–14357 (2000).
Shakya, M. et al. High expression of TACC2 in hepatocellular carcinoma is associated with poor prognosis. Cancer Biomark. 22, 611–619 (2018).
Onodera, Y. et al. TACC2 (transforming acidic coiled‐coil protein 2) in breast carcinoma as a potent prognostic predictor associated with cell proliferation. Cancer Med. 5, 1973–1982 (2016).
Cheng, S., Douglas-Jones, A., Yang, X., Mansel, R. E. & Jiang, W. G. Transforming acidic coiled-coil-containing protein 2 (TACC2) in human breast cancer, expression pattern and clinical/prognostic relevance. Cancer Genom. Proteom. 7, 67–73 (2010).
Nagai, A. et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Akiyama, M. et al. Characterizing rare and low-frequency height-associated variants in the Japanese population. Nat. Commun. 10, 4393 (2019).
UK10K Consortium, Huang, J. et al. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nat. Commun. 6, 8111 (2015).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529 (2009).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287 (2016).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164 (2010).
Visscher, P. M., Hill, W. G. & Wray, N. R. Heritability in the genomics era–concepts and misconceptions. Nat. Rev. Genet 9, 255–266 (2008).
Benner, C. et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016).
Ito S. Shuji2022/Code: v1.0.0. Zenodo https://doi.org/10.5281/zenodo.10675274 (2024).
Acknowledgements
We thank all the participants of the Shimane CoHRE Study and the Bunkyo Health Study. Furthermore, we would like to express our gratitude to members of the studies for their skillful assistance and data collection. This work is supported by JSPS KAKENHI (18H03184, 19H03996, 20H00462, 22H03207) and the Strategic Research Foundation at Private Universities (S1411006) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Mizuno Sports Promotion Foundation, the Mitsui Life Social Welfare Foundation, AMED (JP21ek0109555, JP21tm0424220 and JP21ck0106642, JP23ek0410114, JP23tm0424225), and Takeda Hosho Grants for Research in Medicine.
Author information
Authors and Affiliations
Contributions
S.Ito, C.T., and S.Ikegawa designed the study. S.Ito analyzed the data with the help of Y.I., X.L., N.O., H.S., Y.K., K.H., K.T., S.Ikegawa, and C.T.; S.Ito wrote the manuscript. Y.M. and K.O. performed the genotyping for the GWAS. S.Ito, H.T., S.Kakehi, Y.S., H.Kaga, N.K., S.Kuwata, T.W., M.K., S.Y., T.A., M.T., M.Isomura, T.N., H.K., M.Ishijima, R.K., H.W., Y.T. and Y.U. collected and managed DNA samples and clinical data. All authors critically reviewed and revised the manuscript draft and approved the final version for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Chiea Chuen Khor, George Inglis and Johannes Stortz. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ito, S., Takuwa, H., Kakehi, S. et al. A genome-wide association study identifies a locus associated with knee extension strength in older Japanese individuals. Commun Biol 7, 513 (2024). https://doi.org/10.1038/s42003-024-06108-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06108-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
