
Abstract
PRMT5, a type II arginine methyltransferase, is involved in transcriptional regulation, RNA processing and other biological processes and signal transduction. Secondary metabolites are vital pharmacological compounds in Ganoderma lucidum, and their content is an important indicator for evaluating the quality of G. lucidum. Here, we found that GlPRMT5 negatively regulates the biosynthesis of secondary metabolites. In further in-depth research, GlPP2C1 (a type 2C protein phosphatase) was identified out as an interacting protein of GlPRMT5 by immunoprecipitation-mass spectrometry (IP-MS). Further mass spectrometry detection revealed that GlPRMT5 symmetrically dimethylates the arginine 99 (R99) and arginine 493 (R493) residues of GlPP2C1 to weaken its activity. The symmetrical dimethylation modification of the R99 residue is the key to affecting GlPP2C1 activity. Symmetrical demethylation-modified GlPP2C1 does not affect the interaction with GlPRMT5. In addition, silencing GlPP2C1 clearly reduced GA content, indicating that GlPP2C1 positively regulates the biosynthesis of secondary metabolites in G. lucidum. In summary, this study reveals the molecular mechanism by which GlPRMT5 regulates secondary metabolites, and these studies provide further insights into the target proteins of GlPRMT5 and symmetric dimethylation sites. Furthermore, these studies provide a basis for the mutual regulation between different epigenetic modifications.
Similar content being viewed by others

Introduction
Protein arginine methyltransferase 5 (PRMT5), a type II arginine methyltransferase member with clear functions, mainly participates in transcription regulation, RNA processing and other biological processes and signal transduction by symmetrically dimethylating target proteins1. For instance, AtPRMT5 mutants exhibit distinct phenotypic changes including decreased plant height, altered leaf morphology, and degeneration of floral organs in Arabidopsis2. Moreover, PRMT5 contributes to secondary metabolite pigment biosynthesis in Penicillium expansum3. Studies have found that the main functional mode of PRMT5 is to directly methylate histones and nonhistones, thereby affecting a variety of physiological processes. Most studies have focused on the function of PRMT5 in affecting histone methylation4,5,6, and there are fewer studies on nonhistone methylation. As research progressed, some nonhistone proteins (enzymes or functional proteins) modified via PRMT5-mediated symmetric dimethylation were found to be involved in signal transduction pathways. For example, PRMT5-mediated symmetric dimethylation of the VP1 arginine (R) 426 residue impaired apparently VP1 polymerase activity, resulting in impaired viral replication7. PRMT5-mediated symmetric dimethylation of the R124 residue in cyclic GMP-AMP synthetase (cGAS) blocks the DNA-binding ability of cGAS to attenuate cGAS-mediated antiviral immune responses8. Crucially, there are few reports on PRMT5 target proteins in fungi via methylation modification. Exploring the downstream proteins modified by PRMT5 through nonhistone methylation will help elucidate the function of PRMT5 and fill gaps in this field.
Type 2C protein phosphatases (PP2Cs) are a large family of Mg2+/Mn2+-dependent phosphatases9. Published research clearly shows that PP2C plays a crucial role in many signal transduction pathways and physiological functions and responds to various environmental stresses. For example, overexpressing AtPP2CF1 enhanced plant biomass production by activating cell proliferation and expansion to accelerate inflorescence stem growth in Arabidopsis10. Overexpression of OsPP108 (a PP2C from rice) resulted in improved tolerance of transgenic Arabidopsis plants with better physiological parameters for fresh weight, chlorophyll content and photosynthetic potential under salt, mannitol and drought stress11. In addition, research on protein phosphatases of the PP2C family in microorganisms has been reported successively. For instance, Saccharomyces cerevisiae cells lacking Ptc1 (a type 2C protein phosphatase) showed high sensitivity to exogenous stresses such as high pH, LiCl, CaCl2, ZnCl2, CFW, caffeine, and rapamycin12. Fusarium oxysporum Ptc6 (type 2C protein phosphatase) mutants showed increased sensitivity to membrane (SDS), cell wall (CFW), and oxidative (menadione) stress compounds13. Most studies have reported the downstream mechanism of PP2C as a phosphatase regulator14,15. In addition, the upstream mechanism of PP2C regulation has also been reported. On the one hand, the expression of PP2C is inhibited at the transcriptional level to inhibit its activity. As an illustration, H2O2 produced by the ABA-induced NADPH oxidase RbohB/E inhibits PP45 (a type 2C protein phosphatase in rice) activity by inhibiting the expression of PP4516. OsbZIP12/OsABF1 and OsbZIP46/OsABF2 (the bZIP family transcription factors in rice) directly bind to the OsPP2C09 (a type 2C protein phosphatase in rice) promoter and mediate rapid induction of its expression after exogenous ABA treatment17. On the other hand, posttranslational modifications affect PP2C activity. As evidence, ABA affects PP45 activity by oxidizing cysteine (Cys-350 and Cys-428) residues to form intermolecular dimers of PP4516. However, it is worth noting that the molecular mechanisms regulating the protein posttranslational modification of PP2C family protein phosphatases have remained poorly investigated thus far. Exploring the mechanism of PP2C regulation will not only help fully elucidate its function but also provide a reference for other species.
Ganoderma lucidum, a traditional Chinese medicinal and food macrofungus, has a variety of pharmacological effects18,19,20,21. Based on its remarkable physiological activity, ganoderic acid (GA) is an important index to evaluate its quality, but its low content limits the application and development of G. lucidum. Furthermore, as a secondary metabolite, the biosynthesis of GA is subject to environmental regulation. Therefore, studying the biosynthetic mechanism of GA is valuable in analyzing the environmental factors that govern the biosynthesis of secondary metabolites. At present, based on developments in genomics and proteomics in recent years and the progress of genetic research methods, there are an increasing number of reports on the mechanism of GA biosynthesis. Previous reports have shown that signaling molecules (reactive oxygen, nitric oxide, calcium ions) and environmental factors (high temperature, nutrients, chemicals) in the GA biosynthesis play an important role in GA biosynthesis22,23,24,25,26,27,28, indicating that GA biosynthesis is driven by many factors coregulated. Importantly, research on these mechanisms is far from sufficient to reveal the regulatory network of the GA biosynthesis mechanism. Research on the function of epigenetic modification in G. lucidum is very limited. Dissecting epigenetic modifications will enrich the regulatory network of the GA biosynthetic mechanism.
In this study, we found that GlPRMT5 negatively regulates the biosynthesis of the secondary metabolite GA in G. lucidum. In contrast, GlPP2C1 positively regulates the biosynthesis of secondary metabolites in G. lucidum. The activity of the protein phosphatase GlPP2C1 is regulated by arginine methyltransferase GlPRMT5-mediated symmetric dimethylation at R99, a critical residue for its enzymatic activity. In conclusion, on the one hand, we elucidated the target of GlPRMT5 symmetric methylation and its function of modifying proteins by symmetric methylation in large medicinal fungi. On the other hand, we demonstrated that GlPP2C1 is involved in GlPRMT5-mediated GA biosynthesis, providing a mechanism by which PRMT5 regulates secondary metabolites.
Results
Gl PRMT5 silencing increases GA content in G. lucidum
Previous studies have reported that PRMT5 is involved in regulating the biosynthesis of secondary metabolites3. Of note, the specific mechanism underlying this regulation remains unclear. To explore the role of GlPRMT5 in the biosynthesis of GA, one of the most important secondary metabolites in G. lucidum, PRMT5-silenced strains were constructed by cloning PRMT5-silenced sequences from cDNA into RNA interference (RNAi) constructs using cassette plasmids as previously described29. Previous studies used real-time reverse transcription PCR (qRT-PCR) to screen out two PRMT5-silenced strains (PRMT5i-31 and PRMT5i-35) with silencing efficiencies of 68% and 72% respectively30. Here, we screened a PRMT5-silenced strain (PRMT5i-4) with a silencing efficiency of 54% by qRT-PCR (Fig. 1a). The expression of PRMT5 protein in the WT, CK (empty vector control), and PRMT5-silenced strains was further detected by Western blot analysis (Fig. 1b). The results showed that the PRMT5-silenced strains effectively reduced PRMT5 protein levels compared with the WT and CK strains. These results confirmed the effectiveness of the PRMT5-silenced strains. In addition, the GA content was determined in the WT, CK and PRMT5-silenced strains. We found that compared with the WT and CK strains, the GA content of the PRMT5i-4, PRMT5i-31 and PRMT5i-35 strains increased by 1.4-fold, 1.47-fold and 1.48-fold, respectively (Fig. 1c). The results showed that GlPRMT5 silencing promoted GA accumulation in G. lucidum.

a qRT–PCR analysis of the expression of GlPRMT5 in the tested strains. b The PRMT5 protein content in the WT, CK and PRMT5-silenced strains was detected by Western blot analysis. c The GA content in the tested strains. The data are presented as the means ± SDs based on three independent experiments (****P < 0.0001 by one-way ANOVA).
GlPRMT5 interacts with GlPP2C1 in vivo and in vitro
Previous studies have reported that PRMT5 regulates physiological functions by directly binding to nonhistone proteins7,8. To study the molecular mechanism by which GlPRMT5 regulates GA biosynthesis, immunoprecipitated proteins were analyzed by mass spectrometry using an anti-PRMT5 antibody. A total of 98 interacting proteins were found by mass spectrometry analysis, among which Gl22901 (GlPP2C1 protein phosphatase) had the highest score (Score: the protein matching degree, the higher the score, the higher the reliability, Supplementary Table 1). To investigate the interaction between endogenous GlPRMT5 and GlPP2C1, immunoprecipitation was conducted. After immunoprecipitation with rabbit anti-PRMT5 or normal rabbit IgG beads, the IP product was treated with anti-PRMT5 antibody and subjected to Western blot analysis to assess the accuracy of the experiment. Furthermore, the IP product was also treated with anti-GlPP2C1 antibody to examine the interaction with endogenous GlPRMT5. The results showed that endogenous GlPRMT5 forms a physical complex with endogenous GlPP2C1 in G. lucidum (Fig. 2a). Previous studies reported that the cDNA length of GlPP2C1 is 1545 bp, consisting of a nonconserved C-terminal (CT, 1-378 bp) domain and a conserved N-terminal (NT, 379-1545 bp) PP2C_SIG domain31. To identify the interaction between GlPRMT5 and GlPP2C1 and identify the specific domain of interaction, the GlPP2C1 gene was truncated into CT and NT according to the domain (Fig. 2b), and a yeast two-hybrid assay (Y2H) was performed. As shown in Fig. 2c, the yeast strains cotransformed with pGBKT7-PRMT5 and full-length pGADT7-PP2C1 or pGADT7-PP2C1 CT grew well on selective media in the presence of the X-α-D-galactoside (X-α-gal) indicator, which is blue on the plate. The truncated form of pGADT7-PP2C1 NT did not grow on plates containing X-α-gal. This indicated that GlPRMT5 interacts with the full length of GlPP2C1 in yeast and interacts with the CT of PP2C1, rather than the NT of PP2C1. In addition, we further determined the interaction between the GlPRMT5 and GlPP2C1 through a bimolecular fluorescence complementation (BiFC) assay. According to the domain structure, the GlPP2C1 gene was truncated (Fig. 2d). Yeast strain SFY2620 cotransformed with PVN-PRMT5 and full-length PVC-PP2C1 and PVC-PP2C1 CT fusion vectors exhibited strong green fluorescent protein (GFP) fluorescence signals (Fig. 2e). Notably, the truncated form of PVC-PP2C1 NT showed no signal. Taken together, these results suggested that GlPRMT5 interacts with GlPP2C1 in vivo and in vitro.

a Co-IP detection of the interaction between endogenous GlPRMT5 and GlPP2C1. Immunoprecipitation of mycelial lysates with control rabbit IgG or anti-PRMT5 antibody. The IP products were detected by Western blotting with an anti-PRMT5 antibody to assess the accuracy of the experiment. Additionally, the IP product was probed with an anti-GlPP2C1 antibody to specifically detect the interaction between GlPP2C1 and endogenous GlPRMT5. Whole cell extracts (Input) show the results of Western blot with anti-PRMT5 antibody and anti-GlPP2C1 antibody as controls. b Schematic representation of the N-terminal (NT) and C-terminal (CT) structures of GlPP2C1 used for Y2H analysis. c Y2H assay detection of the interaction between GlPRMT5 and GlPP2C1. pGBKT7-PRMT5, pGADT7-PP2C1, pGADT7-PP2C1 NT, and pGADT7-PP2C1 CT were cotransformed into the Y2H strain. SD-Leu-Trp medium was used for testing successful mating, and SD-Ade-His-Leu-Trp/X-α-gal medium was used for testing interactions. The combination of pGBKT7-53 and pGADT7-T was used as the positive control. d Schematic representation of the NT and CT structures of GlPP2C1 used for BiFC analysis. e BiFC verified the interaction between GlPRMT5 and GlPP2C1. PVN-PRMT5 containing the GFP tag, PVC-PP2C1, PVC-PP2C1 CT, and PVC-PP2C1 NT were cotransformed into the SFY2620 yeast strain (scale bar = 100 μm). DIC, yeast cell morphology under the normal white field of view; Venus, yeast cell morphology under green fluorescence.
Three-dimensional structure and molecular docking of the GlPRMT5 and GlPP2C1 proteins
To observe the three-dimensional structure of GlPRMT5 and GlPP2C1 more intuitively, SWISS-MODEL was used. The protein sequence of GlPRMT5 was uploaded to SWISS-MODEL, and the protein with the highest sequence identity (PDB ID: A0A5C3P5V3.1.A, GMQE: 0.85) was selected as a template (Fig. 3a). Similarly, the protein sequence of GlPP2C1 was uploaded to SWISS-MODEL, and the protein with the highest sequence identity (PDB ID: A0A4Q9NLU0, GMQE: 0.81) was selected as a template (Fig. 3b). Molecular docking is one of the most important methods of molecular simulation, helping explore the binding mode and interaction between proteins. To explore the protein interaction between GlPRMT5 and GlPP2C1, molecular docking of GlPRMT5 and GlPP2C1 was performed (Fig. 3c). Using GlPRMT5 as the receptor and GlPP2C1 as the ligand, all functional residues were found by protein‒protein interaction analysis in PyMol. In the hydrogen bond interaction, there are multiple groups of residues used to form hydrogen bonds between GlPRMT5 and GlPP2C1, such as the hydrogen bond formed by HIS608 of GlPRMT5 and GLU373 of GlPP2C1. Under the effect of these interaction forces, GlPRMT5-GlPP2C1 scored −711 and performed better. These results further suggested the interaction of GlPRMT5 and GlPP2C1.

a The protein three-dimensional structure of GlPRMT5 by SWISS-MODEL. b The protein three-dimensional structure of GlPP2C1 by SWISS-MODEL. c Molecular docking of the GlPRMT5 and GlPP2C1 proteins by AutoDockTools-1.5.7 and PyMol. In PyMol, GlPRMT5 is represented as a dark blue cartoon model, GlPP2C1 is shown as a cyan cartoon model, and their binding sites are shown as stick structures of corresponding colors. When focusing on a binding region, the binding site is displayed with a representation of the protein to which it belongs.
GlPRMT5 negatively regulates GlPP2C1 protein activity
Numerous studies have shown that protein‒protein interactions can plainly affect the activity of target proteins32,33. To observe the changes in GlPP2C1 protein activity, GlPP2C1 protein phosphatase activity was detected in the WT, CK and PRMT5i-silenced strains. The data showed that GlPP2C1 activity was evidently increased in the PRMT5-silenced strains, and a 62% increase in GlPP2C1 activity was found in the PRMT5-silenced strains compared with the WT and CK strains (Fig. 4a). This result suggested that silencing GlPRMT5 increases GlPP2C1 activity.

a Determination of GlPP2C1 enzyme activity in the WT, CK and PRMT5-silenced strains. b Determination of GlPP2C1 enzyme activity in the WT, CK, PP2C1-silenced, PRMT5-silenced and PRMT5-PP2C1 cosilenced strains. The data are presented as the means ± SDs based on three independent experiments (ns, not significant; ****P < 0.0001 by one-way ANOVA).
In addition, PP2C1-silenced strains and PRMT5-PP2C1 cosilenced (PRMT5i-PP2C1i) strains were constructed using the silencing vector of the dual-promoter system previously described by our laboratory29. Previous studies used qRT-PCR to screen out two PP2C1-silenced strains (PP2C1i-1 and PP2C1i-3) with silencing efficiencies of 65% and 70%, respectively31. Here, we screened a PP2C1-silenced strain (PP2C1i-14) with a silencing efficiency of 62% by qRT-PCR (Supplementary Fig. 1a). The expression of PP2C1 protein in the WT, CK and PP2C1-silenced strains was further detected by Western blot analysis (Supplementary Fig. 1b). The results showed that the PP2C1-silenced strains effectively reduced PP2C1 protein levels compared with the WT and CK strains. Moreover, the structure of the PRMT5i-PP2C1i vector constructed for silencing the expression of PRMT5-PP2C1 is shown in Supplementary Fig. 1c. Three strains (PRMT5i-PP2C1i-6, PRMT5i-PP2C1i-21 and PRMT5i-PP2C1i-27) were screened by qRT‒PCR among the 38 gene silencing candidate strains (Supplementary Fig. 1d). The expression of PRMT5 and PP2C1 protein in the WT, CK and PRMT5-PP2C1 cosilenced strains was further detected by Western blot analysis (Supplementary Fig. 1e). The results showed that the PRMT5-PP2C1 cosilenced strains obviously reduced PRMT5 and PP2C1 protein levels compared with the WT and CK strains. Taken together, these results confirmed the effectiveness of the PP2C1-silenced strains and the PRMT5-PP2C1 cosilenced strains.
To further observe the changes in GlPP2C1 activity, GlPP2C1 protein phosphatase activity was determined in the WT, CK, PRMT5-silenced, PP2C1-silenced and PRMT5-PP2C1 cosilenced strains (Fig. 4b). The results showed that GlPP2C1 activity in the PRMT5-silenced strains was obviously higher (by 62%) than that in the WT and CK strains, while GlPP2C1 activity in the PP2C1-silenced strains was evidently lower (by 77%) than that in the WT and CK strains. However, GlPP2C1 activity in the PRMT5-PP2C1 cosilenced strains was between that of the PRMT5-silenced strains and PP2C1-silenced strains, and there was no difference between the WT and CK strains. The above results indicate that GlPRMT5 negatively regulates GlPP2C1 protein activity.
GlPRMT5 symmetrically dimethylates GlPP2C1
This is the most fundamental mechanism by which PRMT5 participates in the signal transduction pathway through symmetric dimethylation of arginine residues in target proteins. To verify the molecular mechanism by which GlPRMT5 regulates GlPP2C1 activity, the pColdI-PRMT5 recombinant protein and pColdI-PP2C1 recombinant protein were expressed and purified by a prokaryotic expression system, and the symmetrical dimethylation of arginine (sDMA) modification status of GlPP2C1 was further analyzed. In the presence of S-adenosyl-methionine (SAM, a methyl donor), GlPRMT5 can symmetrically dimethylate GlPP2C1. In the absence of SAM, when GlPRMT5 was coincubated with GlPP2C1, no signal was detected with the sDMA antibody. Furthermore, no signal was detected when SAM was incubated with GlPRMT5 or GlPP2C1 single protein (Fig. 5a). These results suggested that GlPRMT5 can symmetrically dimethylate GlPP2C1. To further explore potential sites of GlPRMT5-mediated symmetric dimethylation in the His-PP2C1 recombinant protein, the samples after coincubation of GlPRMT5 and GlPP2C1 were analyzed by LC‒MS/MS. The data results showed a series of high-mass-accuracy y ions and b ions identified symmetric dimethylation on arginine 99 (Arg99/R99) and arginine 493 (Arg493/R493) of GlPP2C1 (Fig. 5b, c). The above results indicated that the R99 and R493 sites of the GlPP2C1 protein can be specifically symmetrically dimethylated via GlPRMT5.

a Purified His-PRMT5 fusion protein was incubated with His-PP2C1 in the presence or absence of SAM. Symmetric dimethylation of His-PP2C1 was detected via Western blot by anti‐SDMA antibody and anti-His, and total amounts of proteins were visualized by Coomassie Blue staining. b Mass spectrum of GlPP2C1 Arg 99, AAVSLEGNLDR. c Mass spectrum of GlPP2C1 Arg 493, YRDDMTVLVVFFAEEDGR.
GlPRMT5 catalyzes arginine symmetric dimethylation of GlPP2C1 at the Arg99 and Arg493 residues
Since R99 and R493 are the targets of symmetric dimethylation of the GlPP2C1 protein, the symmetric dimethylation modification of these arginine residues may interfere with the GlPP2C1 activity regulated by GlPRMT5. A site-directed mutagenesis kit was used to mutate arginine modified by symmetrical dimethylation into a lysine (R99K and R493K), which cannot be symmetrically dimethylated via GlPRMT5. In addition, a double mutant form (R99/493K) of the recombinant protein was also constructed. The in vitro methylation test was performed on these different forms of recombinant proteins again, and the results showed that the sDMA levels of the single mutant recombinant proteins of His-PP2C1 R99K and His-PP2C1 R493K were evidently lower than those of the unmutated protein His-PP2C1. No signal was detected for the R99/493K double mutant recombinant protein (Fig. 6a–c). In addition, to highlight changes in the structure of GlPP2C1 modified by symmetric dimethylation, Schrödinger analysis was performed. Compared with unmodified GlPP2C1 (Fig. 6d), the R99 and R493 residues of symmetrically dimethylated GlPP2C1 had two symmetrical methyl groups added (Fig. 6e). Changes in protein structure are accompanied by changes in activity. GlPP2C1 activity in different mutant recombinant protein bodies was further determined by the Serine/Threonine Phosphatase Assay System, and the standard curve of absorbance of free phosphate concentration at 630 nm is shown in Supplementary Fig. 2a. In the presence of equal amounts of serine/threonine phosphopeptide substrates, the absorbance gradually increased with increasing amounts of His-PP2C1 protein (Supplementary Fig. 2b), demonstrating that GlPP2C1 possesses phosphatase activity in vitro. In addition, the results showed that the addition of GlPRMT5 plainly reduced GlPP2C1 activity (by 62%) compared to the untreated GlPP2C1 control (Fig. 6f). However, there was a marked increase in GlPP2C1 activity in the single mutant proteins GlPP2C1 R99K body (1.9-fold) and GlPP2C1 R493K body (1.4-fold). Interestingly, GlPP2C1 activity in the double mutant protein body GlPP2C1 R99/493K was consistent with the activity in the control group where only a single GlPP2C1 protein was present. Therefore, the symmetrical dimethylation modification of GlPP2C1 obviously inhibits PP2C activity, and R99 of GlPP2C1 is the key site for its activity. In summary, the above results indicated that GlPRMT5-mediated symmetric dimethylation reduces GlPP2C1 activity, and the modification of the R99 site is the key to affecting GlPP2C1 activity.

Purified His-PRMT5 fusion protein was incubated with His-PP2C1, His-PP2C1 R99K (a), His-PP2C1 R493K (b) or His-PP2C1 R99/493K (c) in the presence or absence of SAM. Symmetric dimethylation of His-PP2C1 was detected via Western blot by anti‐SDMA antibody and total amounts of proteins were visualized by Coomassie Blue staining. d Unmodified local structure of GlPP2C1 residues R99 and R493 by Schrödinger analysis. e Structure of GlPP2C1 R99 and R493 residues modified by symmetric dimethylation via Schrödinger. Gray sticks represent H, cyan sticks represent N, dark blue sticks represent C, and red sticks represent O. f Different GlPP2C1 proteins were determined using the Serine/Threonine Phosphatase Assay System as directed by the manufacturer. After the reaction was complete, the absorbance was measured on a microplate reader at a wavelength of 630 nm. The data are presented as the means ± SD based on three independent experiments (ns not significant; **P < 0.01; ****P < 0.0001 by one-way ANOVA).
Symmetrical dimethylation of GlPP2C1 does not affect the interaction with GlPRMT5
Symmetrical dimethylation modification of GlPP2C1 reduces its activity via GlPRMT5. To test whether this modification affects the binding between GlPRMT5 and GlPP2C1, GlPP2C1 single mutants (R99K, R493K) and double mutants (R99/493K) were inserted into pGBKT7 and PVC1 (Fig. 7a, c), and further Y2H and BiFC were performed. As shown in Fig. 7b, two single mutant full-length (pGADT7-PP2C1 R99K, pGADT7-PP2C1 R493K) or double mutant full-length (pGADT7-PP2C1 R99/493K) cotransformed with pGBKT7-PRMT5 grew well on the selective medium and appeared blue on the plate containing X-α-gal. In addition, the truncated form of pGADT7-PP2C1 R99K CT, a cotransformed in yeast strain with pGBKT7-PRMT5, grew well on selective medium and appeared blue on plates containing X-α-gal, while the truncated form of pGADT7-PP2C1 R493K NT did not grow on plates containing X-α-gal. In addition, BiFC was conducted to further determine the interaction between GlPRMT5 and GlPP2C1 modified by symmetric dimethylation. Yeast strain SFY2620 cotransformed with PVN-PRMT5 and two single mutant full-length (PVC-PP2C1 R99K, PVC-PP2C1 R493K) or double mutant full-length (PVC-PP2C1 R99/493K) fusion vectors exhibited strong GFP fluorescence signals (Fig. 7d). In addition, the truncated form of PVC-PP2C1 R99K CT, cotransformed in yeast strain SFY2620 with PVN-PRMT5, exhibited strong GFP fluorescence signals. However, the truncated form of PVC-PP2C1 R493K NT showed no signal. These results indicated that GlPP2C1 modified by symmetrical dimethylation did not affect the interaction with GlPRMT5.

a Schematic representation of NT and CT structures of GlPP2C1 single mutant (R99K, R493) and double mutant (R99/493K) for Y2H analysis. b pGBKT7-PRMT5, pGADT7-PP2C1 R99K, pGADT7-PP2C1 R493K, pGADT7-PP2C1 R99/493K, pGADT7-PP2C1 R99K CT, and pGADT7-PP2C1 R493K NT were cotransformed into the Y2H strain. SD-Leu-Trp medium was used for testing successful mating, and SD-Ade-His-Leu-Trp/X-α-gal medium was used for testing interactions. The combination of pGBKT7-53 and pGADT7-T was used as the positive control. c Schematic representation of NT and CT structures of GlPP2C1 single mutant (R99K, R493) and double mutant (R99/493K) for BiFC analysis. d PVN-PRMT5 containing the GFP tag, PVC-PP2C1 R99K, PVC-PP2C1 R493K, PVC-PP2C1 R99/493K, PVC-PP2C1 R99K CT, and PVC-PP2C1 R493K NT were cotransformed into the SFY2620 yeast strain (scale bar = 100 μm). DIC, yeast cell morphology under the normal white field of view; Venus, yeast cell morphology under green fluorescence.
Silencing Gl PP2C1 results in reduced GA content
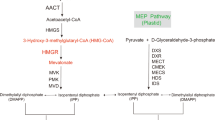
GA is the most important secondary metabolite in G. lucidum. To explore whether GlPP2C1 is involved in regulating the GA biosynthesis, the GA content was determined in the WT, CK and PP2C1-silenced strains. We found that the GA content in the PP2C1-silenced strains was plainly decreased (by approximately 33%), compared with that in the WT and CK strains (Fig. 8a). This result indicated that GlPP2C1 played a positive regulatory role in the GA biosynthetic pathway. To elucidate the role of GlPP2C1 in GlPRMT5-regulated GA biosynthesis, the GA content was examined in the WT, CK, PP2C1-silenced, PRMT5-silenced and PRMT5-PP2C1 cosilenced strains (Fig. 8b). The results showed that the GA content in the PRMT5-PP2C1 cosilenced strains was between the PRMT5-silenced strains and the PP2C1-silenced strains, consistent with the content in the WT and CK strains. Previous studies have suggested that GA is synthesized via the mevalonate/isoprenoid pathway, wherein hydroxy-3-methylglutaryl-CoA reductase (hmgr), 2,3-oxidosqualene-lanosterol cyclase (osc) and squalene synthase (sqs) play crucial roles in this pathway34,35,36,37,38. The key genes expression levels of GA biosynthesis (hmgr, osc and sqs) were detected in the WT, CK and PP2C1-silenced strains. The results revealed that the expression of hmgr and osc in the PP2C1-silenced strains were not obviously different from those in the WT and CK strains (Fig. 8c, d), while the expression of sqs exhibited a decrease of 70% in the PP2C1-silenced strains in comparison to the WT and CK strains (Fig. 8e). The above results suggested that silencing GlPP2C1 results in reduced GA content.

a Determination of GA content in the WT, CK and PP2C1-silenced strains. b Determination of GA content in the WT, CK, PP2C1-silenced, PRMT5-silenced and PRMT5-PP2C1 cosilenced strains. c Relative expression levels of hmgr in the WT, CK and PP2C1-silenced strains. d Relative expression levels of osc in the WT, CK and PP2C1-silenced strains. e Relative expression levels of sqs in the WT, CK and PP2C1-silenced strains. The data are presented as the means ± SDs based on three independent experiments (ns not significant; *P < 0.1; ***P < 0.001; ****P < 0.0001 by one-way ANOVA).
Discussion
PRMT5, a type II arginine methyltransferase, can perform symmetric dimethylation on the arginine sites of histones and nonhistones39. Compared with the research on histones, there are very few studies on the regulation of PRMT5 on nonhistones. With the development of science, technology and instruments, a series of target nonhistones of PRMT5 have been gradually identified. For example, MCM7 (minichromosome maintenance-7) was identified as a direct PRMT5 binding partner via Co-IP and MS analysis40. MTHFD1 (methylenetetrahydrofolate dehydrogenase, cyclohydrolase and formyltetrahydrofolate synthetase 1) can specifically bind PRMT541. Discovering target proteins of PRMT5 is the key to exploring its function. Here, we report for the first time the target protein GlPP2C1 of GlPRMT5 by MS analysis in G. lucidum. In addition, PRMT5 was shown to affect the activity, localization, and function of target proteases by binding to nonhistone proteins and undergoing symmetrical dimethylation modification42. For example, PRMT5 promotes AKT activation by symmetrically dimethylating AKT protein kinase to control tumorigenesis33. AtPRMT5-mediated AtLCD (l-cysteine desulfhydrase) methylation increases its enzymatic activity, thereby enhancing endogenous H2S signaling and ultimately improving plant tolerance to Cd2+ stress in Arabidopsis43. In this study, it was found that GlPRMT5-mediated symmetric dimethylation of GlPP2C1 reduced GlPP2C1 enzymatic activity. The interaction of GlPRMT5 and GlPP2C1 may regulate the biosynthesis of secondary metabolites.
A large number of existing studies have reported that type 2C protein phosphatases are involved in signal transduction and biological processes. For example, overexpression of PtrHAB2 (a type 2C protein phosphatase) leads to an increased plant growth rate, increased trunk height, and altered leaf morphogenesis in Populus trichocarpa44. Genome-wide expression analysis in rice revealed that transcripts of several PP2C genes were obviously altered during critical stages of reproductive development45. Biosynthesis of the secondary metabolite deoxynivalenol is noticeably reduced in 11 phosphatase deletion mutants of Fusarium graminearum46. Aspergillus fumigatus protein phosphatase PpzA is involved in affecting secondary metabolites47. Type 2C protein phosphatases PTC1 and PTC2 in Aspergillus flavus positively regulate the content of secondary metabolite aflatoxin15. In our study, silencing GlPP2C1 resulted in a remarkable decrease in GA content, suggesting that GlPP2C1 positively regulates GA biosynthesis. This indicates that protein phosphatase generally has the function of regulating fungal secondary metabolism. It was further found that the transcription level of key genes for GA biosynthesis (sqs) was notably decreased in the PP2C1-silenced strains. This indicates that GlPP2C1 may affect the biosynthesis of secondary metabolites of G. lucidum by regulating the key genes expression level of GA biosynthesis. Until now, PP2C has also been reported to regulate the biosynthesis of secondary metabolites in various ways. For instance, type 2C protein phosphatase regulates the stability of ACS7, the rate-limiting enzyme in ethylene biosynthesis in Arabidopsis48. Type 2C phosphatases Ptc1 and Ptc2 participate in autophagy and mitochondrial pyruvate metabolism through dephosphorylation of phosphoglycerate kinase 1 (PGK1) in A. flavus, thereby regulating aflatoxin synthesis15. Type 2C protein phosphatase GhDRP1 in cotton affects ROS scavenging enzymes and the accumulation of proline by regulating the flavonoid biosynthetic pathway49. This indicates that GlPP2C1 may also affect the secondary metabolism and biosynthesis of G. lucidum through other modes of action. Furthermore, it has been widely reported that type 2C protein phosphatases perform their main function by dephosphorylating substrate protein kinases. For example, Ptc1, a type 2C Ser/Thr phosphatase, inactivates the HOG pathway by dephosphorylating the mitogen-activated protein kinase (MAPK) in S. cerevisiae50. The PP2C enzyme deactivates MAPKs through dephosphorylation, and thus blocks the downstream regulation of the signaling cascade51. Therefore, it is necessary to study the molecular mechanism by which GlPP2C1 regulates GA biosynthesis in G. lucidum.
Most studies have screened the downstream proteins of PP2C14,15, but there are still few studies on the proteins regulating PP2C. Upon light exposure, SAUR50 binds and inhibits PP2C-D1 activity, inducing general cell expansion and ultimately leading to hook and cotyledon opening and cotyledon enlargement in Arabidopsis52. In addition, regulatory mechanisms of type 2 A protein phosphatases have been extensively reported53,54. SBI1, encodes a leucine carboxylmethyltransferase (LCMT) that methylates PP2A, thereby promoting its association with activated BRI1 in A. thaliana55. PRMT5 negatively regulates the enzymatic activity of PP2A56. Therefore, it is necessary to explore the mechanism regulating type 2C protein phosphatase activity. It has been reported that the activity of type 2C protein phosphatase is regulated through protein post-translational modification. For instance, the H2O2 produced by aba-induced NADPH oxidase RbohB/E inhibits PP45 activity by oxidizing Cys-350 and Cys-428 residues of PP4516. Our study showed that GlPRMT5 mediates symmetrical dimethylation of GlPP2C1 at residues R99 and R493, resulting in decreased GlPP2C1 activity. The study identifies the upstream protein of GlPP2C1, providing more meaningful insights into the molecular mechanism of PP2C in other species.
With recent advances in proteomics technology, especially mass spectrometry, a large number of studies have shown that many proteins are cotargeted by many different types of posttranslational modifications to regulate protein function. For example, PRMT5 symmetrically dimethylates ASK1 (apoptosis signal-regulating kinase 1) at the arginine 89 residue, thereby promoting the interaction between ASK1 and Akt and phosphorylating ASK1 at the serine 83 residue to negatively regulate its activity57. PRMT5-mediated symmetric dimethylation of KLF5 (an oncogenic factor) at R57 antagonizes GSK3β-mediated KLF5 phosphorylation and Fbw7-mediated KLF5 ubiquitination to promote basal-like breast cancer (BLBC)58. It is worth noting that PRMT5 has been widely reported to have diverse functions and can participate in the same biological process through multiple target proteins. For instance, PRMT5 interacts with and methylates Mxi1, thereby promoting the binding of β-Trcp ligase to Mxi1 and promoting the ubiquitination and degradation of Mxi1 in lung cancer59. PRMT5 methylates the oncogenic factor Krüppel-like factor 5 (KLF5) to prevent its degradation, thereby promoting the maintenance and proliferation of lung cancer cells60. In our study, we found that GlPRMT5-mediated symmetric dimethylation of GlPP2C1 markedly decreased its activity and GA content. Furthermore, the GA content and GlPP2C1 activity in the PRMT5-PP2C1 cosilenced strains were higher than those in the PP2C1 alone-silenced strains, indicating that in addition to GlPP2C1, GlPRMT5 may have other pathways to regulate GA biosynthesis. This may be the result of the crosstalk formed by methylation and dephosphorylation jointly regulating GA biosynthesis. This will also be the focus of our further research.
Collectively, our research results showed that GlPP2C1, the interaction target protein of GlPRMT5, has been identified. The interaction between GlPRMT5 and GlPP2C1 was verified in vivo and in vitro. Furthermore, on the one hand, our data suggested that GlPRMT5-mediated symmetric dimethylation negatively regulates GlPP2C1 activity. On the other hand, it was demonstrated that GlPP2C1 is involved in GlPRMT5-mediated GA biosynthesis. GlPRMT5-mediated symmetric dimethylation of residues R99 and R493 in GlPP2C1 clearly attenuated GlPP2C1 activity. Further data indicated that the R99 residue in GlPP2C1 is the key to affecting GlPP2C1 activity. In addition, the symmetrical dimethylation modification did not affect the interaction between GlPP2C1 and GlPRMT5. These data establish a framework for the regulation of secondary metabolites via GlPRMT5 in macrofungi. The identification of modified proteins and characteristic sites is conducive to a comprehensive understanding of the molecular mechanism and regulatory functions of GlPRMT5-mediated symmetric dimethylation.
Methods
Strains and culture conditions
The wild-type (WT) strain was selected from G. lucidum ACCC53264 provided by the Agricultural Culture Collection of China. The PP2C1-silenced strains (PP2C1i-1 and PP2C1i-3) and PRMT5-silenced strains (PRMT5i-31 and PRMT5i-35) were established in our previous study30,31. The strains were fermented in CYM broth for 7 days at 28 °C in a shaking incubator at 150 rpm to measure GA content.
Construction of RNAi strains
G. lucidum cDNA was used as a template to amplify the GlPRMT5 gene and GlPP2C1 gene fragments, and PP2C1i-F (GGGGTACACTCGCATTCCCGCTT), PP2C1i-R (CGCCTCTCCGTTCATTGCCCTGTGCTTT), PRMT5i-F (AAAGGCACCAGGCACATGAAACGGGCG) and PRMT5i-R (GACTAGTAGCGTGGGTATGTGGG) were used for joint PCR to construct fungal RNAi vectors61. The RNAi silencing vector pAN7-dual-PRMT5i-PP2C1i was electroporated into G. lucidum, and three independent strains with the highest silencing efficiency were selected for subsequent experiments25. The empty vector control was named CK.
Gene expression analysis
Using the cDNAs of different transformants screened as templates and the primers listed in Supplementary Table 2, quantitative RT‒qPCR analysis was performed using Eppendorf Mastercycler Ep Realplex 2.2 software. The transcript level was analyzed for gene-specific mRNAs using 18 S rRNA as the housekeeping gene as previously described62. The relative gene expression levels were determined using the 2−ΔΔCT method.
Extraction and detection of GA
GA was extracted and detected as previously described22. In brief, dried mycelial powder (200 mg) was ultrasonically extracted in 10 mL of 95% ethanol for 2 h. The mixture was centrifuged at 4000 rpm for 10 min to obtain the supernatant. Next, the supernatant was dried with a rotary evaporator to obtain the crude extract. The crude extract was then resuspended in 0.5 mL methanol and analyzed with ultraperformance liquid chromatography (UPLC).
Expression and purification of recombinant proteins and antibody preparation
GlPRMT5 and GlPP2C1 sequences from G. lucidum were deposited in GenBank (OP360010 and OP251201, respectively). The expression vectors pColdI-PRMT5, pColdI-PP2C1, pColdI-PP2C1 R99K, pColdI-PP2C1 R493K and pColdI-PP2C1 R99/493K were transformed into E. coli strain BL21 (DE3), protein expression was induced with 500 µM isopropyl-β-D-thiogalactopyranoside (IPTG), and the strains were grown for 16 h at 16 °C. Purification of recombinant proteins was performed using nickel-nitrilotriacetate (Ni-NTA) agarose columns (Sangon, C600033). In addition, the purified His-PP2C1 recombinant protein (Supplementary Fig. 3) was sent to a professionally qualified antibody preparation company for immunization of rabbits (Chemgen Biotech)63.
Phosphatase activity assay
Protein was extracted from PRMT5-silenced strains or PRMT5-PP2C1 cosilenced strains with phosphatase storage buffer (10 mM Tris, pH 7.5, 1 mM EDTA, 0.02% [w/v] sodium azide). The assay was then performed using the Serine/Threonine Phosphatase Assay System (Promega, Cat# V2460) as directed by the manufacturer. After the reaction was complete, the absorbance was measured on a microplate reader at a wavelength of 630 nm. The purified protein (pColdI-PP2C1, pColdI-PP2C1 R99K, pColdI-PP2C1 R493K and pColdI-PP2C1 R99/493K) was also measured as above.
In vitro methylation assay
The purified pColdI-PRMT5 in pColdI-PP2C1, pColdI-PP2C1 R99K, pColdI-PP2C1 R493K and pColdI-PP2C1 R99/493K were incubated with 50 μL of reaction buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1 mM PMSF, 1 mM DTT), with or without 16 mmol/L methyl donor S-adenosyl-methionine (SAM, Solarbio, S9990). Then, the cells were incubated at 30 °C for 90 min. The reactions were stopped by the addition of sample loading buffer, followed by immunoblotting with anti-sDMA antibody (anti-dimethyl-arginine, symmetric, SYM11, Millipore, 07-413).
Mass spectrometry (MS) analysis
After incubating the purified pColdI-PRMT5, pColdI-PP2C1 and SAM in the above 50 μL reaction buffer, they were treated at 30 °C for 90 min and separated on a 12% (wt/vol) SDS‒PAGE gel. The GlPP2C1 protein was cleaved, digested with trypsin in the gel, and analyzed by LC‒MS/MS analysis of polypeptides. Peptide identification was performed using MS/MS spectra of the GlPP2C1 protein.
Yeast two-hybrid assay
The full-length GlPRMT5 gene was inserted into pGBKT7 to construct the bait vector. We inserted the full-length GlPP2C1 gene and the truncated versions of PP2C1 CT and PP2C1 NT into pGADT7 to construct the prey vector. The bait and prey vectors were cotransformed into the yeast Y2HGold strain and cultured in SD/-Leu/-Trp medium and SD/-Ade-His-Leu-Trp/X-α-Gal medium at 30 °C for 3 to 5 days. Assessment of protein interaction is based on the development of blue colonies on the plate. Y2H interactions between two single mutant full-length proteins (pGADT7-PP2C1 R99K, pGADT7-PP2C1 R493K), double mutant full-length proteins (pGADT7-PP2C1 R99/493 K), the truncated form of pGADT7-PP2C1 R99K CT and pGADT7-PP2C1 R493K NT and pGBKT7-PRMT5 were detected by the above method.
BiFC assay
Bimolecular fluorescence complementation (BiFC) assays were performed as follows64. Briefly, the full-length GlPRMT5 gene was inserted into pVN1, and full-length GlPP2C1 and truncated forms of PP2C1 CT and PP2C1 NT were inserted into pVC1. The recombinant constructs were subsequently cotransformed into yeast strain SFY2620 and incubated in SD/-Leu-Ura medium at 30 °C for 3 days. Yeast strains were evaluated for green fluorescence by confocal microscopy. The BiFC interaction between two single mutant full-length proteins (PVC-PP2C1 R99K, PVC-PP2C1 R493K), double mutant full-length proteins (PVC-PP2C1 R99/493K), the truncated forms of PVC-PP2C1 R99K CT and PVC-PP2C1 R493K NT and PVN-PRMT5 were detected by the above method.
Coimmunoprecipitation assay
Coimmunoprecipitation was carried out according to a previously reported method in the laboratory61. Briefly, G. lucidum mycelia powder (0.15 g) was lysed in lysis buffer containing 100 mM NaCl, 20 mM Tris–HCl pH 7.6, 0.1% Triton X-100, 0.5% protease inhibitor cocktail (Sigma), and 1 mM phenylmethanesulfonyl fluoride (Sangon) for 1 h. The supernatants (200 µL) were immunoprecipitated with protein A/G agarose (25 µL, Thermo Scientific, 88802), followed by incubation with control rabbit IgG (Solarbio) or PRMT5 antibody (Abcam, ab109451) at 4 °C overnight. The immunoprecipitated proteins were was washed (5 times) with lysis buffer, and the protein lysate was further eluted with 5× SDS loading buffer. Western blotting was further performed using anti-PRMT5 antibody and anti-GlPP2C1 polyclonal antibody. In addition, using the same method to immunoprecipitate interacting proteins with PRMT5, the proteins were separated on a 12% (wt/vol) SDS‒PAGE gel and stained with Coomassie blue. Bands were excised, followed by digestion and plastid analysis.
Homology modeling and molecular docking
The protein sequence information of GlPRMT5 and GlPP2C1 was uploaded to the online website SWISS-MODEL to construct the three-dimensional structure of GlPRMT5 and GlPP2C1. The protein with the highest sequence identity (GMQE value close to 1, high reliability) was selected as the template. Using GlPRMT5 as the receptor and GlPP2C1 as the ligand, AutoDockTools-1.5.7 was used for molecular docking65. The default hydrogen bond distance is 2.5 Å, but when the two forces are weak, the distance is set to 3.0 Å, and when the two forces are strong, the distance is set to 2.0 Å. Then, the docking server (GRAMM) was used for protein‒protein docking66,67. The obtained protein‒protein complexes were also optimized with AutoDockTools-1.5.7 for dehydration and hydrogenation. Finally, PyMol was used to predict protein interactions and generate protein‒protein interaction maps. Structures of the unmodified GlPP2C1 and symmetric dimethylation modified GlPP2C1 R99 and R493 residues were illustrated using Schrödinger.
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism 8 on the data presented in this article, which were obtained from at least three independent samples. Error bars indicate the standard deviation (SD) of triplicate means. Differences in means were analyzed by one-way or two-way analysis of variance (ANOVA) for differences between groups using GraphPad Prism. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 are statistically significant. NS indicates not significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files) or are available from the corresponding author on reasonable request. The source data underlying the graphs in the figure are shown in Supplementary Data 1. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD048799. Uncropped western blots and gel are in Supplementary Figs. 5–9.
References
Guccione, E. & Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 20, 642–657 (2019).
Pei, Y. et al. Mutations in the Type II protein arginine methyltransferase AtPRMT5 result in pleiotropic developmental defects in Arabidopsis. Plant Physiol. 144, 1913–1923 (2007).
Xu, X. D., Chen, Y., Li, B. Q. & Tian, S. P. Arginine methyltransferase PeRmtC regulates development and pathogenicity of via mediating key genes in conidiation and secondary metabolism. J. Fungi. 7, ARTN 80710.3390/jof7100807 (2021).
Andreu-Perez, P. et al. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci. Signal. 4, ra58 (2011).
Dacwag, C. S., Ohkawa, Y., Pal, S., Sif, S. & Imbalzano, A. N. The protein arginine methyltransferase Prmt5 is required for myogenesis because it facilitates ATP-dependent chromatin remodeling. Mol. Cell. Biol. 27, 384–394 (2007).
Tae, S. et al. Bromodomain protein 7 interacts with PRMT5 and PRC2, and is involved in transcriptional repression of their target genes. Nucleic Acids Res. 39, 5424–5438 (2011).
Hu, X. F. et al. PRMT5 facilitates infectious bursal disease virus replication through arginine methylation of VP1. J. Virol. 97, https://doi.org/10.1128/jvi.01637-22 (2023).
Ma, D. P. et al. Arginine methyltransferase PRMT5 negatively regulates cGAS-mediated antiviral immune response. Sci. Adv. 7, ARTN eabc183410.1126/sciadv.abc1834 (2021).
Xia, C. X., Gong, Y. S., Chong, K. & Xu, Y. Y. Phosphatase OsPP2C27 directly dephosphorylates OsMAPK3 and OsbHLH002 to negatively regulate cold tolerance in rice. Plant Cell Environ. 44, 491–505 (2021).
Sugimoto, H. et al. Overexpression of a novel Arabidopsis PP2C isoform, AtPP2CF1, enhances plant biomass production by increasing inflorescence stem growth. J. Exp. Bot. 65, 5385–5400 (2014).
Singh, A., Jha, S. K., Bagri, J. & Pandey, G. K. ABA inducible rice protein phosphatase 2C confers ABA insensitivity and abiotic stress tolerance in Arabidopsis. Plos One 10, ARTN e012516810.1371/journal.pone.0125168 (2015).
Arino, J., Casamayor, A. & Gonzalez, A. Type 2C protein phosphatases in fungi. Eukaryot Cell 10, 21–33 (2011).
Nunez-Rodriguez, J. C., Ruiz-Roldan, C., Lemos, P., Membrives, S. & Hera, C. The phosphatase Ptc6 is involved in virulence and MAPK signalling in Fusarium oxysporum. Mol. Plant Pathol. 21, 206–217 (2020).
Guo, Y. et al. The clade F PP2C phosphatase ZmPP84 negatively regulates drought tolerance by repressing stomatal closure in maize. New Phytol. 237, 1728–1744 (2023).
Zhu, Z. et al. PP2C phosphatases Ptc1 and Ptc2 dephosphorylate PGK1 to regulate autophagy and aflatoxin synthesis in the pathogenic fungus Aspergillus flavus. mBio, e0097723 https://doi.org/10.1128/mbio.00977-23 (2023).
Ni, L. et al. Abscisic acid inhibits rice protein phosphatase PP45 via H2O2 and relieves repression of the Ca2+/CaM-dependent protein kinase DMI3. Plant Cell 31, 128–152 (2019).
Miao, J. et al. OsPP2C09, a negative regulatory factor in abscisic acid signalling, plays an essential role in balancing plant growth and drought tolerance in rice. New Phytol. 227, 1417–1433 (2020).
Cao, Y., Wu, S.-H. & Dai, Y.-C. Species clarification of the prize medicinal Ganoderma mushroom “Lingzhi. Fungal Divers. 56, 49–62 (2012).
Yu-Cheng, D. et al. Diversity and systematics of the important macrofungi in Chinese forests. Mycosystema 40, 770–805 (2021).
Paterson, R. R. Ganoderma—a therapeutic fungal biofactory. Phytochemistry 67, 1985–2001 (2006).
Zhou, L. W. et al. Global diversity of the Ganoderma lucidum complex (Ganodermataceae, Polyporales) inferred from morphology and multilocus phylogeny. Phytochemistry 114, 7–15 (2015).
Han, X. F. et al. Phospholipase D and phosphatidic acid mediate regulation in the biosynthesis of spermidine and ganoderic acids by activating GlMyb in Ganoderma lucidum under heat stress. Environ. Microbiol. https://doi.org/10.1111/1462-2920.16211 (2022).
Lian, L. D. et al. GCN4 regulates secondary metabolism through activation of antioxidant gene expression under nitrogen limitation conditions in Ganoderma lucidum. Appl. Environ. Microb. 87, ARTN e00156-2110.1128/AEM.00156-21 (2021).
Liu, R. et al. SA inhibits complex III activity to generate reactive oxygen species and thereby induces GA overproduction in Ganoderma lucidum. Redox Biol. 16, 388–400 (2018).
Liu, R. et al. GSNOR regulates ganoderic acid content in Ganoderma lucidum under heat stress through S-nitrosylation of catalase. Commun. Biol. 5, ARTN 3210.1038/s42003-021-02988-0 (2022).
Liu, R. et al. Nitric oxide regulates ganoderic acid biosynthesis by the S-nitrosylation of aconitase under heat stress in Ganoderma lucidum. Environ. Microbiol. 23, 682–695 (2021).
Tian, J. L. et al. Hydrogen sulfide, a novel small molecule signalling agent, participates in the regulation of ganoderic acids biosynthesis induced by heat stress in Ganoderma lucidum. Fungal Genet. Biol. 130, 19–30 (2019).
Zhang, G. et al. Functional analysis of an APSES transcription factor (GlSwi6) involved in fungal growth, fruiting body development and ganoderic-acid biosynthesis in Ganoderma lucidum. Microbiol. Res. 207, 280–288 (2018).
Mu, D. et al. The development and application of a multiple gene co-silencing system using endogenous URA3 as a reporter gene in Ganoderma lucidum. PLoS One 7, e43737 (2012).
Liu, R. et al. PRMT5 regulates the polysaccharide content by controlling the splicing of thaumatin-like protein in Ganoderma lucidum. Microbiol. Spectr. e0290623 https://doi.org/10.1128/spectrum.02906-23 (2023).
Wang, Z. et al. GlPP2C1 Silencing Increases the Content of Ganodermalingzhi Polysaccharide (GL-PS) and Enhances Slt2 Phosphorylation. J. Fungi (Basel) 8, https://doi.org/10.3390/jof8090949 (2022).
Huang, L. et al. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat. Commun. 13, 3955 (2022).
Yin, S. et al. PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nat. Commun. 12, 3444 (2021).
Chen, S. et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 3, 913 (2012).
Goldstein, J. L. & Brown, M. S. Regulation of the mevalonate pathway. Nature 343, 425–430 (1990).
Shang, C. H., Shi, L., Ren, A., Qin, L. & Zhao, M. W. Molecular cloning, characterization, and differential expression of a lanosterol synthase gene from Ganoderma lucidum. Biosci. Biotechnol. Biochem. 74, 974–978 (2010).
Shang, C. H. et al. Cloning and characterization of a gene encoding HMG-CoA reductase from Ganoderma lucidum and its functional identification in yeast. Biosci. Biotechnol. Biochem. 72, 1333–1339 (2008).
Zhao, M. W. et al. Cloning and characterization of squalene synthase (SQS) gene from Ganoderma lucidum. J. Microbiol. Biotechnol. 17, 1106–1112 (2007).
Jarrold, J. & Davies, C. C. PRMTs and arginine methylation: cancer’s best-kept secret? Trends Mol. Med. 25, 993–1009 (2019).
Li, X., Wang, X., Zhao, J., Wang, J. & Wu, J. PRMT5 promotes colorectal cancer growth by interaction with MCM7. J. Cell Mol. Med. 25, 3537–3547 (2021).
Meng, Q. et al. Arginine methylation of MTHFD1 by PRMT5 enhances anoikis resistance and cancer metastasis. Oncogene 41, 3912–3924 (2022).
Kim, H. & Ronai, Z. A. PRMT5 function and targeting in cancer. Cell Stress 4, 199–215 (2020).
Cao, H. et al. AtPRMT5-mediated AtLCD methylation improves Cd2+ tolerance via increased H2S production in Arabidopsis. Plant Physiol. 190, 2637–2650 (2022).
Rigoulot, S. B., Petzold, H. E., Williams, S. P., Brunner, A. M. & Beers, E. P. Populus trichocarpa clade A PP2C protein phosphatases: their stress-induced expression patterns, interactions in core abscisic acid signaling, and potential for regulation of growth and development. Plant Mol. Biol. 100, 303–317 (2019).
Singh, A., Giri, J., Kapoor, S., Tyagi, A. K. & Pandey, G. K. Protein phosphatase complement in rice: genome-wide identification and transcriptional analysis under abiotic stress conditions and reproductive development. Bmc Genom. 11, Artn 43510.1186/1471-2164-11-435 (2010).
Yun, Y. et al. Functional analysis of the Fusarium graminearum phosphatome. New Phytol. 207, 119–134 (2015).
Manfiolli, A. O. et al. Aspergillus fumigatus protein phosphatase PpzA is involved in iron assimilation, secondary metabolite production, and virulence. Cell Microbiol. 19, https://doi.org/10.1111/cmi.12770 (2017).
Marczak, M. et al. Protein phosphatases type 2C group A interact with and regulate the stability of ACC synthase 7 in Arabidopsis. Cells 9, https://doi.org/10.3390/cells9040978 (2020).
Chen, Y. et al. A type-2C protein phosphatase (GhDRP1) participates in cotton (Gossypium hirsutum) response to drought stress. Plant Mol. Biol. 107, 499–517 (2021).
Warmka, J., Hanneman, J., Lee, J., Amin, D. & Ota, I. Ptc1, a type 2C Ser/Thr phosphatase, inactivates the HOG pathway by dephosphorylating the mitogen-activated protein kinase Hog1. Mol. Cell Biol. 21, 51–60 (2001).
Smekalova, V., Doskocilova, A., Komis, G. & Samaj, J. Crosstalk between secondary messengers, hormones and MAPK modules during abiotic stress signalling in plants. Biotechnol. Adv. 32, 2–11 (2014).
Wang, J. et al. SAUR17 and SAUR50 differentially regulate PP2C-D1 during Apical Hook development and cotyledon opening in Arabidopsis. Plant Cell 32, 3792–3811 (2020).
Li, Y. et al. Coupling to short linear motifs creates versatile PME-1 activities in PP2A holoenzyme demethylation and inhibition. Elife 11, https://doi.org/10.7554/eLife.79736 (2022).
Rasool, R. U. et al. Loss of LCMT1 and biased protein phosphatase 2A heterotrimerization drive prostate cancer progression and therapy resistance. Nat. Commun. 14, 5253 (2023).
Wu, G. et al. Methylation of a phosphatase specifies dephosphorylation and degradation of activated brassinosteroid receptors. Sci. Signal. 4, ra29 (2011).
Otani, Y. et al. Inhibiting protein phosphatase 2A increases the antitumor effect of protein arginine methyltransferase 5 inhibition in models of glioblastoma. Neuro Oncol. 23, 1481–1493 (2021).
Chen, M. et al. Cross-talk between Arg methylation and Ser phosphorylation modulates apoptosis signal-regulating kinase 1 activation in endothelial cells. Mol. Biol. Cell 27, 1358–1366 (2016).
Wang, X. et al. Arginine methyltransferase PRMT5 methylates and stabilizes KLF5 via decreasing its phosphorylation and ubiquitination to promote basal-like breast cancer. Cell Death Differ. 28, 2931–2945 (2021).
Yang, X. et al. Arginine methyltransferase PRMT5 methylates and destabilizes Mxi1 to confer radioresistance in non-small cell lung cancer. Cancer Lett. 532, 215594, https://doi.org/10.1016/j.canlet.2022.215594 (2022).
Zhou, H. et al. PRMT5 activates KLF5 by methylation to facilitate lung cancer. J. Cell Mol. Med. https://doi.org/10.1111/jcmm.17856 (2023).
Hu, Y. R. et al. In Ganoderma lucidum, Glsnf1 regulates cellulose degradation by inhibiting GlCreA during the utilization of cellulose. Environ. Microbiol. 22, 107–121 (2020).
Wang, Z., Chen, J., Ding, J., Han, J. & Shi, L. GlMPC activated by GCN4 regulates secondary metabolism under nitrogen limitation conditions in Ganoderma lucidum. mBio, e0135623, https://doi.org/10.1128/mbio.01356-23 (2023).
Vautard-Mey, G., Cotton, P. & Fevre, M. Expression and compartmentation of the glucose repressor CRE1 from the phytopathogenic fungus Sclerotinia sclerotiorum. Eur. J. Biochem. 266, 252–259 (1999).
Lian, L. et al. GCN4 enhances the transcriptional regulation of AreA by interacting with SKO1 to mediate nitrogen utilization in Ganoderma lucidum. Appl. Environ. Microbiol. 88, e0132222, https://doi.org/10.1128/aem.01322-22 (2022).
Morris, G. M., Huey, R. & Olson, A. J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. Chapter 8, Unit 8 14, https://doi.org/10.1002/0471250953.bi0814s24 (2008).
Katchalski-Katzir, E. et al. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA 89, 2195–2199 (1992).
Vakser, I. A. Long-distance potentials: an approach to the multiple-minima problem in ligand-receptor interaction. Protein. Eng. 9, 37–41 (1996).
Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20200556), the Young Elite Scientists Sponsorship Program by CAST (2022QNRC001), the National Natural Science Foundation of China (grants 32000056), and the China Agriculture Research System of MOF and MARA (grant CARS20).
Author information
Authors and Affiliations
Contributions
R.L., Z.W. and M.W.Z. designed the study. R.L., Z.W., H.Q., and Y.F.L. carried out experiments and analyzed data. All authors gave input and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Gang Liu, Jia-Xun Feng and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Dario Ummarino.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Z., Qiu, H., Li, Y. et al. GlPRMT5 inhibits GlPP2C1 via symmetric dimethylation and regulates the biosynthesis of secondary metabolites in Ganoderma lucidum. Commun Biol 7, 241 (2024). https://doi.org/10.1038/s42003-024-05942-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-05942-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
