
Abstract
Aspergillus fumigatus is a pathogenic fungus with a global distribution. The emergence of azole-resistant A. fumigatus (ARAf) other than the TR-mutants is a problem in Japan. Additionally, the genetic diversity of A. fumigatus strains in Japan remains relatively unknown. Here we show the diversity in the A. fumigatus strains isolated in Japan as well as the complexity in the global distribution of the pathogenic strains. First, we analyzed the genome sequences of 171 strains from Japan as well as the antifungal susceptibility of these strains. Next, we conducted a population analysis of 876 strains by combining the available genomic data for strains isolated worldwide, which were grouped in six clusters. Finally, a genome-wide association study identified the genomic loci associated with ARAf strains, but not the TR-mutants. These results highlight the complexity of the genomic mechanism underlying the emergence of ARAf strains other than the TR-mutants.
Similar content being viewed by others

Introduction
The filamentous fungus Aspergillus fumigatus, which is distributed worldwide, is the most important pathogenic fungus among Aspergillus species associated with aspergillosis1,2. Azoles, such as voriconazole (VRCZ) and itraconazole (ITCZ), are the main antifungal compounds used to treat A. fumigatus infections3.
The number of azole-resistant A. fumigatus (ARAf) strains that have been identified has continued to increase over the past decade4,5, resulting in serious clinical implications6. It is widely accepted that the azole resistance of A. fumigatus was acquired through the use of medication (patient route) and the application of azole fungicides in the environment (environmental route)7,8,9,10,11. The mechanisms underlying the azole resistance of ARAf strains have been characterized on the basis of mutations in cyp51A (erg11), which encodes a 14-alpha sterol demethylase targeted by azole antifungal compounds. Specifically, several point mutations (i.e., G54, G138, P216, M220, and G448) may be associated with gene structural changes7,8,9. Moreover, a 34 bp tandem repeat (TR34) in the promoter region of cyp51A along with a nucleotide change that results in the substitution of leucine 98 to histidine (TR34/L98H) as well as TR46/Y121F/T289A lead to gene overexpression12,13. The TR-type mutants are prevalent in Europe and the US14. In a previous study, 6.7% of the strains from soil samples were identified as ARAf in the UK15. The genetic diversity of the TR-mutants is low. Additionally, they have been grouped in a single population because they propagate through asexual reproduction5,16. In contrast to the situation in Europe, only a few TR-type mutants have been isolated in clinical and environmental settings in Japan17. A growing concern in Japan is the spread of ARAf through floriculture products, including tulip bulbs imported from the Netherlands18,19,20.
Notably, 43% of ARAf strains lack mutations in cyp51A21. Similarly, the surveillance in Germany and the US reported 47.1% and 65% of resistant isolates harboring the wild type cyp51A without any mutation, respectively22,23. Hence, the resistance mechanisms that do not involve a mutated cyp51A are currently being characterized. For example, the high basal expression of cdr1B, which encodes an ABC transporter, and mutations in hmg1, which encodes a hydroxymethylglutaryl-CoA (HMG-CoA) reductase (rate-determining enzyme in ergosterol biosynthesis), contribute to azole resistance10,24,25.
To investigate the heterogeneity of A. fumigatus genomes and ARAf resistance mechanisms, population genomics and pan-genomic analyses of a subset of A. fumigatus isolates collected worldwide have been conducted. There are many reports describing A. fumigatus genomes, including 30026 and 26027 genomes from A. fumigatus strains collected worldwide, 76 genomes from A. fumigatus strains from Japan28, 179 genomes from A. fumigatus strains from the US14, and 218 genomes from A. fumigatus strains collected across the UK and Ireland29. Moreover, microbial genome-wide association studies (GWAS) have been performed to identify mutations, including non-cyp51A mutations associated with azole resistance28,29,30. Zhao et al. detected mutations related to ITCZ sensitivity and validated the function of the candidate gene28. Although 17 strains31,32 and 76 strains susceptible to azoles28 obtained across Japan have been analyzed, the available information regarding the genetic diversity of A. fumigatus strains from Japan remains limited.
In this study, to explore the emergence of ARAf strains in Japan, we analyzed 171 strains (160 clinical strains, 10 environmental strains, and 1 strain from an unknown source), including previously reported strains8,10,25,28,31,33. First, we assessed the antifungal susceptibility of these strains, which revealed 22 strains, including 11 newly analyzed strains, with minimum inhibitory concentration (MIC) values ≥ 2 µg/mL. Next, to clarify the genetic diversity of A. fumigatus strains from Japan, we conducted a population analysis and a phylogenetic analysis using the genome sequences of 876 strains from the UK and Ireland, the US, Germany, Canada, Spain34, and the Netherlands in addition to the 171 strains from Japan, including 92 newly sequenced strains. We identified six clusters in the A. fumigatus population, with almost all of the strains from Japan assigned to Clusters 1, 2, and 4. Furthermore, using 628 strains in these three clusters, we performed a GWAS and detected the genomic loci associated with the azole resistance of the ARAf strains other than the TR-mutants. Finally, a ridge regression analysis revealed the complexity of the genomic mechanism underlying the emergence of ARAf. This study has elucidated the development of ARAf strains other than the TR-mutants, while also clarifying the genomic diversity of A. fumigatus strains from Japan.
Results
Characterization of ARAf strains
A total of 173 strains were used, including 171 strains from Japan8,10,25,28,31,33 and the laboratory strains Af29335 and Afs3536. Most strains (83%; 134 clinical strains and 8 environmental strains) were isolated from the Kanto region (Chiba, Ibaraki, and Tokyo) in Japan (Fig. 1a). We determined the susceptibility of the strains to ITCZ on the basis of our analysis as well as the results of earlier studies (Supplementary Data 1). Twenty-two strains (13%) (21 clinical strains and 1 environmental strain) had MIC values ≥ 2 µg/mL. Accordingly, they were designated as ARAf (Table 1). Eleven of these 22 strains had not previously been identified as ARAf. To investigate the ITCZ resistance mechanisms, we confirmed the sequences of the cyp51A and hmg1 alleles. Twenty strains, including serially isolated strains from seven patients (patient I, IFM 57543-2 and IFM 59984-1; patient II, IFM 60237 and IFM 65468; patient III, IFM 62103 and IFM 62105-1; patient IV, IFM 63240, IFM 63537-2, IFM 63714-1, and IFM 64173; patient V, IFM 63768 and IFM 63772; patient VI, IFM 64258, IFM 63805, and IFM 64259-1; and patient VII, IFM 63559-1 and IFM 63560-1), had mutations in cyp51A (G54R, G54W, G138C, H147Y, P216L, M220K, and G448S) and/or hmg1 (S269F, S269Y, G307D, and F390Y). All identified variants were consistent with known alleles, indicating these mutations may be associated with azole resistance. Although IFM 62103 and IFM 62105-1 were isolated from the same patient (patient III), their mutation profiles differed. Two strains (IFM 62628 and IFM 63772) lacked mutated cyp51A and hmg1 genes. Notably, the MIC values of IFM 63537-2 and IFM 63537 were 2 and 8 µg/mL, respectively10. Moreover, IFM 63537-2 was re-isolated from IFM 63537 via single colony isolation.

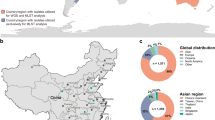
a Map showing the strains from Japan included in this study. A total of 165 strains, not including six strains isolated from an unknown region, were mapped. More specifically, 142 strains (16 resistant strains) were from the Kanto region (Chiba, Ibaraki, and Tokyo), 8 strains (2 resistant strains) were from the Chubu region (Aichi, Gifu, Ishikawa, and Nagano), 7 strains (3 resistant strains) were from the Kinki region (Kyoto and Osaka), 4 strains were from the Kyushu region (Kagoshima, Kumamoto, and Fukuoka), 3 strains (1 resistant strain) were from the Hokkaido region, and 1 strain was from the Tohoku region (Iwate). Resistant and susceptible strains are indicated in red and gray, respectively. b DAPC scatterplot of the 876 strains. The optimal number of principal components (PC = 5) was estimated using the optim.a.score function implemented in the adegenet package. c Population structure plot (K = 6). The fastStructure analysis determined K = 6. d Overrepresentation of geographic distributions of the strains for each cluster.
Among the strains from a single patient, IFM 62103 and IFM 62105-1 from patient III harbored different mutations in cyp51A and hmg1, even though the short tandem repeat patterns were the same25. We identified 377 mutations between IFM 62103 and IFM 62105-1 (Supplementary Data 2). In addition to the missense variants in cyp51A and hmg1, 99 other missense variants were detected.
Population structure of A. fumigatus strains in Japan
To clarify the population structure of the 171 strains from Japan, we analyzed 876 strains (31%; 183 resistant strains), including the 171 strains from Japan, 2 laboratory strains (Af293 and Afs35), 8 strains isolated from a single tulip bulb in Japan18,20, 212 strains from the UK and Ireland29, 12 strains from the Netherlands37,38,39, 256 strains from Germany26, 27 strains from Spain34, 10 strains from Canada30, and 178 strains (excluding AFIS1704) from the US14. We did not include AFIS1704 because its estimated genome size (64 Mb) differed considerably from the genome size (29 Mb) of Af293 (Supplementary Fig. 1).
Using 68,816 loci, we estimated the optimal number of populations on the basis of the discriminant analysis of principal components (DAPC). According to the Bayesian information criterion (BIC) with five principal components retained, K = 6 was the most likely number of populations (Fig. 1b, Supplementary Fig. 2 and Supplementary Data 3). In addition, fastStructure was used to estimate the number of populations. Because the marginal likelihood values increased until K = 6 (Supplementary Fig. 3), six clusters were supported by fastStructure (Fig. 1c and Supplementary Data 4). Cluster 4 was the largest with 241 strains (13%; 31 ARAf strains), followed by Cluster 1 with 214 strains (21%; 44 ARAf strains), Cluster 2 with 185 strains (15%; 27 ARAf strains), Cluster 5 with 175 strains (45%; 78 ARAf strains), Cluster 6 with 36 strains (89%; 32 ARAf strains), and Cluster 3 with 25 strains (8%; 2 ARAf strains) (Supplementary Data 5). Among the 171 strains from Japan, 58, 73, 33, and 7 strains were assigned to Clusters 1, 2, 4 and 5, respectively (i.e., no strains assigned to Clusters 3 and 6).
We assessed the geographic distributions of six clusters by Fisher’s exact test (Fig. 1d, Supplementary Fig. 4 and Supplementary Table 1). The six clusters were characterized by particular geographic regions. Clusters 1, 2, 3, and 4 were significantly overrepresented for the strains from the US (FDR corrected p = 5.85 × 10−6), Japan (FDR corrected p = 4.52 × 10−11), Spain (FDR corrected p = 2.18 × 10−12), Germany (FDR corrected p = 2.96 × 10−12), respectively. Clusters 5 and 6 were significantly overrepresented for the strains from the UK and Ireland (FDR corrected p = 7.16 × 10−15, p = 6.60 × 10−15).
The profiles of Tajima’s D values varied among Clusters 1, 2, 3, 4, 5 and 6 (Supplementary Fig. 5). The average Tajima’s D values for Clusters 1, 2, 3, 4, 5, and 6 were 0.65, 0.49, −0.56, 0.81, 0.99, and −1.31, respectively (Supplementary Table 2). The signature of positive selection was highest and lowest for Clusters 5 and 6, respectively (Supplementary Fig. 5), indicating the populations in Clusters 5 and 6 comprising TR-mutants may be under high evolutionary pressure, which is consistent with the use of azole fungicides in the field. Interestingly, the average Tajima’s D values and signature of positive selection differed among the chromosomes. More specifically, among the six clusters, Tajima’s D value for chromosome 6 was highest in Cluster 4, whereas Tajima’s D value for chromosome 8 was highest in Cluster 1, suggestive of the six cluster membership of the A. fumigatus population.
The comparison of the DAPC and fastStructure results revealed the cluster assignments were generally consistent. 26 strains were the exceptions (Supplementary Data 3 and 4). The differences between the strains in the cluster were evaluated by the numbers of pairwise single nucleotide polymorphisms (SNPs). Clusters 3 and 6 exhibited lower diversities (Supplementary Fig. 6 and Supplementary Table 3). Furthermore, we calculated D-statistics to test the admixture based on four clusters. Among 45 four-cluster comparisons, 9 and 20 comparisons exhibited significant D-statistics with Z-score > 3 and Z-score < −3, respectively, indicating that the admixture between most of clusters (Supplementary Fig. 7 and Supplementary Table 4).
To compare the recombination hot spots of clusters, we estimated the recombination rates for each cluster using LDhat analysis. The recombination rates for Clusters 1, 2, 3, 4, 5, and 6 were 0.2036/bp−1, 0.1215/bp−1, 0.0228/bp−1, 0.1320/bp−1, 0.1568/bp−1, 0.0078/bp−1, respectively. Cluster 1 exhibited the largest recombination rate. The greater numbers of recombination hot spots of Clusters 1, 2, 4 and 5 were detected than those of Clusters 3 and 6 (Supplementary Fig. 8). This is consistent with the numbers of pairwise SNPs (Supplementary Fig. 6), indicating that Clusters 3 and 6 could be highly clonal.
Phylogenetic analysis
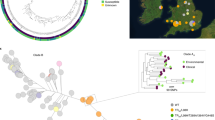
We conducted a phylogenetic analysis using the maximum likelihood method (Fig. 2a and Supplementary Data 5). By mapping six clusters on the phylogenetic tree, almost all strains were consistently assigned to their corresponding cluster. According to DAPC, 29 strains had a posterior probability of cluster membership <85%, indicating these strains may have been derived from the admixture between strains in two or more of the clusters.

a Phylogenetic tree of 876 strains. RAxML was used to construct the unrooted phylogenetic tree. The metadata rings on the outside of the tree indicate the cluster, ITCZ resistance, and county. b Principal component analysis of 876 strains. The x-axis and y-axis correspond to principal components (PCs) 1 and 2, respectively; PC1 and PC2 explained 35% and 16% of the variance, respectively.
On the basis of the principal component analysis (PCA), two populations (A and B) designated by Sewell et al5. were observed along with the first principal component (40% variation) (Fig. 2b). Indeed, Clusters 1, 2, and 4 were observed along with the second principal component (16% variation), while Clusters 5 and 6 were observed along with the first principal component (35% variation). The clusters defined by DAPC were the subclusters of populations A and B. Clusters 1, 2, and 4 were the subclusters of population B, whereas Clusters 5 and 6 were the subclusters of population A. Among the 131 TR-type mutants, 119 strains (93%) were assigned to Clusters 5 and 6. In contrast, the other TR-mutants were assigned to Clusters 1 (C87 and C91 from the UK and Ireland, and 698-L-3-11-2 from Germany), 2 (B11982, B11978, B11957, B11943, B11930, and B11927 from the US) and 4 (AB01_C43_NRZ-2018-313, AB01_C40_NRZ-2018-290, and AB01_C19_NRZ-2017-214 from Germany), but were positioned between populations A and B. The TR-mutants B11927, B11930, B11943, B11957, B11978, and B11982 had a 46% probability of belonging to Cluster 5 according to fastStructure.
Genome-wide association study of the ITCZ resistance of A. fumigatus
The ARAf strains other than the TR-mutants were mainly obtained in clinical settings in Japan. These strains were assigned to Clusters 1, 2, and 4 of population B. To explore the genomic loci of the ARAf strains other than the TR-mutants (i.e., high-risk population), we performed a GWAS involving 628 strains from Clusters 1, 2, and 4, of which 165 strains were from Japan. Among these 628 strains, 92 were ARAf strains, including 22 strains from Japan (Table 1), 22 strains from the UK and Ireland, and 14 strains from Germany (excluding the TR-mutants 698-L-3-11-2, AB01_C43_NRZ-2018-313, AB01_C40_NRZ-2018-290, and AB01_C19_NRZ-2017-214), 2 strains from Spain, 32 strains from the US (excluding the TR-mutants B11927, B11930, B11943, B11957, B11978, and B11982). Moreover, 46 strains had mutations in cyp51A, whereas the other 46 strains (50%) had no mutations in cyp51A. Only six strains had mutations in hmg1. The mixed linear model (MLM) analysis was conducted using TASSEL 5 (Fig. 3a and Supplementary Fig. 9). The azole resistance of the ARAf strains was treated as a binary trait. A total of 90,648 loci were filtered by allowing 10% missing values. The following 12 SNPs were significantly (p < 10−4) associated with ARAf: 1 missense variant, 1 synonymous variant, 1 intron variant and 9 intergenic variants (Table 2). Because 47 ARAf strains from the UK and Ireland, the US, Germany and Spain had no mutations in cyp51A, we screened for mutations in hmg1. We found seven strains with the mutations in hmg1, that is, E105K in C162, P309Q in C165 and CM7510, I419N in 106-C-1-72s-2, AB01_C6_NRZ-2016-108, and 313-H-1-15-2, and S541G in C4.

a Manhattan plot. The azole resistance of ARAf was treated as a binary trait. The x-axis presents 90,648 loci, whereas the y-axis presents the p-values determined by TASSEL 5. b Fitting by ridge regression. The MIC value was treated as a quantitative trait. The x-axis presents log2-transformed MIC values, whereas the y-axis presents the predicted log2-transformed MIC values.
The PCA of 628 strains revealed the overlap between the populations of ARAf and susceptible strains (Supplementary Fig. 10). This was consistent with the lack of strong signals detected by GWAS (Fig. 3a). Because GWAS assumes the phenotype can be explained by particular loci, we performed a genomic selection analysis via ridge regression to evaluate the effectiveness of each locus. The MIC values predicted by the ridge regression model were consistent with the observed MIC values (R2 = 0.76) (Fig. 3b), indicating that the MIC values could be explained by genomic loci. In accordance with the GWAS results, the coefficients of genomic loci determined by the ridge regression model contributed evenly to the MIC values (i.e., ITCZ resistance) (Supplementary Fig. 11). Considered together, these results indicate the azole resistance of the ARAf strains in Clusters 1, 2, and 4 may be associated with multiple loci, which is in contrast to the TR-mutants with a mutated cyp51A allele (L98H).
Discussion
In this study, we analyzed 171 A. fumigatus strains isolated in Japan in terms of their susceptibility to ITCZ and their genomic sequences. By incorporating publicly available sequence data, we conducted a population analysis for the largest dataset, which resulted in the identification of six clusters. Furthermore, we conducted a GWAS to explore the genomic loci related to the azole resistance of the ARAf strains.
Among the 171 strains from Japan, 22 (13%) were ARAf strains, including non-cyp51A ARAf strains. In addition, 11 were newly identified, whereas 11 were previously reported strains (Table 1). Moreover, 14 strains (64%) harbored mutations in cyp51A. In contrast, seven strains (32%) harbored mutations in hmg1. Both IFM 62628 and IFM 63772 lacked mutations in these two genes. Interestingly, although the ARAf strains IFM 62103 and IFM 62105-1 were isolated from patient III and were clustered together (i.e., relatively close phylogenetic relationship), they differed regarding the cyp51A and hmg1 mutations (Table 1 and Fig. 2a). Strain IFM 62103 harbored mutations in cyp51A (H147Y), whereas IFM 62105-1 harbored mutations in both cyp51A (M220K) and hmg1 (G307D). Among the tested strains, IFM 62105-1 is reportedly the most resistant to ITCZ, VRCZ, and posaconazole25. By comparing the IFM 62103 and IFM 62105-1 genomes, we detected 377 variants, including 102 missense variants (Supplementary Data 2), suggesting that these strains may have other phenotypic differences in addition to the diversity in their susceptibility to azoles. Considering the similarity in the duration of azole treatments25, the mutations in different strains likely vary. Thus, various strains, including different ARAf strains, may have co-infected the lungs of patient III (i.e., heterogeneous population). Because heterogeneity may be advantageous for survival, heterogeneous populations are likely to emerge in human lungs. In addition, we detected a missense variant (I433N) in clcA, which encodes a Zn2-Cys6 transcription factor influencing hyphal growth, conidiogenesis, and adaptation to copper stress40. Mutations in clcA have been identified in both laboratory-based evolutionary analyses as well as in clinical settings8,40, suggesting that environmental stresses may induce clcA mutations that lead to adaptive responses.
We used 876 genomes, including the genomes of 854 strains with ITCZ susceptibility (MIC values or binary traits), to investigate the population structure of the strains from Japan (Supplementary Data 5). By analyzing the genomic context, we determined that the A. fumigatus strains can be divided into six clusters (Fig. 1b, c). The DAPC and fastStructure analyses supported the classification of the A. fumigatus strains in six clusters. The two A. fumigatus populations proposed by Sewell et al.5 were population A, which consists of TR-mutants, and population B, which typically does not include TR-mutants. For 212 strains by Rhodes et al.29, 116 (97%) and 89 strains (96%) were populations A and B, respectively. For 178 strains Etienne et al.14, 160 (100%) and 12 strains (67%) were populations A and B, respectively. The classification of two populations were consistent. In the current study, 165 strains from Japan were assigned to Clusters 1, 2, and 4 (i.e., subclusters of population B). Seven strains were assigned to Cluster 5 (i.e., subcluster of population A), but these strains were not TR-mutants. The population B could be abundant in Japan. In earlier studies, the number of populations ranged from two to seven26,27,28,29,41. We determined that PC1 explained 35% of the variation (Fig. 2b), which is less than the value (62%) reported by Etienne et al.14. This implies the subclusters were likely correct because expanding a strain set, especially Clusters 1, 2, and 4, may improve the resolution of the population structure. The optimal K value (i.e., 4) calculated by Zhao et al.28 for the strains from Japan was consistent with the results of the current study. In accordance with Clade 3 by Lofgren et al.27, Cluster 3 comprising of the strains from Germany and Spain was far from other clusters (Fig. 1b, c). The recombination analysis and Tajima’s D values indicated the high clonality of strains in Cluster 3 (Supplementary Figs. 5 and 8). Since the strains from Spain harbored unique cyp51A-3SNPs42, the mechanisms of ARAf strains belonging to Cluster 3 could be different. In addition, among the 53 non-cyp51A ARAf strains, 13 had mutations in hmg1, reflecting the importance of analyzing the hmg1 allele as well as cyp51A. Especially, P309Q and I419N in hmg1 are located in PF12349 (i.e., “sterol-sensing domain of SREBP cleavage-activation”), similar to S269F, suggesting that these alleles may be associated with azole resistance. Because the azole resistance mechanisms of the ARAf lacking cyp51A mutations remain unexplained, additional studies are required. Notably, the laboratory strains Af293 and Afs35 were assigned to different clusters, namely Cluster 1 of population B and Cluster 5 of population A, respectively (Fig. 1b), suggesting that these two laboratory strains may be useful for future research (depending on the study objectives). We excluded AFIS1704 from the population study because of the substantial difference in its estimated genome size (approximately 64 Mb) (Supplementary Fig. 1). Indeed, we confirmed the presence of two mat1-2 and cyp51A genes on different contigs, suggesting that AFIS1704 may be an allodiploid hybrid strain (e.g., Aspergillus latus)43.
Strains from Clusters 1, 2, and 4 were included in the GWAS performed to explore the genomic loci related to azole resistance because they represent a high-risk population for the emergence of ARAf strains with mutations in cyp51A and/or hmg1 (but are not TR-mutant strains). We identified 12 significant SNPs (p < 10−4), but there were no strong signals. These candidate SNPs were not overlapped with previous studies. Possibly, differently from GWAS for TR-mutants29 and azole resistance for all populations30, the GWAS for particular populations (Clusters 1, 2 and 4) could propose the novel SNPs in ARAf. In addition, we detected an overlap between the cyp51A ARAf strains and the susceptible strains in Clusters 1, 2, and 4 (Supplementary Fig. 10). Finally, a ridge regression analysis was conducted. The regression model explained the MIC values (Fig. 3b), but no significant loci were detected, consistent with the GWAS results. These findings suggest the phenotype of ARAf strains may be explained by multiple loci. The emergence of ARAf strains during azole treatments may occur randomly, regardless of the genomic background. In the GWAS analysis, clinical strains were overrepresented among 628 strains (Fisher’s exact test; p-value = 2.2 × 10−8). Since the genomic and metabolic differences between clinical and environmental strains have been reported26,44, the GWAS results might be potentially missing the aspects of environmental strains.
The results of this study revealed the diversity in the A. fumigatus strains isolated in Japan as well as the complexity in the global distribution of the pathogenic strains by using the largest dataset. Moreover, our findings complement the results of a previous study on the population structure of the isolates from Japan by Zhao et al.28. Furthermore, we identified significant loci related to ARAf strains, but not to TR-mutants. These candidate loci and their sequence data are relevant for future investigations conducted to conclusively determine how ARAf strains emerge in patients treated with azole-based antifungal compounds.
Methods
Strains and culture conditions
The strains used in this study (Supplementary Data 1) were isolated from various patients and environments in Japan from 1987 to 2018. All of the strains (IFM strains) are stored and maintained at the Medical Mycology Research Center, Chiba University in Japan. To prepare fresh conidia, the strains were grown on potato dextrose agar (BD Difco, Franklin Lakes, NJ) for 5–7 days at 37 °C.
Antifungal susceptibility analysis
Antifungal susceptibility analyses were conducted using ITCZ in RPMI 1640 medium (pH 7.0) at 35 °C according to the Clinical and Laboratory Standards Institute reference broth microdilution method (document M38; 3rd edition)45,46 with minor modifications. Specifically, dried plates were used for evaluating antifungal susceptibility (Eiken Chemicals, Tokyo, Japan). The strains with a MIC value ≥ 2 µg/mL were defined as ARAf.
Sequencing cyp51A and hmg1 genes
The mutations in the cyp51A and hmg1 genes were analyzed on the basis of a PCR amplification and sequencing using appropriately designed primers25. Sequence variants were detected via a comparison with reference sequences in GenBank (i.e., AF338659 for cyp51A and AFUB_020770 for hmg1).
DNA extraction and whole-genome sequencing
Genomic DNA was extracted from mycelia derived from an overnight culture according to a published phenol-chloroform method40. Genomic DNA libraries of the A. fumigatus strains were constructed using the NEBNext Ultra DNA Library Prep Kit (New England BioLabs, Ipswich, MA). The 150-bp paired-end sequencing was performed using an Illumina HiSeq 4000 system (Illumina, San Diego, CA) by GENEWIZ (Saitama, Japan) or BGI (Shenzhen, China). An Illumina MiSeq system was used to generate 300-bp paired-end sequences of IFM 63345, IFM 63666, and IFM 63768.
Single nucleotide polymorphism analysis
The raw genomic reads of all samples were screened for quality and trimmed using fastp v.0.20.147. The filtered reads were aligned with the Af293 reference genome retrieved from AspGD (genome version: s03-m05-r04)48 using BWA-MEM v.0.7.17-r118849. The mitochondrial genome was excluded for the analysis. SNPs were analyzed using GATK v.4.1.2.050. According to the best practice workflow for ‘Germline short variant discovery’ of GATK20,26,28,42, the sorted BAM file for each sample was recalibrated using ‘BaseRecalibrator’ and known SNVs from FungiDB (release 56)51 as well as ‘ApplyBQSR’. Next, ‘HaplotypeCaller’ with ‘--sample-ploidy 1’ and the recalibrated BAM file for each sample were used to call short variants (SNPs and INDELs), after which ‘GenotypeGVCFs’ was used to combine the vcf files. Only SNPs were extracted from the joint-called variant file using ‘SelectVariants’. To eliminate false positives, ‘VariantFiltration’ was used with the following parameters as described in the GATK document: ‘QUAL < 30.0 || QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0 || SOR > 3.0’.
The pairwise comparison of the SNPs in IFM 62103 and IFM 62105-1 was performed using mpileup in SAMtools v.1.1052. The pileup vcf files were generated, and the consensus SNPs were excluded if they did not meet a minimum coverage of 10× or if the variant was present in <90% of the base calls by in-house scripts8,53,54.
Phylogenetic analysis of whole-genome sequencing SNP data
The SNP sites with a minor allele frequency ≥5% and no missing data were filtered using VCFtools v.0.1.16 with the options ‘--maf 0.05 --max-missing 1’55. A phylogenetic tree was constructed using the multithreaded version of RAxML v.8.2.1256, the GTRCAT model, and 1,000 bootstrap replicates. The phylogenetic tree was visualized using the ggtree package57. Tajima’s D values were calculated using VCFtools with the option ‘--TajimaD 10000’. The numbers of pairwise SNPs between the strains of each cluster were calculated using snp-dists (https://github.com/tseemann/snp-dists).
Population structure analysis
DAPC implemented in the adegenet package v.2.1.1058 was performed to assign the strains according to 68,816 loci. The vcfR package v.1.14.059 was used for reading and parsing the vcf file. The function optim.a.score was iteratively used to determine the number of principal components used. Additionally, fastStructure v.1.060 was used to estimate the population structure. The marginal likelihood values for each number of populations (K = 1–15) were calculated using 30 independent seeds. PCA was conducted using plink v.1.9061.
Overrepresented and underrepresented countries of each cluster were identified using Fisher’s exact test. The one-tailed Fisher’s exact p-value corresponding to overrepresentation and underrepresentation of a particular country have been calculated based on counts in 2 × 2 contingency tables. The p-values were corrected by the FDR method62.
D-statistic is a statistical test for admixture based on a four-cluster comparison63. The D-statistics were calculated using the f4 function implemented in the admixtools package v.2.0.064.
Recombination analysis was performed using LDhat v.2.2a65. The interval program was used to estimate the recombination rates for each cluster, following generation of the lookup table by the lkgen program using “lk_n320_t0.01” for 320 sequences with theta = 0.01 (https://zenodo.org/records/3934350). The program was executed for 2 million iterations with sampling every 200 iterations after a 20,000-iteration burn-in period. The stat program was used for summarizing the results.
Genome-wide association study and genomic selection on the basis of ridge regression
The 90,648 SNP sites that satisfied certain criteria (i.e., minor allele frequency ≥5% and ≤10% missing data) were used for the GWAS. MLM analysis was completed using TASSEL v.566. Multidimensional scaling (MDS) and Kinship matrices were used as covariates to control the population structure. The ridge regression analysis was performed using the glmnet package v.4.1-867,68,69. The log2-transformed MIC values were predicted according to genomic loci through a 5-fold cross validation using the function cv.glmnet. The SNPs were annotated using SnpEff v.5.1d70 and the annotated A. fumigatus Af293 reference genome.
Determination of mating type idiomorphs
The mitochondrial genomes were assembled using GetOrganelle v.1.6.471. To filter the mitochondrial reads, the reads were aligned with the mitochondrial genome using BWA. The mapped reads were filtered using SAMtools and SeqKit v.0.10.172. The nuclear genomes were assembled using VelvetOptimiser v.2.2.673. blastn v.2.5.0+74 was used for identifying MAT types, with MAT1-1 (AY89866.1) and MAT1-2 (Afu3g06170) serving as query sequences. The AFIS1704 genome size was estimated using GenomeScope75 with 21 k-mers.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw reads have been deposited in the DDBJ BioProject database (BioProject accession number PRJDB16281). Source data underlying Fig. 1c can be found in Supplementary Data 4. The newick file underlying Fig. 2a can be found in Supplementary Data 6. Other source data underlying Figs. can be found in Supplementary Data 7.
Code availability
Codes for analysis and visualization are available from the authors upon request.
References
Bodey, G. P. & Vartivarian, S. Aspergillosis. Eur. J. Clin. Microbiol. Infect. Dis. 8, 413–437 (1989).
Latgé, J. P. & Chamilos, G. Aspergillus fumigatus and aspergillosis in 2019. Clin. Microbiol. Rev. 33, e00140–18 (2019).
Jenks, J. D. & Hoenigl, M. Treatment of aspergillosis. J. Fungi (Basel) 4, 98 (2018).
Fisher, M. C., Hawkins, N. J., Sanglard, D. & Gurr, S. J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 360, 739–742 (2018).
Sewell, T. R. et al. Nonrandom distribution of azole resistance across the global population of Aspergillus fumigatus. mBio 10, e00392-19 (2019).
Lestrade, P. P. et al. Voriconazole resistance and mortality in invasive aspergillosis: a multicenter retrospective cohort study. Clin. Infect. Dis. 68, 1463–1471 (2019).
Camps, S. M. T. et al. Rapid induction of multiple resistance mechanisms in Aspergillus fumigatus during azole therapy: a case study and review of the literature. Antimicrob. Agents Chemother. 56, 10–16 (2012).
Hagiwara, D. et al. Whole-genome comparison of Aspergillus fumigatus strains serially isolated from patients with aspergillosis. J. Clin. Microbiol. 52, 4202–4209 (2014).
Toyotome, T., Hagiwara, D., Takahashi, H., Watanabe, A. & Kamei, K. Emerging antifungal drug resistance in Aspergillus fumigatus and among other species of Aspergillus. Curr. Fungal Infect. Rep. 12, 105–111 (2018).
Hagiwara, D. et al. Non-cyp51A azole-resistant Aspergillus fumigatus isolates with mutation in HMG-CoA reductase. Emerg. Infect. Dis. 24, 1889–1897 (2018).
Schoustra, S. E. et al. Environmental hotspots for azole resistance selection of Aspergillus fumigatus, the Netherlands. Emerg. Infect. Dis. 25, 1347–1353 (2019).
Verweij, P. E., Mellado, E. & Melchers, W. J. G. Multiple-triazole-resistant aspergillosis. N. Engl. J. Med. 356, 1481–1483 (2007).
Mellado, E. et al. A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51A alterations. Antimicrob. Agents Chemother. 51, 1897–1904 (2007).
Etienne, K. A. et al. Genomic Diversity of azole-resistant Aspergillus fumigatus in the United States. mBio 12, e0180321 (2021).
Sewell, T. R. et al. Elevated prevalence of azole-resistant Aspergillus fumigatus in urban versus rural environments in the United Kingdom. Antimicrob. Agents Chemother. 63, e00548-19 (2019).
Klaassen, C. H. W., Gibbons, J. G., Fedorova, N. D., Meis, J. F. & Rokas, A. Evidence for genetic differentiation and variable recombination rates among Dutch populations of the opportunistic human pathogen Aspergillus fumigatus. Mol. Ecol. 21, 57–70 (2012).
Toyotome, T. et al. First clinical isolation report of azole-resistant Aspergillus fumigatus with TR34/L98H-type mutation in Japan. J. Infect. Chemother. 23, 579–581 (2017).
Hagiwara, D. Isolation of azole-resistant Aspergillus fumigatus from imported plant bulbs in Japan and the effect of fungicide treatment. J. Pestic. Sci. 45, 147–150 (2020).
Nakano, Y. et al. Characteristics of azole-resistant Aspergillus fumigatus attached to agricultural products imported to Japan. J. Infect. Chemother. 26, 1021–1025 (2020).
Takahashi, H., Oiki, S., Kusuya, Y., Urayama, S. I. & Hagiwara, D. Intimate genetic relationships and fungicide resistance in multiple strains of Aspergillus fumigatus isolated from a plant bulb. Environ. Microbiol. 23, 5621–5638 (2021).
Bueid, A. et al. Azole antifungal resistance in Aspergillus fumigatus: 2008 and 2009. J. Antimicrob. Chemother. 65, 2116–2118 (2010).
Bader, O. et al. cyp51A-based mechanisms of Aspergillus fumigatus azole drug resistance present in clinical samples from Germany. Antimicrob. Agents Chemother. 57, 3513–3517 (2013).
Pham, C. D., Reiss, E., Hagen, F., Meis, J. F. & Lockhart, S. R. Passive surveillance for azole-resistant Aspergillus fumigatus, United States, 2011-2013. Emerg. Infect. Dis. 20, 1498–1503 (2014).
Fraczek, M. G. et al. The cdr1B efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in Aspergillus fumigatus. J. Antimicrob. Chemother. 68, 1486–1496 (2013).
Arai, T. et al. Hmg1 mutations in Aspergillus fumigatus and their contribution to triazole susceptibility. Med. Mycol. 59, 980–984 (2021).
Barber, A. E. et al. Aspergillus fumigatus pan-genome analysis identifies genetic variants associated with human infection. Nat. Microbiol. 6, 1526–1536 (2021).
Lofgren, L. A., Ross, B. S., Cramer, R. A. & Stajich, J. E. The pan-genome of Aspergillus fumigatus provides a high-resolution view of its population structure revealing high levels of lineage-specific diversity driven by recombination. PLoS Biol. 20, e3001890 (2022).
Zhao, S., Ge, W., Watanabe, A., Fortwendel, J. R. & Gibbons, J. G. Genome-wide association for itraconazole sensitivity in non-resistant clinical isolates of Aspergillus fumigatus. Front. Fungal Biol. 1, 617338 (2021).
Rhodes, J. et al. Population genomics confirms acquisition of drug-resistant Aspergillus fumigatus infection by humans from the environment. Nat. Microbiol. 7, 663–674 (2022).
Fan, Y., Wang, Y., Korfanty, G. A., Archer, M. & Xu, J. Genome-wide association analysis for triazole resistance in Aspergillus fumigatus. Pathogens 10, 701 (2021).
Takahashi-Nakaguchi, A. et al. Genome sequence comparison of Aspergillus fumigatus strains isolated from patients with pulmonary aspergilloma and chronic necrotizing pulmonary aspergillosis. Med. Mycol. 53, 353–360 (2015).
Hagiwara, D., Takahashi, H., Takagi, H., Watanabe, A. & Kamei, K. Heterogeneity in pathogenicity-related properties and stress tolerance in Aspergillus fumigatus clinical isolates. Med. Mycol. J. 59, E63–E70 (2018).
Toyotome, T. et al. Azole susceptibility in clinical and environmental isolates of Aspergillus fumigatus from eastern Hokkaido, Japan. J. Infect. Chemother. 22, 648–650 (2016).
Garcia-Rubio, R., Monzon, S., Alcazar-Fuoli, L., Cuesta, I. & Mellado, E. Genome-wide comparative analysis of Aspergillus fumigatus strains: the reference genome as a matter of concern. Genes (Basel) 9, 363 (2018).
Pain, A. et al. Insight into the genome of Aspergillus fumigatus: analysis of a 922 kb region encompassing the nitrate assimilation gene cluster. Fungal Genet. Biol. 41, 443–453 (2004).
Wagener, J. et al. The putative alpha-1,2-mannosyltransferase AfMnt1 of the opportunistic fungal pathogen Aspergillus fumigatus is required for cell wall stability and full virulence. Eukaryot Cell 7, 1661–1673 (2008).
Abdolrasouli, A. et al. Genomic context of azole resistance mutations in Aspergillus fumigatus determined using whole-genome sequencing. mBio 6, e00536 (2015).
Puértolas-Balint, F. et al. Revealing the virulence potential of clinical and environmental Aspergillus fumigatus isolates using whole-genome sequencing. Front. Microbiol. 10, 1970 (2019).
Ballard, E. et al. Raw genome sequence data for 13 isogenic Aspergillus fumigatus strains isolated over a 2 year period from a patient with chronic granulomatous disease. Data Brief 25, 104021 (2019).
Kusuya, Y., Bian, C., Hagiwara, D., Ban, S. & Takahashi, H. A novel Zn2-Cys6 transcription factor clcA contributes to copper homeostasis in Aspergillus fumigatus. Curr. Genet. 68, 605–617 (2022).
Zhao, S. & Gibbons, J. G. A population genomic characterization of copy number variation in the opportunistic fungal pathogen Aspergillus fumigatus. PLoS One 13, e0201611 (2018).
Majima, H. et al. Genetic differences between Japan and other countries in cyp51A polymorphisms of Aspergillus fumigatus. Mycoses 64, 1354–1365 (2021).
Steenwyk, J. L. et al. Pathogenic allodiploid hybrids of Aspergillus fungi. Curr. Biol. 30, 2495–2507.e7 (2020).
Mirhakkak, M. H. et al. Genome-scale metabolic modeling of Aspergillus fumigatus strains reveals growth dependencies on the lung microbiome. Nat. Commun. 14, 4369 (2023).
Kikuchi, K. et al. Antifungal susceptibility of Aspergillus fumigatus clinical isolates collected from various areas in Japan. J. Infect. Chemother. 20, 336–338 (2014).
CLSI. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi. In: CLSI standard M38, 3rd edn. Clinical and Laboratory Standards Institute, Wayne, PA. (2017).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Cerqueira, G. C. et al. The Aspergillus Genome Database: multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res. 42, D705–D710 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Amos, B. et al. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 50, D898–D911 (2022).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Holt, K. E. et al. Detecting SNPs and estimating allele frequencies in clonal bacterial populations by sequencing pooled DNA. Bioinformatics 25, 2074–2075 (2009).
Gillece, J. D. et al. Whole genome sequence analysis of Cryptococcus gattii from the Pacific Northwest reveals unexpected diversity. PLoS One 6, e28550 (2011).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 69, e96 (2020).
Jombart, T. & Ahmed, I. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011).
Knaus, B. J. & Grünwald, N. J. vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53 (2017).
Raj, A., Stephens, M. & Pritchard, J. K. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573–589 (2014).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc.: Series B (Methodol.) 57, 289–300 (1995).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Maier, R. et al. On the limits of fitting complex models of population history to f-statistics. Elife 12, e85492 (2023).
McVean, G. A. T. et al. The fine-scale structure of recombination rate variation in the human genome. Science 304, 581–584 (2004).
Bradbury, P. J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Simon, N., Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J. Stat. Softw. 39, 1–13 (2011).
Tay, J. K., Narasimhan, B. & Hastie, T. Elastic net regularization paths for all generalized linear models. J. Stat. Softw. 106, 1 (2023).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Jin, J. J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241 (2020).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11, e0163962 (2016).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Numbers JP21K07001, JP21K18217, and JP22H04925 (PAGS) to HT as well as AMED Grant Numbers JP19fm0208024, JP23wm0325035 and JP23gm1610004 to HT. Some of the computational analysis was performed using the NIG supercomputer at the ROIS National Institute of Genetics. We thank Machiko Zen for assisting with the data analyses. We also thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
X.H. and Y.K. conceived the study, participated in designing and coordinating the study, and wrote the manuscript. D.H., T.T., and A.W. coordinated the study and wrote the manuscript. T.A., C.B., M.N., and S.S. coordinated the study. H.T initiated and supervised the project and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
: Communications Biology thanks Jian-Ping Xu, Johanna Rhodes and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Matteo Dell’Acqua and Tobias Goris.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, X., Kusuya, Y., Hagiwara, D. et al. Genomic diversity of the pathogenic fungus Aspergillus fumigatus in Japan reveals the complex genomic basis of azole resistance. Commun Biol 7, 274 (2024). https://doi.org/10.1038/s42003-024-05902-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-05902-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
