
Abstract
The transcription factor Growth Factor Independence 1B (GFI1B) recruits Lysine Specific Demethylase 1 A (LSD1/KDM1A) to stimulate gene programs relevant for megakaryocyte and platelet biology. Inherited pathogenic GFI1B variants result in thrombocytopenia and bleeding propensities with varying intensity. Whether these affect similar gene programs is unknow. Here we studied transcriptomic effects of four patient-derived GFI1B variants (GFI1BT174N,H181Y,R184P,Q287*) in MEG01 megakaryoblasts. Compared to normal GFI1B, each variant affected different gene programs with GFI1BQ287* uniquely failing to repress myeloid traits. In line with this, single cell RNA-sequencing of induced pluripotent stem cell (iPSC)-derived megakaryocytes revealed a 4.5-fold decrease in the megakaryocyte/myeloid cell ratio in GFI1BQ287* versus normal conditions. Inhibiting the GFI1B-LSD1 interaction with small molecule GSK-LSD1 resulted in activation of myeloid genes in normal iPSC-derived megakaryocytes similar to what was observed for GFI1BQ287* iPSC-derived megakaryocytes. Thus, GFI1B and LSD1 facilitate gene programs relevant for megakaryopoiesis while simultaneously repressing programs that induce myeloid differentiation.
Similar content being viewed by others


Introduction
Blood stem cells develop into a variety of hematopoietic cells, including platelet producing megakaryocytes, through a complex process that requires intricate regulation of different transcription factors. The transcription factor Growth Factor Independent 1B (GFI1B) and its paralogue GFI1 are essential for adult hematopoietic stem cells maintenance1. GFI1 is expressed throughout different lymphoid differentiation stages and plays an essential role in myeloid development2,3. In parallel, GFI1B plays an essential role in the formation of erythrocytes and megakaryocytes4. GFI1/1B control gene expression through the recruitment of the chromatin modifying CoREST complex. This is a large protein complex that represses its target genes through deacetylation and demethylation5,6. CoREST consists of RCOR and HDAC proteins6, as well as Lysine Specific Demethylase 1 (LSD1/KDM1A)7. LSD1 was first identified as a demethylase, but recent evidence shows that it also acts as a CoREST scaffolding protein in blood cells4,8,9,10,11. It links GFI1/1B to other CoREST members through interaction with the N-terminal Snail/GFI1 (SNAG) domain of GFI1/1B to form a functional multi-protein complex9,12. In turn, GFI1/1B recruit the complex to the DNA through its C-terminal zinc fingers three, four, and five, leading to downregulation of its target genes.
The GFI1B-LSD1 interaction can be disturbed using LSD1 inhibitors like GSK-LSD17,9,12,13,14,15. They occupy the catalytic domain of LSD1 which blocks the interaction with GFI1B in hematopoietic cells10,13,16. Lsd1 knockdown in mice impaired differentiation of megakaryocytes, erythrocytes, and granulopoiesis, while monopoiesis was stimulated17. Consequently, a LSD1 inhibitor, Bomedemstat, is currently being evaluated in clinical trials to reduce platelet counts in patients with thrombocytosis and myeloproliferative disorders18. Inherited variants in GFI1B itself play a role in bleeding disorders with varying intensity. Truncating mutations in the most C-terminal DNA-binding zinc finger cause thrombocytopenia, moderate to severe bleedings, and various megakaryocyte and platelet abnormalities13,19,20,21,22. In mice, truncating mutations are linked to a milder phenotype and missense mutations in non-DNA binding zinc fingers are linked to milder disease phenotypes in humans19,23,24,25,26.
Currently, it remains unclear how different GFI1B mutations affect the transcriptome in developing megakaryocytes. Here, we elucidated transcriptomic changes caused by one dominant-negative C-terminal truncating mutation (GFI1BQ287*) and three dominant-negative upstream missense mutations (GFI1BT174N, GFI1BH181Y, GFI1BR184P) in megakaryocytes. Among the missense variants, the GFI1BT174N variant may not be pathogenic27. We observed that each variant has different effects on the megakaryocyte transcriptome. Remarkably, GFI1BQ287* expression resulted in derepression of myeloid genes in megakaryocytes. Treatment with an LSD1 inhibitor recapitulated these findings. These data indicate that GFI1B and LSD1 act together to repress myeloid genes during megakaryopoiesis.
Results and discussion
GFI1BQ287* fails to repress innate immunity genes in megakaryoblast-like cells
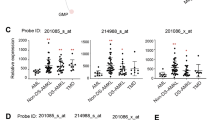
We assessed transcriptomic changes induced by four GFI1B variants linked to bleeding disorders to varying degrees. (Fig. 1). To that extent, we retrovirally expressed GFI1BT174N, GFI1BH181Y, GFI1BR184P, GFI1BQ287*, wild type GFI1B, or GFP in megakaryoblast MEG01 cells followed by RNA-sequencing. This resulted in comparable (2−3 fold) GFI1B induction in all samples compared to empty-vector transduced MEG01 cells, while LSD1 expression remained stable (Supplementary Fig. 1). Principal component analysis showed all replicates of a sample clustering together. Furthermore, GFI1B and its variants were separated from GFP by PC2, albeit with little variance (Fig. 2). Together, this shows that GFI1B variants are expressed at similar levels across samples and that they have distinct effects on the transcriptome compared to control. Identifying genes that characterize each condition proved difficult due to the number of comparisons needed for classical differential gene expression analysis, since this reduced statistical power considerably. Therefore, we utilized Weighted Gene Co-Expression Network Analysis28 that merges co-expressed genes into gene modules and correlates them to each condition. This yielded 12 modules containing between 22 and 1005 genes (Supplementary Data 1). 10 modules significantly correlated with at least one condition except for empty vector (GFP) MEG01 cells. They did not show significant correlation with any module (Fig. 3a). Earlier studies showed that alteration of GFI1B by either upregulation or depletion resulted in dysfunctional erythropoiesis11,29, indicating that GFI1B dosage is important. This is in line with our findings showing that forced expression of any variant or wild type GFI1B induced transcriptomic changes. We performed non-supervised hierarchical clustering of the module gene expression data to confirm that module correlations in GFI1B variants correspond to higher gene expression in these samples. This grouped all replicates of a condition together and confirmed identified module activities on a per gene basis (Fig. 3b). All tested GFI1B conditions associated with unique sets of modules that converge to different biological pathways. For instance, normal GFI1B caused inhibition of modules 9 and 11 genes, which are implicated in stress response and the immune system, respectively (Fig. 3a, Supplementary Data 2). Module 9 genes were active downstream of GFI1BH181Y, indicating that this variant failed to repress this gene program. Module 11 genes were suppressed by GFI1BT174N but activated downstream of GFI1BR184P and GFI1BQ287* (Fig. 3a). This suggests that regulation of this module is preserved by the GFI1BT174N variant but not by GFI1BR184P or GFI1BQ287*. Thus, the repression of certain gene programs downstream of normal GFI1B appears to be lost by different GFI1B mutants. The strongest correlation was observed for module 10 that was uniquely active in GFI1BQ287* replicates (Fig. 3a). This module contained 39 genes related to innate immunity, such as SPI1, GFI1, and CEBPA, that normally stimulate myeloid differentiation30,31,32 (Fig. 3c, Supplementary Data 2). These data indicate that only GFI1BQ287* fails to repress innate immunity genes which may contribute to disturbed megakaryopoiesis and impaired platelet production and function resulting in bleedings in affected cases.

a The schematic overview for GFI1B includes the N-terminal Snail/GFI1 domain (SNAG), the intermediate domain and the six C-terminal zinc fingers, labeled 1 to 6. In total four GFI1B variants were investigated: Three miss-sense mutations in the first zinc finger and one truncating mutation in zinc finger 5. b Clinical data overview for the 4 variants investigated. GFI1BH181Y, GFI1BR184P, and GFI1BQ287* resulted in bleedings with varying intensity. Bleeding tendencies for GFI1BT174N has not been reported publicly. GFI1BR184P and GFI1BQ287* have been reported to be likely pathogenic and pathogenic in ClinVAR, respectively. Information obtained from GnomAD (accessed 19.12.2023)52, ClinVAR (accessed 19.12.2023), refs. 19,20.

RNA sequencing of MEG01 cells with forced retroviral expression of different GFI1B variants, wild type GFI1B and empty vector control (GFP) was done. PCA showed that replicates cluster per sample.

a Weighted Gene Co-Expression Network Analysis revealed 12 modules, of which 10 modules were significant in at least one condition (see Supplementary Data 1). Module 12 contains all genes that were not be assigned to a module and module 6 did not return a significant correlation with any condition. Every condition had at least one significantly correlating module except for GFP. The strongest and most significant correlation was found between module 10 and GFI1BQ287*. b Expression analysis of all module genes showed that high module correlation indeed corresponded to high expression of those genes in that condition. Unsupervised hierarchical clustering grouped all replicates of a condition together. c REACTOME enrichment for module 10 using g:Profiler. Most genes in module 10 belonged to pathways that are involved in the innate immune system and homeostasis.
GFI1B represses myeloid differentiation during megakaryopoiesis
Patients with the GFI1BQ287* mutation suffer from megakaryocyte abnormalities and bleeding tendencies. To investigate whether GFI1BQ287* also fails to repress myeloid genes during megakaryopoiesis, we studied the single-cell transcriptome of megakaryocytes differentiated from patient-derived GFI1BQ287* induced pluripotent stem cells (iPSCs) and compared them to the single-cell transcriptome from wild type megakaryocytes. Single cell transcriptome analysis showed that wild type iPSCs predominantly formed megakaryocytes and megakaryocyte progenitors (75%) and smaller populations of erythroblasts (10%), myeloid and myeloid progenitors (13%), and lymphoid-myeloid progenitors (1%). The megakaryocyte/myeloid ratio was 75%/13% = 5.7. While GFI1BQ287* iPSCs formed erythroblasts at a similar rate as wild type (7%), the lymphoid-myeloid progenitors population increased to 9% and the megakaryocyte/myeloid cell ratio dropped to 1.3 (Fig. 4a, b). This indicates that GFI1BQ287* severely affects iPSC-derived megakaryocyte formation. Importantly, differential gene expression analysis revealed upregulation of myeloid genes within GFI1BQ287* megakaryocytes compared to wild type megakaryocytes (Fig. 5; Supplementary Data 3). Together, this shows that GFI1BQ287* fails to repress myeloid gene expression which likely contributes to impaired megakaryopoiesis.

UMAP showing iPSC-derived wild type (a), GFI1BQ287* (b), and GSK-LSD1 inhibitor treated (c) cells after megakaryocyte differentiation. Cell type annotation revealed erythroblasts (red), megakaryocytes (dark green), megakaryocyte progenitors (light green), myeloid cell (dark blue), myeloid cell progenitors (light green), and lymphoid-myeloid progenitors (gold). The megakaryocytes and megakaryocyte progenitors to myeloid and myeloid progenitor cell ratio was 5.7, 1.3 and 2.7 for the wild type, GFI1BQ287* and LSD1i conditions, respectively. d Heatmap showing the activity of gene programs that are regulated by a transcription factor (regulons). SPI1, ETV5 and CEBPA were active in myeloid cells in all conditions. Regulons controlled by megakaryocytic transcription factors such as GATA1, TAL1, and NFE2 were not active in myeloid cells of any condition. e The GATA1, TAL1, and NFE2 regulons were active in 50% of all wild type megakaryocytes or more. NFE2 was not found to be active in GFI1BQ287* megakaryocytes and GATA1 and TAL1 were active in 22% and 29% of GFI1BQ287* megakaryocytes, respectively. Myeloid SPI1 and ETV5 were not active in wild type megakaryocytes but showed activity in LSD1i and GFI1BQ287* megakaryocytes. f Regulon activity was projected onto our UMAP data. The myeloid-related SPI1 regulon was active in myeloid cells in the wild type condition and in all cell types in the GFI1BQ287* and LSD1i conditions including megakaryocytes. g SCENIC revealed that the ETV5 regulon was only active in myeloid cells in the wild type condition. In contrast, it was active in all cell types in the GFI1BQ287* and LSD1 inhibitor-treated condition. h The erythro-megakaryocytic GATA1 regulon was active in wild type megakaryocytes. Its activity was reduced in GFI1BQ287* and LSD1i megakaryocytes. i SCENIC revealed that TAL1 was active in wild type megakaryocytes. The activity of the TAL1 regulon was reduced in LSD1 inhibitor-treated megakaryocytes and almost absent in GFI1BQ287* megakaryocytes.

Platelet-related genes were upregulated in the wild type sample. Genes that are also differentially expressed when comparing wild type and GFI1BQ287* megakaryocytes are annotated in orange (also upregulated in GSK-LSD1 inhibitor (LSD1i) treated megakaryocytes) and blue (also upregulated in wild type megakaryocytes compared to LSD1i megakaryocytes). For full list see Supplementary Data 3.
GFI1B represses myeloid differentiation through SPI1 and other transcription factors
Next, we sought to identify transcription factors downstream of GFI1B that are implicated in repression of myeloid traits using the Single-Cell rEgulatory Network Inference and Clustering (SCENIC) algorithm. SCENIC determines active regulons, which are defined as sets of genes that are regulated as a unit by one transcription factor. As expected, the myeloid-associated SPI1 and ETV5 regulons were active in wild type myeloid cells while the megakaryocytic-erythrocytic GATA1 and TAL1 regulons were active in wild type developing megakaryocytes. Strikingly, SPI1 and ETV5 regulons were not restricted to GFI1BQ287* myeloid cells. Instead, they were active in most GFI1BQ287* cells, including megakaryocytes. This is in line with earlier studies showing that GFI1B suppressed myeloid differentiation of CD34+ cells through SPI1 inhibition33. Furthermore, GATA1 and TAL1 regulons showed reduced activity in GFI1BQ287* megakaryocytes (Fig. 4d−i). Our findings indicate that GFI1B facilitates megakaryopoiesis by driving megakaryocyte-specific regulons and repressing myeloid regulons at the same time.
GFI1B is a major interactor of LSD1 in K562 cells
GFI1B has been found to interact with LSD1 in several different cell types, including MEG01, HEL, and K562 cells7,12,13,34. To confirm this interaction, we tagged the LSD1 gene with GFP at its C-terminus in the GFI1B expressing megakaryocyte-erythrocyte progenitor cell line K562. Subsequent GFP immunoprecipitation followed by label-free mass-spectrometry identified 22 interacting proteins, including LSD1, confirming the interaction with established core CoREST members RCOR1/2/3 and HDAC1/2, as well as PHF21A and HMG20A/B. Additionally, it showed interaction with the CtBP complex members ZMYM2/3, RREB1, ZNF217 and CTBP1 (Fig. 6a). It is known that the CoREST complex is part of the CtBP containing complex35,36,37, which our data confirms. LSD1 interacts with the SNAG domain of transcription factors and our mass-spectrometry analysis revealed interaction with the SNAG-domain containing transcription factor GFI1B (Fig. 6a). Because the interaction between GFI1B and LSD1 has been described in detail in hematopoietic systems, we wondered if LSD1 inhibition resulted in a similar impaired megakaryocyte development as GFI1BQ287*.

a The endogenous LSD1 gene was tagged with GFP in K562 cells and mass-spectrometry was performed after GFP pulldown. We identified LSD1-interacting proteins after pulldown, showing that the CoREST members RCOR1/2/3, HDAC1/2, PHF21A, HMG20A/B, and GFI1B interacted with LSD1. Additionally, members of the CtBP complex were found to interact with LSD1 (solid dots). b MEG01 cells were treated with the small molecule GSK-LSD1 inhibitor and RNA sequenced. Module 10 genes that were identified to be highly expressed in GFI1BQ287* MEG01 cells were also highly expressed after LSD1 inhibition. c Differential gene expression of ISPC-derived scRNA-seq data showed that LSD1 inhibition leads to the upregulation of myeloid genes in megakaryocytes. Genes that were also upregulated in GFI1BQ287* megakaryocytes are highlighted in orange, while genes also upregulated in wild type megakaryocytes are highlighted in blue. d Wild type iPSCs were differentiated to megakaryocytes and treated with an LSD1 inhibitor for 72 h before harvesting. FACS analysis showed that LSD1 inhibition resulted in induced expression of CD86 and CD34 in megakaryocytes compared to untreated wild type cells. CD42b expression was absent in treated cells compared to untreated (n = 2 independent experiments, bars denote mean, points are individual data points).
GFI1B and LSD1 repress similar myeloid traits during megakaryocyte development
We inhibited the GFI1B-LSD1 interaction using GSK-LSD113 to determine whether this interaction is required to inhibit myeloid traits in megakaryocytes. To this end, MEG01 cells were exposed to GSK-LSD1 for 72 h, followed by bulk RNA-sequencing. Analysis of module 10 innate immunity genes that were identified in GFI1BQ287* expressing MEG01 cells showed that the vast majority were also active following LSD1 inhibition (Fig. 6b). This indicates that the GFI1B-LSD1 interaction is required to suppress these genes in MEG01 cells. Next, we investigated whether LSD1 inhibition also fails to repress myeloid traits during iPSC-derived megakaryopoiesis. To this end, wild type iPSCs were differentiated towards megakaryocytes and treated with GSK-LSD1 for only 24 h prior to single-cell RNA-sequencing. Similar to GFI1BQ287* iPSC-derived megakaryopoiesis, the megakaryocyte/myeloid cell ratio dropped from 5.7 in wild type cells to 2.7 after LSD1 inhibition (Fig. 4a, c). Additionally, myeloid genes were upregulated in LSD1-inhibitor treated megakaryocytes compared to wild type megakaryocytes, some of which were also upregulated in GFI1BQ287* megakaryocytes (Figs. 5, 6c; Supplementary Data 3). Prolonged treatment of wild type iPSC-derived cells with GSK-LSD1 revealed the absence of the megakaryocyte marker CD42b and an increase of the myeloid marker CD86 (Fig. 6d). Thus, the GFI1B-LSD1 interaction is fundamental to megakaryocyte development. Finally, we determined which regulons were activated upon LSD1 inhibition using single-cell RNA sequencing of iPSC-derived megakaryocytes. Again, SPI1 and ETV5 regulons were found to be active in all cell types including megakaryocytes. GATA1 and TAL1 regulon activities were reduced compared to non-treated cells, comparable to the effects of GFIBQ287* (Fig. 4d−i). Additionally, some regulons were affected by LSD1 inhibition but not the GFI1BQ287* mutation, such as ATF4 (Fig. 4d, e). Recently, it was shown that ATF4 is downregulated upon LSD1 inhibition potentially through a loss of the CREBBP-LSD1 interaction in glioblastoma cells38. In a lung carcinoma cell line, ATF4 was downregulated through increased H3K9me2 levels after LSD1 inhibition39. On the contrary, we found induction of ATF4 regulon activity upon LSD1 inhibition, suggesting that the mechanisms of action following LSD1 inhibition are different in our model system. More detailed studies that determine early chromatin binding, epigenetic, and transcriptional changes following GFI1BQ287* expression and LSD1 inhibition may reveal which activities are at play during megakaryopoiesis.
Summarizing, our data demonstrate that different inherited GFI1B mutations affect the megakaryocyte gene programs in unique ways. The most severe bleedings in patients caused by the GFI1BQ287* mutation could be explained by the loss of GFI1B-LSD1 mediated suppression of myeloid traits during megakaryopoiesis, potentially through derepression of SPI1 and ETV5. SPI1 is a direct GFI1B target and loss of GFI1B-mediated SPI1 suppression has been linked to myelomonocytic differentiation33. LSD1 has been found to be required for terminal differentiation of various hematopoietic cell types except monocytes17. Our own data confirms and expands on these findings. We propose that GFI1B and LSD1 work together to suppress SPI1 and other myeloid transcription factor programs during megakaryopoiesis. Loss of GFI1B or LSD1 function deregulates these programs inducing myeloid differentiation. This could explain perturbed megakaryocyte development and platelet production in GFI1BQ287* patients20 and reduced platelet formation in patients treated with the LSD1 inhibitor Bomedemstat18.
Methods
MEG01 RNA sequencing and network analysis
MEG01 cells were cultured at 5% CO2 and 37 °C in RPMI 1640 (GIBCO) supplemented with 10% heat-inactivated fetal calf serum. Cells were transduced using pMIGR1-GFI1B_variant-IRES-GFP constructs and FACS-sorted for GFP (BD FACSAria). RNA was isolated using the NucleoSpin RNA plus kit (Macherey-Nagel). Library generation was performed on 100 ng RNA using the KAPA RNA HyperPrep Kit with RiboErase (HMR;Kapa Biosystems), with a RNA fragmentation of approximately 300 bp fragments for 6 min at 94 °C. Library size distribution was measured using High Sensitivity DNA analysis on an Agilent 2100 Bioanalyzer (Agilent) and its corresponding software (version B.02.08.SI648). Libraries with average sizes between 300 and 400 bp were sequenced on the NextSeq 500 (Illumina). Data was quality-controlled, mapped, and count matrices were generated using nextflow pipelines. Count matrices were loaded into R (v.4.1.1), and variance-stabilized counts were generated using DESeq2 (v1.32). The top 5% most variable genes were used as input for Weighted Gene Co-expression Network analysis28 (v1.71). The softpower threshold was set to 12 and the networks were calculated using signed-hybrid networks. Resulting modules were merged using a cutting height of 0.2. All module genes were uploaded to gProfiler40 (version e109_eg56_p17_1d3191d, accessed 04.08.2023). REACTOME enrichment was performed as a multi-query using all expressed genes (cumulative count per gene > 10 and expressed in at least 50% of all samples) as a statistical domain scope.
MEG01 and induced pluripotent stem cells were treated with 4 µM of the GSK-LSD1 inhibitor (Merck) for 24 h, 48 h or 4 days, as indicated.
Induced pluripotent stem cell maintenance and differentiation towards megakaryocytes
Induced pluripotent stem cells were maintained according to previously described protocols on Matrigel (Corning), E8 medium (Thermofisher Scientific), and mTESR1 medium (Stemcell Technologies)41,42. The medium was regularly changed and cells were passaged with TrypLE Select (Thermofisher Scientific). One day prior to differentiation, the cells were clump passaged using ReLeSR (Stemcell Technologies). Cells were differentiated towards hematopoietic stem cells using the STEMdiff Hematopoietic kit (Stemcell technologies). From day 10 onwards, hematopoietic stem cells were harvested in modified Iscove modified Dulbecco medium (HEMAdef)43 according to protocol and to stimulate megakaryocyte differentiation supplemented with a cytokine cocktail of 10 ng/ml rhIL-3, 10 ng/ml rhIL-6, 10 ng/ml rhIL-9, 10 ng/ml rhIL-11, 10 ng/ml VEGF, 10 ng/ml BMP4, 1% Stem Cell Factor (Immunotools), 50 ng/ml Thrombopoietin (Peprotech), and 1% Insulin Transferrin Selenium (Thermofisher Scientific)13,44.
Cell analysis by flow cytometry
Flow cytometric analyses were performed on a Gallios flow cytometer (Beckman Coulter) and analyzed using Kaluza software v.2.1.2 (Beckman Coulter). Differentiated iPSCs were stained using the surface marker antibodies CD34-561 Brilliant Violet 421 (Biolegend), CD86-IT2.2 PE (Biolegend), and CD42b-HIP1 Brilliant Violet 510 (Biolegend). Cells were gated for live cells based on forward/side scatter area and singlets were gated based on forward scatter area/side scatter area (Supplementary Fig. 2) For single cell RNA sequencing, living cells were sorted using 7AAD (Sigma) as a viability marker on the FACS Aria (BD biosciences) using a 100 µm nozzle (BD Biosciences).
Single cell RNA sequencing data processing and analysis
Single-cell RNA-sequencing was performed using the Chromium Single Cell 3’ kit (v3.1 Chemistry; 10X genomics) according to protocol. Sorted cells were loaded at a density of ~1200 cells/µl with a desired recovery of 10.000 cells per sample. Quality and size of the generated libraries were verified using the Qubit 1x dsDNA HS assay kit (Invitrogen) and the Bioanalyzer (Agilent). Sequencing was performed on the Nextseq500 (Illumina) at an average sequencing depth of ~266 million reads. Raw data were processed using Cellranger v.6.0.0 (10x Genomics) and aligned against the GRCh38 transcriptome, and feature-barcoded matrices were created. Empty droplets were filtered using the emptydrops function from the DropletUtils package (v1.10.3)45 using 30,000 iterations and FDR of 0.01 and 100 UMIs as the lower bound UMI threshold for non-empty droplets. The Seurat package (v.4.0.1)46 was used for downstream analysis and quality checks within the R environment (v.4.0). Dimensional reduction was performed on retained droplets, and a shared nearest neighbor graph was constructed to identify clusters using Louvain clustering. Doublets were filtered using the DoubletFinder package (v.2.0.3)47. Clusters were identified using the shared nearest neighbor (SNN) approach and selecting the Louvain clustering method. Clustree (v.0.4.1)48 was used to identify the optimal clustering resolution. High mitochondrial gene content indicates that cells were damaged during sample preparation, because cytoplasmic RNA dissipates from the damaged cell while mitochondria are retained. Clusters with a high proportion of mitochondrial genes and no megakaryocyte marker expression were removed from the analysis (cluster 6 and 19 for LSD1i sample, cluster 14, 16, 23 for GFI1BQ287* and none for wild type). After filtering, 7470 wild type cells, 4554 GSK-LSD1-treated (LSD1i) cells and 2824 GFI1BQ287* cells passed quality control (Supplementary Fig. 3). Samples were normalized using SCTransform and integrated using the FindIntegrationAnchors function. Cell cycle was assessed using CellCycleScoring. Dimensional reduction using Uniform Manifold Approximation and Projection (UMAP) showed that cells were clustering according to their cell cycle which is a confounder in our analysis (Supplementary Fig. 3f). Therefore, we regressed out cell cycle during sample integration to exclude any effect based on cell cycle. Following the cell cycle regression, the samples were re-integrated based on the FindIntegrationAnchors function. Clusters were identified using Louvain clustering, and resolution 0.3 was the optimal cluster separation (Supplementary Fig. 4a, b). Clusters were annotated using two independent annotation tools. First, we performed single cell gene set enrichment analysis with the escape package (v.1.0.1). For single cell gene set enrichment analysis, the C8 cell type signature gene set was downloaded from the MSigDB website (Database v7.4). Secondly, we used Azimuth in combination with the publicly available Azimuth PBMC data set46,49. We merged both annotations obtained by each tool, manually renamed each cluster and determined to top expressed genes per cluster (Supplementary Fig. 4c−e). Differential expressed genes were identified using the FindMarkers function with a minimal expression percentage of 25 in at least one cluster. Genes were considered differentially expressed with log2 fold change <−0.69 or >0.69 and adjusted p < 0.05.
SCENIC analysis
We calculated the gene regulatory network for each sample independently, using the python SCENIC implementation and Arboreto allowing for multi-processing50. We ran the pipeline 100 times for each sample to account for random number generation. The iteration outputs were merged per sample, and regulons (i.e., transcription factors and its associated target genes) that were found in at least 95% of the iterations were retained. We calculated the regulon activity using AUCell50. Activity scores were binarized for each sample to annotate the On/Off state for every regulon in a sample. This enabled between sample comparison. AUC thresholds for regulon binarization were calculated automatically, and curve fit was checked manually as described by the authors of (py)SCENIC. Binary regulon score for each regulon in each cell are supplied as Supplementary Data 4.
CRISpainting LSD1 and mass-spectrometry
K562 cells were GFP CRISPainted at the endogenous LSD1 locus according to Schmid-Burgks protocol51. 680 × 106 K562 cells with endogenously tagged LSD1 were harvested for mass-spectrometry. Nuclei were isolated and GFP pulldown was performed using GFP nano-trap beads (Chromotek). Beads were combined with 2 mg nuclear extract in buffer C (300 mM NaCl, 20 mM Hepes KOH pH 7.9, 20% v/v glycerol, 2 mM MgCl2, 0.2 mM EDTA, 0.1% NP40, complete protease inhibitors w/o EDTA, 0.5 mM DTT) + 50 µg/ml EtBr and incubated (90 min at 4 degrees). Then, beads were washed two times with buffer C, two times with PBS + 0.25% NP40 and two times with PBS. Liquid was removed and resuspended in elution buffer (2 M Urea in 100 mM Tris pH 8 and 10 mM DTT) at RT and incubated for 20 minutes while shaking. Proteins were digested on beads with trypsin (2 M urea, 100 mM Tris/HCl pH8.5, 10 mM DTT, 0.25 µg Protease). Digested protein were then STAGE tipped. Peptides were measured using a reverse phase column connected to an Easy-nLC1000 (ThermoFisher) coupled to a Thermo Orbitrap Exploris 480 (ThermoFisher Scientific). Data were analyzed using MaxQuant 1.6.6.0.
Statistics and reproducibility
Statistical analysis for WGCNA and SCENIC has been built into the model as part of the standard workflow. RNA seq samples were done in triplicate based on one biological sample. scRNA analysis and mass-spectrometry was done based one biological sample. Flow data was analyzed based on two biological samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequencing data have been submitted to GEO under accession numbers GSE244609 and GSE244756. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD050401.
References
Khandanpour, C. et al. Evidence that growth factor independence 1b regulates dormancy and peripheral blood mobilization of hematopoietic stem cells. Blood 116, 5149–5161 (2010).
Yucel, R., Kosan, C., Heyd, F. & Moroy, T. Gfi1:green fluorescent protein knock-in mutant reveals differential expression and autoregulation of the growth factor independence 1 (Gfi1) gene during lymphocyte development. J. Biol. Chem. 279, 40906–40917 (2004).
Hock, H. et al. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity 18, 109–120 (2003).
Saleque, S., Cameron, S. & Orkin, S. H. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev. 16, 301–306 (2002).
Lee, M. G., Wynder, C., Cooch, N. & Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437, 432–435 (2005).
You, A., Tong, J. K., Grozinger, C. M. & Schreiber, S. L. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 98, 1454–1458 (2001).
Saleque, S., Kim, J., Rooke, H. M. & Orkin, S. H. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 27, 562–572 (2007).
Casey, M. J. et al. The scaffolding function of LSD1/KDM1A reinforces a negative feedback loop to repress stem cell gene expression during primitive hematopoiesis. iScience 26, 105737 (2023).
Ravasio, R. et al. Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid-induced differentiation. Sci. Adv. 6, eaax2746 (2020).
Sacilotto, N. et al. Comprehensive in vitro characterization of the LSD1 small molecule inhibitor class in oncology. ACS Pharmacol .Transl. Sci. 4, 1818–1834 (2021).
Osawa, M. et al. Erythroid expansion mediated by the Gfi-1B zinc finger protein: role in normal hematopoiesis. Blood 100, 2769–2777 (2002).
Yamamoto, R. et al. Selective dissociation between LSD1 and GFI1B by a LSD1 inhibitor NCD38 induces the activation of ERG super-enhancer in erythroleukemia cells. Oncotarget 9, 21007–21021 (2018).
van Oorschot, R. et al. Molecular mechanisms of bleeding disorderassociated GFI1B(Q287*) mutation and its affected pathways in megakaryocytes and platelets. Haematologica 104, 1460–1472 (2019).
Kerenyi, M. A. et al. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife 2, e00633 (2013).
Thambyrajah, R. et al. GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol. 18, 21–32 (2016).
Ishikawa, Y. et al. A novel LSD1 inhibitor T-3775440 disrupts GFI1B-Containing complex leading to transdifferentiation and impaired growth of AML Cells. Mol. Cancer Ther. 16, 273–284 (2017).
Sprussel, A. et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 26, 2039–2051 (2012).
Gill, H. et al. A Phase 2 Study of the LSD1 Inhibitor Bomedemstat (IMG-7289) for the Treatment of Essential Thrombocythemia (ET). Blood 140, 1784–1787 (2022).
van Oorschot, R. et al. Inherited missense variants that affect GFI1B function do not necessarily cause bleeding diatheses. Haematologica 104, e260–e264 (2019).
Monteferrario, D. et al. A dominant-negative GFI1B mutation in the gray platelet syndrome. N. Engl. J. Med. 370, 245–253 (2014).
Marneth, A. E. et al. Platelet CD34 expression and alpha/delta-granule abnormalities in GFI1B- and RUNX1-related familial bleeding disorders. Blood 129, 1733–1736 (2017).
Van Bergen, M. et al. Specific proteome changes in platelets from individuals with GATA1-, GFI1B-, and RUNX1-linked bleeding disorders. Blood 138, 86–90 (2021).
Beauchemin, H., Shooshtharizadeh, P., Pinder, J., Dellaire, G. & Moroy, T. Dominant negative Gfi1b mutations cause moderate thrombocytopenia and an impaired stress thrombopoiesis associated with mild erythropoietic abnormalities in mice. Haematologica 105, 2457–2470 (2020).
Chen, L. et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science 345, 1251033 (2014).
Ferreira, C. R. et al. Combined alpha-delta platelet storage pool deficiency is associated with mutations in GFI1B. Mol. Genet. Metab. 120, 288–294 (2017).
Rabbolini, D. J. et al. Thrombocytopenia and CD34 expression is decoupled from alpha-granule deficiency with mutation of the first growth factor-independent 1B zinc finger. J. Thromb. Haemost. 15, 2245–2258 (2017).
van Bergen, M. et al. S847 distinct and common deregulated pathways in RUNX1, GATA1 AND GFI1B associated bleeding disorders. HemaSphere 3, 378 (2019).
Zhang, B. & Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, https://doi.org/10.2202/1544-6115.1128 (2005).
Randrianarison-Huetz, V. et al. Gfi-1B controls human erythroid and megakaryocytic differentiation by regulating TGF-beta signaling at the bipotent erythro-megakaryocytic progenitor stage. Blood 115, 2784–2795 (2010).
Person, R. E. et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat. Genet. 34, 308–312 (2003).
Rosmarin, A. G., Yang, Z. & Resendes, K. K. Transcriptional regulation in myelopoiesis: hematopoietic fate choice, myeloid differentiation, and leukemogenesis. Exp. Hematol. 33, 131–143 (2005).
Radomska, H. S. et al. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol. Cell Biol. 18, 4301–4314 (1998).
Anguita, E. et al. A somatic mutation of GFI1B identified in leukemia alters cell fate via a SPI1 (PU.1) centered genetic regulatory network. Dev. Biol. 411, 277–286 (2016).
McClellan, D. et al. Growth factor independence 1B-mediated transcriptional repression and lineage allocation require lysine-specific demethylase 1-dependent recruitment of the BHC complex. Mol. Cell Biol. 39 https://doi.org/10.1128/MCB.00020-19 (2019).
Shi, Y. et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738 (2003).
Cowger, J. J., Zhao, Q., Isovic, M. & Torchia, J. Biochemical characterization of the zinc-finger protein 217 transcriptional repressor complex: identification of a ZNF217 consensus recognition sequence. Oncogene 26, 3378–3386 (2007).
Hayakawa, T. & Nakayama, J. Physiological roles of class I HDAC complex and histone demethylase. J. Biomed. Biotechnol. 2011, 129383 (2011).
Faletti, S. et al. LSD1-directed therapy affects glioblastoma tumorigenicity by deregulating the protective ATF4-dependent integrated stress response. Sci. Transl. Med. 13, eabf7036 (2021).
Du, L. et al. Inhibition of LSD1 induces ferroptosis through the ATF4-xCT pathway and shows enhanced anti-tumor effects with ferroptosis inducers in NSCLC. Cell Death Dis. 14, 716 (2023).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Hansen, M. et al. Generation and characterization of human iPSC line MML-6838-Cl2 from mobilized peripheral blood derived megakaryoblasts. Stem Cell Res. 18, 26–28 (2017).
Hansen, M. et al. Generation and characterization of a human iPSC line SANi005-A containing the gray platelet associated heterozygous mutation p.Q287* in GFI1B. Stem Cell Res. 25, 34–37 (2017).
Migliaccio, G. et al. Humanized culture medium for clinical expansion of human erythroblasts. Cell Transpl. 19, 453–469 (2010).
Hansen, M. et al. Efficient production of erythroid, megakaryocytic and myeloid cells, using single cell-derived iPSC colony differentiation. Stem Cell Res. 29, 232–244 (2018).
Lun, A. T. L. et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 20, 63 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e324 (2019).
Zappia, L. & Oshlack, A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience 7 https://doi.org/10.1093/gigascience/giy083 (2018).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Schmid-Burgk, J. L., Honing, K., Ebert, T. S. & Hornung, V. CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism. Nat. Commun 7, 12338 (2016).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Acknowledgements
This work was supported by the Landsteiner Foundation for Blood Transfusion Research (project 1531) and the Radboud Research Institute for Medical Innovation. The Vermeulen lab is part of the Oncode Institute, which is partly funded by the Dutch Cancer Society (KWF).
Author information
Authors and Affiliations
Contributions
M.G.J.M.V.B., J.V., J.H.A.M. and B.A.V.D.R. designed the research project. M.G.J.M.V.B., J.V. and S.B. performed the experiments and analyzed the data. L.W. performed the (sc)RNA sequencing and D.G. ran the bioinformatic pipelines and provided expert knowledge for the bioinformatic analysis. C.G.S. ran the mass-spectrometry experiments and analyzed the raw data under supervision of M.V. E.V.D.A. provided the iPSC cell lines and expert knowledge for culturing and differentiation. J.H.J. critically evaluated the results and research progression. The manuscript was written by J.V., M.G.J.M.V.B. and B.A.V.D.R. and was critically evaluated by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Communications Biology thanks Anna Rita Migliaccio and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Eirini Trompouki and Dario Ummarino. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Venhuizen, J., van Bergen, M.G.J.M., Bergevoet, S.M. et al. GFI1B and LSD1 repress myeloid traits during megakaryocyte differentiation. Commun Biol 7, 374 (2024). https://doi.org/10.1038/s42003-024-06090-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06090-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
