
Abstract
The omnipresence of ants is commonly attributed to their eusocial organization and division of labor, however, bacteria in their nests may facilitate their success. Like many other arboreal ants living in plant-provided cavities, Azteca ants form dark-colored “patches” in their nesting space inside Cecropia host plants. These patches are inhabited by bacteria, fungi and nematodes and appear to be essential for ant colony development. Yet, detailed knowledge of the microbial community composition and its consistency throughout the life cycle of ant colonies was lacking. Amplicon sequencing of the microbial 16S rRNA genes in patches from established ant colonies reveals a highly diverse, ant species-specific bacterial community and little variation within an individual ant colony, with Burkholderiales, Rhizobiales and Chitinophagales being most abundant. In contrast, bacterial communities of early ant colony stages show low alpha diversity and no ant species-specific community composition. We suggest a substrate-caused bottleneck after vertical transmission of the bacterial patch community from mother to daughter colonies. The subsequent ecological succession is driven by environmental parameters and influenced by ant behavior. Our study provides key information for future investigations determining the functions of these bacteria, which is essential to understand the ubiquity of such patches among arboreal ants.
Similar content being viewed by others
Introduction
Ants are dominant taxa in terrestrial ecosystems and key ecosystem engineers in tropical rainforests1,2. They influence the chemical and physical soil properties by nesting underground and excavating tunnels3,4,5, and are important in food chains at higher and lower trophic levels6,7. Ants are also involved in plant seed dispersal8 and, in rare cases, also in plant pollination9,10,11.
Commonly known main reasons for the omnipresence of ants are their eusocial organization, with overlapping generations and division of labor, and their well-developed ability to communicate chemically12. Yet, as in many animals, associated bacteria may help ants to overcome unfavorable conditions such as pathogenic pressure13,14 or nitrogen (N)-limitations15,16,17,18,19,20,21. These bacteria are often closely associated with their ant host and can be found on the body surface or in the gut of ants. Ant-associated Actinobacteriota have been shown to produce antibiotics and are thought to protect the larvae, or, in case of leaf-cutter ants, fungus cultivars against pathogens13,14. Furthermore, proteobacterial gut symbionts appear to be particularly associated with herbivorous ants15,16,21. They have been shown to recycle N and synthesize essential amino acids20,21,22, or have been hypothesized to fix atmospheric dinitrogen (N2)15,16,17,18. Thus, bacterial mutualists are thought to enable ants to colonize N-limited tropical canopies7.
Many of the canopy-dwelling ants live in a mutualistic association with plants. The ants defend the host plant against insect herbivores and fungal pathogens23 and often also provide nutrients to the host plant in form of their debris24,25,26,27. In exchange, ants receive plant-provided food supply and nesting space such as hollow stems, branches, or leaf pouches (domatia). The few existing studies on bacterial communities in ants inhabiting plants suggest that not only ant body-associated bacteria, but also free-living bacterial communities may play an essential role for the ant colony. Actinobacteriota (Streptomycetales) isolated from domatia of the Pseudomyrmex penetrator/Tachigali sp. and Petalomyrmex phylax/Leonardoxa africana ant-plant mutualisms, and Proteobacteria (Pseudomonadales) from domatia of the Azteca sp./Cecropia sp. ant-plant mutualism showed antifungal properties14,28. Some strains of Streptomyces identified in the domatia of the P. phylax/L. africana and Crematogaster margaritae/Keetia hispida mutualisms are thought to have the ability to degrade cellulose or to fix N214. Strains of various proteobacterial orders (Rhizobiales, Sphingomonadales, Burkholderiales, Enterobacterales, Pseudomonadales and Xanthomonadales) isolated from domatia of Azteca sp. inhabiting Cecropia trees and Allomerus sp. inhabiting Hirtella physophora were suggested to be involved in substrate preparations for the fungus-cultivation activity of these ants29, in defending against pathogens28,29 or in N2 fixation28,29,30.
Apparently, the bacterial microbiome inside domatia of host trees is considerably influenced by the resident ants. The domatia walls of Azteca and Cephalotes ants inhabiting the same Cordia tree species as a host, contained different bacterial communities, depending on the ant species involved31. The walls of domatia inhabited by Cephalotes consisted mainly of Actinobacteriota and Proteobacteria, in case of Azteca nests also Bacteroidota were abundant31. Moreover, Azteca domatia walls inside the Cecropia host trees harbored unique bacterial communities which differed considerably from those outside the domatia such as the nest entrances or the stem surface of the host plant32. Furthermore, the microbiome varied depending on the purpose of each nest chamber32. These studies support the notion that ants inhabiting host plants are shaping the microbiome of their domatia30,31,32.
Yet, there are still many gaps in our knowledge of the bacterial community inside plants inhabited by mutualistic ants. The studies cited above do not provide details on the consistency of the microbiome of the colonies over time. It is not known whether the microbiome in the domatia of colony founding queens raising her first worker offspring differs from that of the “established ant colonies” that had successfully survived the critical colony-founding phase and had grown to large colonies with numerous workers and often reproduction of new queens and males. It is not known whether microbiome changes occur once the number of workers increases, and more ants forage outside the domatia leading to an increased input of bacteria from the environment. It is not known whether the original bacterial community is retained due to the high concentration of bactericidal and fungicidal chemicals the ants use to keep their nest clean from pathogens12,33,34,35. It is also not yet known whether the microbial community within domatia is specific at the ant genus or even at the ant species level.
To investigate the dynamics of the bacterial community during the life cycle of arboreal ant colonies, we chose one of the most intensively studied ant-plant mutualisms as our model system, the Neotropical Azteca-Cecropia association. These Azteca ants which reside in the hollow stems (domatia) of Cecropia trees, belong to arboreal ant species that consistently form numerous well-defined, dark-colored “patches” in their nesting space, harboring specific Chaetothyriales fungi (Ascomycota)36,37,38,39, bacterivorous nematodes40,41 and also bacteria21,22. Azteca foundress queens have been shown to initially form the first patches by scratching off plant parenchyma and inoculating it with patch material from the mother colony, resulting in a vertical transmission from the mother colonies42. Subsequently, workers form and maintain new patches throughout the plant. It has been shown that larvae from Azteca and various other arboreal ant species take up fungi and other patch material38,42, indicating a significance for the ant colony. We performed amplicon sequencing of the 16S rRNA genes in patches of three ant colony developmental stages of up to three different Azteca ant species to address the following questions: (I) are the bacterial/archaeal community compositions in patches of each ant colony developmental stage Azteca species-specific?; (II) is the composition of the bacterial/archaeal community consistent within the patches of the same established Azteca colony?; and (III) do the bacterial/archaeal community compositions in patches from early to established ant colony developmental stages increase in diversity, as workers may introduce microorganisms from the plant surface into the nesting space of established colonies, in addition to the otherwise expected vertically transmitted bacterial/archaeal community? Such detailed studies, especially of bacterial/archaeal community dynamics inside the nesting space of arboreal ants, are an important step to further understand their role in ant colony developments.
Results
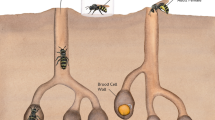
Three stages of ant colony development were defined as follows: initial ant colony with the egg laying queen, young colony in a still sealed internode with few workers only, and established colonies inhabiting many adjacent internodes and patrolling the plant surface (Fig. 1). Similarly, the patches were called “initial patch” (IP), “young colony patch” (YP) and “established colony patch” (EP). In general, bacteria dominated the prokaryotic patch communities of all ant colony developmental stages, accounting for 99.9% of total reads. Without removing archaeal taxa from our datasets, we further refer to the bacterial/archaeal patch microbiome as bacterial patch community, as all found patterns and correlations relate to the detected bacterial community.

a The initial ant colony stage consists of the queen, which entered a single plant internode and resealed the entrance hole, brood and an initial pile of scratched-off and chewed plant parenchyma (“initial ant colony patch”, IP). b In the young ant colony stage, first hatched workers are present in the sealed internode in addition to the queen, brood and patch (“young ant colony patch”, YP). c In established colonies, the ants inhabit many adjoined plant internodes, patrol the tree surface and typically form numerous patches (“established ant colony patch”, EP) throughout their nesting space.
Heterogeneous bacterial community composition in patches of early ant colonization stages independent of ant species
To study if the vertically transmitted bacterial community is ant species-specific and alters as ant colony grows, we investigated first the bacterial patch community composition of two early ant colony developmental stages of Azteca alfari, A. constructor and A. xanthochroa. These closely related ant species were found to colonize Cecropia plants at our study site in Costa Rica. We analyzed patches of 42 initial ant colonies (IPs) surrounded by the founding queen, eggs, brood (Fig. 1a), and from patches of 18 young ant colonies (YPs) surrounded by already hatched workers additionally to the queen, eggs and brood (Fig. 1b). In both cases, each colony was restricted to a single, sealed plant internode.
In the bacterial communities of IPs, Gammaproteobacteria (on average 54.9%), Alphaproteobacteria (23.9%) and Actinobacteria (14.0%) showed highest mean relative read abundances (Fig. 2a). A strong heterogeneity in bacterial community composition across IP samples could be seen on taxonomic order level with Enterobacterales (on average 25.9%, ranging from 1.5–90.1%), Rhizobiales (18.5%, 0.6–60.3%) and Xanthomonadales (13.1%, 0.2–59.2%) showing highest mean relative abundance of reads, followed by Corynebacteriales (11.0%, 0.2–94.5%) and Burkholderiales (10.5%, 0.5–58.2%). The variation in community composition did not correlate with the three ant species (P = 0.178) and thus, communities did not cluster according to ant species in the beta diversity ordination plot (Fig. 2b). Few ASVs accounted for a considerable amount of amplicon reads. Five of around 3400 ASVs were defined as prevalent – ASVs that account for more than 0.4% of reads in more than 50% of respective samples. These prevalent ASVs belonging to Proteobacteria were present in every single IP and accounted for 31.5% of the total IP reads. These ASVs were assigned to Rhizobiales, and Enterobacterales (Table 1).

a Taxonomic bar plot depicting the relative read abundance of orders on the y-axis. On the x-axis, every bar represents the bacterial community composition of one IP. b Principal coordinates analysis plot, displaying bacterial community similarities amongst IPs. The patch samples are grouped according to the Azteca ant species (alf: A. alfari, n = 27; con: A. constructor, n = 5; xan: A. xanthochroa, n = 10).
In YPs, Alphaproteobacteria (on average 40.9%) and Gammaproteobacteria (35.8%) accounted for highest relative read abundances, followed by Bacteroidia (9.8%) and Actinobacteria (8.9%) (Fig. 3a). Compared to IPs, the bacterial communities across YP samples were less heterogeneous. The most prominent orders were Rhizobiales (on average 27.9%, ranging from 1.5–56.2%), Xanthomonadales (12.0%, 0.6–34.4%), Burkholderiales (11.6%, 2.5–28.7%) and Sphingomonadales (10.7%, 1.5–21.7%). The variation in beta diversity of YPs did not correlate with the two ant species (P = 0.740) (Fig. 3b). Nine prevalent ASVs accounted for 36.3% of total YP reads and were assigned to Rhizobiales, Sphingomonadales, Burkholderiales, Enterobacterales and Xanthomonadales (Table 1).

a Taxonomic bar plot depicting the relative read abundance of orders on the y-axis. On the x-axis, every bar represents the bacterial community composition of one YP. b Principal coordinates analysis plot, displaying bacterial community similarities amongst YPs. The patch samples are grouped according to the Azteca ant species (alf: A. alfari, n = 15; con: A. constructor, n = 3).
Little variation in bacterial patch community composition within established ant colonies
Prior to investigating the ant species-specificity of bacterial patch communities across established ant colonies, the variation of bacterial communities within an established ant colony had to be assessed. Within established A. alfari and A. constructor colonies, ant-built patches (EPs) are typically found throughout the nesting space (Fig. 1c). We observed different ages of such patches during field work, which was linked to their spatial location in the Cecropia tree. Patches from the youngest growth zone of the tree (the apical stem internodes) (EP I) were only recently formed. Patches from the middle (EP II) and lower (EP III) part of the ant colonies were gradually older. To investigate the variation of the bacterial community compositions along this age gradient, we repeatedly sampled EPs throughout the nesting space of 17 Azteca ant colonies and sequenced each EP separately. The alpha diversity in EPs was similar and did not significantly differ from younger to older patches (Fig. 4a, P = 0.125; median Shannon index: EP I 4.9, EP II 5.3, EP III 5.2). Also, the variation in community composition did not correlate with the age of the patches (P = 0.995; P = 0.923, if permutations were set within the ant species) and therefore samples of the presumably same age group did not cluster together in beta diversity ordination plots (Fig. 4b). EPs of the same ant colony, but of different age, had a similar community composition (Fig. 4c), which was reflected by the significant correlation between the beta diversity and the ant colony ID (P < 0.001), explaining 73.0% of the variation.

Recently formed patches from apical stem internodes (EP I, n = 14), patches from the middle (EP II, n = 13) and lower (EP III, n = 12) actively colonized stem parts were distinguished, representing an age gradient of patches. a Shannon indices, depicting the alpha diversity in EPs within established ant colonies. Sampling sizes are given in brackets. Letters denote no significant differences between patch types. b Principal coordinates analysis plot, displaying bacterial community dissimilarities between EPs of presumed age. c Distance-based redundancy analysis plot, showing variation of bacterial communities in EPs due to the ant colonies they originated from. Please note that in this analysis, data are not differentiated between ant species.
Ant species-specific bacterial patch communities in established ant colonies
By studying our extended dataset of 34 established ant colonies, we uncovered a high bacterial diversity. We combined the samples of the 17 previously mentioned established ant colonies that were repeatedly sampled with 17 ant colonies of which only one patch sample was taken. Generally, ASVs were assigned to more than 25 phyla and more than 80 classes. Gammaproteobacteria (on average 28.6%), Bacteroidia (23.7%) and Alphaproteobacteria (23.5%) showed the highest relative read abundances in patches of established ant colonies, followed by Verrucomicrobiae (5.2%) and Actinobacteria (5.2%). The most read-abundant orders were Burkholderiales (on average 17.6%), Chitinophagales (12.5%) and Rhizobiales (10.9%) (Fig. 5a). Significant differences in the bacterial community between the ant species could be detected. EPs of A. constructor were significantly more diverse than those of A. alfari (P = 0.003; median Shannon indices: 4.8 A. alfari, 5.4 A. constructor; Fig. 5b). Also, the beta diversity correlated significantly with the ant species, explaining 9.0% of the variation (P < 0.001, Fig. 5c). While the mean relative abundance of several taxonomic orders differed visually between both ant species (Fig. 5a, e.g. Chitinophagales, Rhizobiales, Burkholderiales), some changes were statistically significant. Mainly Rhizobiales (A. alfari: 16.1%, A. constructor: 8.0%), Verrucomicrobiales (7.7% vs. 2.2%), Xanthomonadales (6.6% vs. 4.0%) and Corynebacteriales (1.9% vs. 1.1%) displayed a significantly higher relative read abundance in EPs of A. alfari than of A. constructor (Supplementary Table 1). Fourteen of around 8400 ASVs were prevalent in either A. alfari or A. constructor EPs and made up 18.6% of total EP reads. Despite significant differences in the community composition between ant species, three ASVs were prevalent in both ant species. These ASVs were assigned to Burkholderiales and Sphingomonadales (Table 2). ASVs that reached the prevalence threshold in patches of only one of the two ant species belonged to a variety of different orders: Corynebacteriales, Rhizobiales, Burkholderiales, Xanthomonadales, Verrucomicrobiales, Micrococcales, Chitinophagales and Rhodobacterales.

a Taxonomic bar plot depicting the relative read abundance of orders on the y-axis. On the x-axis, every bar represents the bacterial community composition of one ant colony. b Shannon indices, depicting the alpha diversity in EPs per ant species. Sampling sizes are given in brackets. Letters denote significant differences between sample types. c The distance-based redundancy analysis plot shows the variation of bacterial communities in EPs due to ant species. The patch samples are grouped according to the Azteca ant species (alf: A. alfari, n = 12; con: A. constructor, n = 22). Please note that this dataset comprises average bacterial communities of established ant colonies that were spatially sampled multiple times (Fig. 4), and bacterial communities of established ant colonies that were only sampled once.
Bacterial community composition changed along developmental stages of ant colonies
By combining all bacterial patch communities of initial, young and established ant colonies – one community per colony – sequencing data covered three stages of ant colony development. This allowed us to analyze changes in the bacterial community composition along the life cycle of Azteca colonies. The alpha diversity of patches (regardless of ant species) correlated with the ant colony developmental stages (P < 0.001, x2 = 70.483) and increased significantly from IPs via YPs to EPs (pairwise, all P < 0.001; median Shannon indices: IPs 2.2, YPs 3.5, EPs 5.2; Fig. 6a). Taking the ant species into account, the same pattern was generally visible except for A. constructor, in which IPs and YPs did not significantly differ (Supplementary Fig. 1). Furthermore, the variation in beta diversity was significantly correlating with the ant colony developmental stages explaining 15.4% of the variation (P < 0.001; Fig. 6b). Certain taxonomic classes and orders increased and decreased in the average relative read abundance from IPs via YPs to EPs. Bacteroidia (IP: 2.0%, YP: 9.8%, EP: 22.2%), Clostridia (0.3%, 0.3%, 3.1%) and Verrucomicrobiae (0.7%, 1.4%, 6.0%) increased, while the read relative abundance of Actinobacteria (14.0%, 8.9%, 5.3%) and Gammaproteobacteria (54.9%, 35.8%, 29.4%) decreased (Supplementary Fig. 2). Taxonomic orders tended to increase (e.g. Chitinophagales, Flavobacteriales, Verrucomicrobiales) or decrease (e.g. Enterobacterales, Xanthomonadales, Corynebacteriales) in their relative read abundance from early to late developmental stages (Fig. 6c), of which several shifts from IPs to YPs, or many from YPs to EPs were statistically significant (Table 3).

a Shannon indices, depicting the alpha diversity in patches of all ant colony developmental stages independent of the Azteca ant species. Sampling sizes are given in brackets. Letters denote significant differences between patch types. b Distance-based redundancy analysis plot, showing the variation of bacterial communities in patches due to ant colony developmental stages. c Taxonomic bar plot depicting the relative read abundance of orders on the y-axis. On the x-axis, every bar represents the average bacterial community composition per ant colony developmental stage. Please note that for established ant colonies that were spatially sampled multiple times (Fig. 4) an average bacterial community was calculated. Please also note that displaying average community compositions per ant colony developmental stage (c) is a simplification due to the heterogeneity across patch samples (see Figs. 2a, 3a and 5a).
In total, 20 ASVs were identified as prevalent in either IPs, YPs, or EPs (either A. alfari, or A. constructor) making up 39.8% of total reads. One ASV (assigned to Rhizobiales) reached the prevalence threshold in all three developmental stages (Table 4, Supplementary Fig. 3). Four ASVs assigned to Enterobacterales and Rhizobiales were prevalent in both IPs and YPs. Two ASVs assigned to Sphingomonadales and Xanthomonadales were prevalent in both YPs and EPs. Two and eleven ASVs reached the prevalence threshold only in either YPs or EPs, whereas no ASV was exclusively prevalent in IPs, or exclusively shared between IPs and EPs. The distribution and abundance of these prevalent ASVs showed particular patterns. Many of these prevalent ASVs were most abundant in IPs and YPs, or almost exclusively detected in EPs (Supplementary Fig. 4). Few ASVs were rather equally abundant in both YPs and EPs, but there was a lack of prevalent ASVs being abundant in both IPs and EPs, but not in YPs.
Discussion
Ant-built patches are commonly found in the nesting space of various ant-plant associations36,37,39. They are assumed to play important roles for ant colonies, such as housing bacteria with capabilities against pathogens14,28, or providing nutrients to ant larvae38,42. In addition, the patches potentially enrich the system with N via atmospheric N2-fixing bacteria30. Yet, a detailed analysis of the bacterial community, its consistency during the life cycle of the ant colony and its ant species-specificity was lacking so far.
Bacterial succession along developmental stages of ant colonies
Our study shows that ant species-specificity of the bacterial communities in the patches is determined by the developmental stage of the Azteca colony (Fig. 1). While the bacterial community composition is quite homogeneous across patches of established A. alfari or A. constructor colonies (EPs) and ant species-specific (Fig. 5), at early stages of ant colony development (IPs or YPs), the bacterial community was highly heterogeneous and showed no correlation with the corresponding ant species (Figs. 2 and 3). Although it has been shown that alate founding queens vertically transmit patch organisms when they colonize a new host plant and form the initial patch42, the bacterial community composition of the mother colony does not appear to be maintained. The alpha diversity increased from low diverse IPs, via YPs to highly diverse EPs (Fig. 6a), which was only expected in established ant colonies as workers may introduce surface bacteria43. The community composition in YPs appeared to be an intermediate stage between IPs and EPs (Fig. 6b, c), with several shared ASVs being prevalent in either IPs and YPs, or YPs and EPs, but no ASV being prevalent in both, IPs and EPs (Supplementary Fig. 4). The factors influencing the bacterial community during the ant colony development may be manifold, but the most important impact may be the N-depleted and cellulose-dominated initial patch substrate. This obviously causes a bottleneck during the colony foundation and induces an ecological succession after vertical transmission of the highly diverse bacterial EP community.
The drivers of the succession are primarily biotic and abiotic patch conditions. Personal field observations indicate changing substrate for bacteria when IPs develop into YPs and over time into EPs. While the substrate of early patches (IPs, YPs) exclusively consists of N-poor Cecropia parenchyma (average C/N ratio of around 180, Supplementary Table 2), the substrate in established patches is more diverse. When workers open the sealed domatia entrance and start to forage on the surface of the host plant, they also place items like plant-provided food bodies, parts of mosses and lichens on EPs. While the colony grows, workers die and the fragmented exoskeletons of the dead nest mates are placed on the patches as well. With the latter, the ants of established colonies enrich the patches with a high amount of chitin as a potential N-source which considerably changes the substrate. Additionally, we even visually observed an increase of fungal and nematode biomass from IPs to YPs and to EPs. Fungi and nematodes probably not only introduce their own associated bacteria to the patch community, but also a variety of usable compounds, either by secretion (fungi), feces deposition (nematodes), or decay of their biomass. An important, but often neglected aspect is the behavior of the ants to reduce the pathogen pressure in the nest, which may also impact the bacterial community. This can be done either by fumigating the nesting space with antibiotic compounds from, for example, the metapleural gland44 or by lowering the pH, as shown for leaf cutter ants45,46. The change from a crumbly patch texture in IPs to a denser texture in EPs is also likely to change oxygen and water content30. This certainly affects some members of the bacterial community. In addition to the changing conditions in patches, various other sources of bacteria may alter the bacterial diversity along the ant colony life cycle. While the colony grows, ant workers start foraging on the plant surface and thereby bringing bacteria on the tarsi structure43 into the nest. Workers also colonize new internodes that may house patches with a different microbiome from unsuccessful colony founding attempts where the queens had already died42.
To see if the identified bacterial community reflects a patch condition-driven succession, we identified cultivated strains in GenBank (NBCI) with high 16S rRNA sequence similarity to our prevalent bacterial ASVs (Table 4). Selected ASVs prevalent in IPs and/or YPs, which are parenchyma-dominated and therefore N-depleted, showed 100% 16S rRNA sequence similarity with N2-fixing bacteria (ASV_15, Burkholderiales, Herbaspirillum frisingense47), with cellobiose-utilizing bacteria (ASV_54, Sphingomonadales, Sphingobium yanoikuyae48), or with bacteria generally associated with plant tissue (ASV_26, Rhizobiales, Agrobacterium vitis49). In EPs where the turnover of various patch organisms and dead ant bodies – all commonly containing glycogen as a storage compound – might play a role, ASV_10 (Chitinophagales) was prevalent. The cultivated strain with high sequence similarity to this ASV (Terrimonas aquatica 97.6% 16S similarity) has been shown to use glycogen and cellobiose as carbon sources50. However, further cultivation- or metagenomics-based investigations are needed to determine the metabolic potential and thus probable functions of these bacteria in patches. This might shed light on the question why they are abundant in certain ant colony developmental stages and not in others.
Intraspecific variation of bacterial community composition amongst - but not within - established Azteca colonies
The substrate-driven succession may not only have caused the transformation of the bacterial patch community from initial to established ant colonies, but also may have led to different bacterial communities within an established ant colony. Newly formed EPs in newly grown shoots at the apical end of the plant are comparable to IPs as they consist primarily of plant parenchyma which is still available in uncolonized “new” internodes. We therefore assumed that the bacterial community in the patches of established colonies would undergo comparable transformation processes within the stem of a host plant as between the founding stage and established colony stages, and that the patch community would vary considerably between younger and older EPs. However, we showed that the bacterial communities of all patches within an established ant colony were similar to each other, even though the patches were of different age (Fig. 4). This suggests that workers maintain a specific bacterial community throughout the entire nesting space by inoculating the newly built patches with material from older patches. Field observations with A. constructor colonies (by MN and VEM) showed additional evidence of such patch management. There was a lack of patch material in abandoned internodes. In the oldest, still actively colonized internodes, there were globules of ant-made patch material, probably used to inoculate new patches in the apical part of the nesting space.
Diversity of bacterial patch communities in established Azteca colonies are driven by ant species
We have convincingly demonstrated that the respective ant species plays a major role in shaping the bacterial communities in established Azteca colonies. Previous studies have shown differences in the microbiome between ant colonies of different genera31, and between ants’ nests and their surroundings32. Our study takes a step forward and shows that the bacterial communities associated with the nests differ even among two closely related ant species: A. constructor maintained a significantly different and more diverse bacterial community than A. alfari (Fig. 5). This indicates that the ants influence the bacterial community of a patch through ant species-specific behavior. In established ant colonies, Azteca species form significantly different patch morphologies, which may explain the variation within the bacterial community. The EPs of A. constructor are thought to contain more micro niches due to their typical pile structure which allows the development of moisture and gas gradients, whereas A. alfari EPs are more lawn-like30. Interestingly matching the O2 gradient towards anoxic conditions in EPs of A. constructor EPs30, a cultivated strain in GenBank has been described as facultative anaerobic, which has a high sequence similarity to an ASV frequently found in A. constructor EPs (Table 2) (ASV_37, Micrococcales, 98.4% 16S rRNA gene similarity to Changpingibacter yushuensis51). In addition, the differences in alpha diversity support the idea that the pile-shaped EPs of A. constructor experience gradients of humidity and chemical concentrations, facilitating a more diverse bacterial community than in A. alfari EPs. Though, further research is needed to verify chemical gradients and their influence on the bacterial community composition, or to investigate if ant species-specific chemistry like scent or other excretions may shape the bacterial communities.
In summary, we provide the first evidence of a dynamic bacterial patch community within arboreal ant nests. The bacterial communities in these patches appear to undergo an ecological succession within each ant colony, thus evolving with the ant colony’s life cycle. Following an assumed vertical transmission of bacteria from mother to daughter colonies, the composition of the bacterial community seems to experience a bottleneck caused by the substrate. This bottleneck initially favors bacteria adapted to N-depleted cellulose-dominant substrates in early colonization stages. The subsequent ecological succession is driven by ant behavior, substrate changes and environmental parameters, and results in ant species-specific bacterial communities. We conceptualize patches inside ant-plant associations as complex ecosystems, which are essential for the development of the inhabiting ant colony. This assumption is based on the unexpectedly high bacterial diversity within colonized host plants, and the supposed diverse functional potentials in the patches. Our findings provide crucial insights for future cultivation- and omics-based studies, which can help to determine the role that these patches play in ant colonies.
Material and methods
Study site characteristics and sample collection
Cecropia plants (Urticaceae) colonized by three Azteca species (A. alfari, A. constructor and A. xanthochroa; Formicidae, Dolichoderinae) were investigated between 2015 and 2018 in close vicinity to the Tropical Research Facility La Gamba, Costa Rica (www.lagamba.at; N08˚42’03”, W083˚12’06”, 70 m asl). In total, we investigated 116 patches from 65 Cecropia plants of different sizes – 31 saplings and 34 trees – located at the margins of primary and secondary rain forests, alongside rivers (Rio Bonito, Rio Sardinal) and roads. The sampling procedure has been described in Nepel et al.30. Due to the necessity of destructively cutting Cecropia saplings and trees prior to assessing the presence, developmental stage and species of inhabiting ant colonies, sample sizes vary. We differentiated between three ant colony developmental stages and their corresponding patches (Fig. 1). For claustral colony foundation, the queen had entered a single plant internode, resealed the entrance hole and formed an initial pile of scratched-off and chewed plant parenchyma (“initial ant colony patch”, IP, Fig. 1a). In an intermediate stage, the still sealed internode sheltered a young ant colony consisting of queen, brood and the first hatched workers, next to the patch (“young ant colony patch”, YP, Fig. 1b). In established colonies, the ants inhabited many adjoined internodes of the stem, patrolled the whole tree and typically formed numerous defined dark-colored patches (“established ant colony patch”, EP, Fig. 1c) throughout their nesting space.
We sampled 31 young Cecropia saplings that were recently colonized by Azteca ants, of which 60 single internodes were just inhabited by foundress queens with IPs or optionally first workers, in which case these patches were defined as YPs. From the plant saplings we sampled in total 42 IPs (A. alfari n = 27, A. constructor n = 5, and A. xanthochroa n = 10), and in total 18 YPs (A. alfari n = 15 and A. constructor n = 3). Young and established A. xanthochroa colonies were not found in our study area.
We sampled 34 Cecropia trees, which were inhabited by established A. alfari (n = 12) and A. constructor (n = 22) ant colonies. To test if the bacterial/archaeal community composition remains stable in patches of the same established ant colony, patches from five of the A. alfari and twelve of the A. constructor colonies were collected separately from up to three stem parts representing different ant colony activity and patch age: EPs of the apical plant area recently colonized by the ants and formed with a lot of parenchyma as substrate (EP I, n = 14); EPs of the middle part of the ant colony with the highest ant activity (EP II, n = 13); and EPs of the lower part of the ant colony where many dead nestmates are placed on the patches (EP III, n = 12). Further, we studied if the bacterial/archaeal communities in established ant colonies differ significantly between the two closely related ant species. This required one bacterial/archaeal community composition per ant colony. As there was no significant difference among the different stem parts in established ant colonies, this enabled us to use the average community composition per aforementioned repeatedly sampled established ant colony. These samples were analyzed in combination with seven A. alfari and ten A. constructor colonies, from which one single patch sample per colony was collected. This dataset then resulted in overall 12 A. alfari and 22 A. constructor established ant colonies.
PCR amplification and sequencing
Patch sample collection, preservation and the DNA extraction process have been described in Nepel et al.30. The bacterial and archaeal community composition was investigated by amplifying the 16S rRNA gene for subsequent amplicon sequencing. A two-step multiplexing approach was used to amplify and barcode samples52. DNA-template of 10 ng or at most 4 µL was added per triplicate in the first-step PCR. The 16S rRNA gene was amplified by the primers 515F-mod (5’GTGYCAGCMGCCGCGGTAA’3)53 and 806R-mod (5’GGACTACNVGGGTWTCTAAT’3)54,55 including a 16 bp head sequence for subsequent barcoding52. A no-template control using water instead of template DNA was added in every PCR run as a contamination control. Each PCR reaction was 20 µl in volume and contained 1 × DreamTaq Green Buffer including 2 mM MgCl2, 0.2 mM dNTP mixture, 0.5 U DreamTaq DNA polymerase, 0.08 µg µL−1 bovine serum albumin (all from Thermo Scientific, Waltham, MA, United States), 0.25 µM of each forward and reverse primers and DNA template. The following amplification program was used: 94 °C for 3.5 min followed by 22 (or 25 if DNA template <10 ng) cycles of 94 °C for 30 s, 52 °C for 45 s, 72 °C for 45 s; and final 72 °C for 10 min. Sample preparation for amplicon sequencing was done according to Nepel et al.30. Illumina Truseq library preparation and MiSeq sequencing was performed by Microsynth (Balgach, Switzerland) in the 2 × 300 cycle configuration using the MiSeq Reagent kit V3 (Illumina, San Diego, CA, United States). The raw reads were deposited into the NCBI Short Read Archive under the BioProject accession number PRJNA77700656. Raw MiSeq amplicon reads were processed according to the pipeline described in Herbold et al.52, except for a revised downstream clustering of amplicon sequences. Amplicon sequence variants (ASVs) were inferred using the DADA2 package (version 1.2.0;57) in R (version 4.3.0;58). ASVs were classified against the SILVA database v13859. ASVs assigned to chloroplast and mitochondria were removed, with 8858 ASVs remaining. Investigated 116 patch samples contained at least 1100 and on average around 10,000 16S rRNA reads per sample.
Statistics and reproducibility
All subsequent data management and analyses were performed in R (version 4.3.0;58). Data handled and filtered based on research questions using the package phyloseq (version 1.44.0;60). Analysis functions originated mainly from the package vegan (version 2.6-4;61) unless otherwise mentioned. Graphs were plotted by using the package ggplot2 (version 3.4.2;62).
Absolute sequence reads were converted to relative abundances per sample for microbial community analyses. Community dissimilarity distance matrices were calculated after Bray–Curtis and displayed as principal coordinate analysis by the function capscale(). Permutational multivariate ANOVA (PERMANOVA; function adonis(); 104 permutations;63) was used for testing significant correlations between categorical variables (patch age, ant species, developmental stage) and the variance in the community composition. Significant variables were subsequently included and visualized as constrained distance-based redundancy analysis (dbRDA) by the function capscale(). Shannon indices were determined as alpha diversity measurements by the function phyloseq::estimate_richness() and tested for significant correlations with categorical variables by Wilcoxon (function wilcox.test()) and Kruskal-Wallis (function kruskal.test()) rank sum tests.
As PERMANOVA did not show significant correlations between the bacterial community composition and the assumed patch age in multiple-sampled established ant colonies, for further analyses, one average community composition per ant colony was calculated and merged with the dataset of single-sampled established ant colonies.
For differential abundance analyses testing which taxonomic orders significantly differ between two categories (IP and YP; YP and EP; EPs of A. alfari and A. constructor) ASVs were merged at taxonomic order level (phyloseq::tax_glom()) and subsetted beforehand. Orders, which were present in less than 50% of samples, or which accounted for less than 0.05% of total reads, were dropped before differential abundance analyses. Samples with absolute read counts were centered log-ratio transformed after Monte Carlo (aldex.clr(), package ALDEx2; version 1.32.0;64) and Wilcoxon Rank Sum tests (ALDEx2::aldex.ttest()) were used to test every taxa for a significant shift in abundance. P-values were corrected after Benjamini-Hochberg to account for multiple testing. Only Taxa showing a mean relative read abundance of more than 0.5% across all samples were highlighted in results.
Using the function core() (package microbiome; version 1.22.0;65), prevalent ASVs were extrected from different datasets. Prevalent ASVs were defined as accounting for more than 0.4% of reads in more than 50% of respective samples and were identified by the function microbiome::core_members(). For comparisons between sample types (EP: A. alfari – A. constructor; IP – YP – EP), the lists of prevalent ASVs were then searched for overlaps and displayed as tables. Due to the significant differences in the community composition of EPs by the ant species, we determined prevalent ASVs of A. alfari and A. constructor EPs separately and merged them finally containing all prevalent ASVs in EPs. This list of ASVs in combination with the ones from IPs and YPs were exported as a table and used for the ternary analysis showing the abundance of every prevalent ASV in the three patch types representing developmental stages of ant colonies. For the ternary analysis, the samples of the IP-YP-EP dataset were first merged by sample type (IP, YP, EP_alf, EP_con; phyloseq::merge_sample()) and read abundances were normalized by transforming them to relative abundances (phyloseq::transform_sample_counts()). Then, both EP categories were merged and a final repeated transformation to relative abundance was performed to weigh both ant species in established colonies equally. We depicted the mean community compositions as three artificial samples (IP, YP, EP). Subsequently, the dataset was filtered to contain only the list of prevalent ASVs from the previous analysis and plotted as a ternary graph (ggtern(); package ggtern; version 3.4.2;66). Analysis scripts are available from https://github.com/mnepel/bacteria_in_arboreal_ant_nests67.
Database check of identified prevalent ASVs
To evaluate potential functional capacities of prevalent ASVs and to infer why they may be abundant in certain ant colony developmental stages, 16S rRNA sequences were checked against GenBank (NCBI) database using BLAST68. Subsequently, literature for the cultivated strains with the highest sequence similarity to prevalent ASVs was searched and interpreted.
Evaluating the carbon and nitrogen content of plant tissue used for forming patches
Cecropia plant parenchyma of uncolonized internodes (n = 5) were sampled and dried at 60 °C. The carbon and nitrogen contents of plant parenchyma were determined using an elemental analyzer (EA 110; CE Instruments, Milan, Italy) in Vienna, Austria.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw reads and metadata were deposited into the NCBI Short Read Archive under the BioProject accession number PRJNA77700656. Analysis scripts and input data are available from https://github.com/mnepel/bacteria_in_arboreal_ant_nests67. At this repository, the source data behind the analyses and figures can be retrieved as a text file containing an ASV count table including meta data and the representative sequences, and as a spreadsheet containing Shannon indices.
References
Del Toro, I., Ribbons, R. R. & Pelini, S. L. The little things that run the world revisited: a review of ant-mediated ecosystem services and disservices (Hymenoptera: Formicidae). Myrmecol N. 17, 133–146 (2012).
Parker, J. & Kronauer, D. J. C. How ants shape biodiversity. Curr. Biol. 31, R1208–R1214 (2021).
Frouz, J., Holec, M. & Kalčík, J. The effect of Lasius niger (Hymenoptera, Formicidae) ant nest on selected soil chemical properties. Pedobiologia 47, 205–212 (2003).
Dostál, P., Březnová, M., Kozlíčková, V., Herben, T. & Kovář, P. Ant-induced soil modification and its effect on plant below-ground biomass. Pedobiologia 49, 127–137 (2005).
Frouz, J. & Jilková, V. The effect of ants on soil properties and processes (Hymenoptera: Formicidae). Myrmecol N. 11, 191–199 (2008).
Floren, A., Biun, A. & Eduard Linsenmair, K. Arboreal ants as key predators in tropical lowland rainforest trees. Oecologia 131, 137–144 (2002).
Davidson, D. W., Cook, S. C., Snelling, R. R. & Chua, T. H. Explaining the abundance of ants in lowland tropical rainforest canopies. Science 300, 969–972 (2003).
Lengyel, S., Gove, A. D., Latimer, A. M., Majer, J. D. & Dunn, R. R. Convergent evolution of seed dispersal by ants, and phylogeny and biogeography in flowering plants: a global survey. Perspect. Plant Ecol. Evol. Syst. 12, 43–55 (2010).
Natsume, K., Hayashi, S. & Miyashita, T. Ants are effective pollinators of common buckwheat Fagopyrum esculentum. Agr. For. Entomol. 24, 446–452 (2022).
Luo, C. W., Li, K., Chen, X. M. & Huang, Z. Y. Ants contribute significantly to the pollination of a biodiesel plant, Jatropha curcas. Environ. Entomol. 41, 1163–1168 (2012).
Del-Claro, K., Rodriguez-Morales, D., Calixto, E. S., Martins, A. S. & Torezan-Silingardi, H. M. Ant pollination of Paepalanthus lundii (Eriocaulaceae) in Brazilian savanna. Ann. Bot. 123, 1159–1165 (2019).
Hölldobler, B. & Wilson, E. O. The ants. (Harvard University Press, 1990).
Currie, C. R., Scottt, J. A., Summerbell, R. C. & Malloch, D. Fungus-growing ants use antibiotic-producing bacteria to control garden parasites. Nature 398, 701–704 (1999).
Hanshew, A. S. et al. Characterization of Actinobacteria associated with three ant-plant mutualisms. Micro. Ecol. 69, 192–203 (2015).
Russell, J. A. et al. Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc. Natl Acad. Sci. USA 106, 21236–21241 (2009).
Anderson, K. E. et al. Highly similar microbial communities are shared among related and trophically similar ant species. Mol. Ecol. 21, 2282–2296 (2012).
Sapountzis, P. et al. Acromyrmex leaf-cutting ants have simple gut microbiota with nitrogen-fixing potential. Appl. Environ. Microbiol. 81, 5527–5537 (2015).
van Borm, S., Buschinger, A., Boomsma, J. J. & Billen, J. Tetraponera ants have gut symbionts related to nitrogen-fixing root-nodule bacteria. Proc. R. Soc. B 269, 2023–2027 (2002).
Stoll, S., Gadau, J., Gross, R. & Feldhaar, H. Bacterial microbiota associated with ants of the genus Tetraponera. Biol. J. Linn. Soc. 90, 399–412 (2007).
Hu, Y. et al. Herbivorous turtle ants obtain essential nutrients from a conserved nitrogen-recycling gut microbiome. Nat. Commun. 9, 964 (2018).
Béchade, B. et al. Turtle ants harbor metabolically versatile microbiomes with conserved functions across development and phylogeny. FEMS Microbiol. Ecol. 98, fiac068 (2022).
Feldhaar, H. et al. Nutritional upgrading for omnivorous carpenter ants by the endosymbiont Blochmannia. BMC Biol. 5, 48 (2007).
Heil, M. & McKey, D. Protective ant-plant interactions as model systems in ecological and evolutionary research. Annu Rev. Ecol. Evol. Syst. 34, 425–453 (2003).
Dejean, A., Petitclerc, F., Roux, O., Orivel, J. & Leroy, C. Does exogenic food benefit both partners in an ant-plant mutualism? The case of Cecropia obtusa and its guest Azteca plant-ants. C. R. Biol. 335, 214–219 (2012).
Fischer, R. C., Wanek, W., Richter, A. & Mayer, V. Do ants feed plants? A 15N labelling study of nitrogen fluxes from ants to plants in the mutualism of Pheidole and Piper. J. Ecol. 91, 126–134 (2003).
Gegenbauer, C., Mayer, V. E., Zotz, G. & Richter, A. Uptake of ant-derived nitrogen in the myrmecophytic orchid Caularthron bilamellatum. Ann. Bot. 110, 757–765 (2012).
Chomicki, G. & Renner, S. S. Farming by ants remodels nutrient uptake in epiphytes. N. Phytol. 223, 2011–2023 (2019).
Fukuda, T. T. H. et al. Insights into the ecological role of Pseudomonas spp. in an ant-plant symbiosis. Front. Microbiol. 12, 621274 (2021).
Ruiz-González, M. X. et al. Do host plant and associated ant species affect microbial communities in myrmecophytes? Insects 10, 391 (2019).
Nepel, M. et al. Nitrogen fixation by diverse diazotrophic communities can support population growth of arboreal ants. BMC Biol. 20, 135 (2022).
Pringle, E. G. & Moreau, C. S. Community analysis of microbial sharing and specialization in a Costa Rican ant-plant-hemipteran symbiosis. Proc. R. Soc. B 284, 20162770 (2017).
Lucas, J. M. et al. Azteca ants maintain unique microbiomes across functionally distinct nest chambers. Proc. R. Soc. B 286, 20191026 (2019).
Touchard, A. et al. The biochemical toxin arsenal from ant venoms. Toxins 8, 30 (2016).
Tragust, S. et al. Ants disinfect fungus-exposed brood by oral uptake and spread of their poison. Curr. Biol. 23, 76–82 (2013).
Fernández-Marín, H., Zimmerman, J. K., Rehner, S. A. & Wcislo, W. T. Active use of the metapleural glands by ants in controlling fungal infection. Proc. R. Soc. B 273, 1689–1695 (2006).
Defossez, E. et al. Ant-plants and fungi: a new threeway symbiosis. N. Phytol. 182, 942–949 (2009).
Voglmayr, H. et al. The diversity of ant-associated black yeasts: insights into a newly discovered world of symbiotic interactions. Fungal Biol. 115, 1077–1091 (2011).
Blatrix, R. et al. Plant-ants use symbiotic fungi as a food source: new insight into the nutritional ecology of ant-plant interactions. Proc. R. Soc. B 279, 3940–3947 (2012).
Vasse, M. et al. A phylogenetic perspective on the association between ants (Hymenoptera: Formicidae) and black yeasts (Ascomycota: Chaetothyriales). Proc. R. Soc. B 284, 20162519 (2017).
Esquivel, A., Abolafia, J., Hanson, P. & Pinto-Tomás, A. A. A new species of nematode, Sclerorhabditis neotropicalis sp.n. (Rhabditida), associated with Azteca ants in Cecropia obtusifolia. Nematropica 42, 163–169 (2012).
Morera, J., Mora-Pineda, G., Esquivel, A., Hanson, P. & Pinto-Tomás, A. A. Detection, ultrastructure and phylogeny of Sclerorhabditis neotropicalis (Nematoda: Rhabditidae) nematodes associated with the Azteca ant-Cecropia tree symbiosis. Rev. Biol. Trop. 66, 368–380 (2018).
Mayer, V. E. et al. Transmission of fungal partners to incipient Cecropia-tree ant colonies. PLoS One 13, e0192207 (2018).
Bitar, M. R., Pinto, V. D., Moreira, L. M. & Ribeiro, S. P. Gram-negative bacteria associated with a dominant arboreal ant species outcompete phyllosphere-associated bacteria species in a tropical canopy. Oecologia 195, 959–970 (2021).
Yek, S. H. & Mueller, U. G. The metapleural gland of ants. Biol. Rev. 86, 774–791 (2011).
Ortius-Lechner, D., Maile, R., David Morgan, E. & Boomsma, J. J. Metapleural gland secretion of the leaf-cutter ant Acromyrmex octospinosus: new compounds and their functional significance. J. Chem. Ecol. 26, 1667–1683 (2000).
Vieira, A. S., Morgan, E. D., Drijfhout, F. P. & Camargo-Mathias, M. I. Chemical composition of metapleural gland secretions of fungus-growing and non-fungus-growing ants. J. Chem. Ecol. 38, 1289–1297 (2012).
Kirchhof, G. et al. Herbaspirillum frisingense sp. nov., a new nitrogen-fixing bacterial species that occurs in C4-fibre plants. Int J. Syst. Evol. Microbiol. 51, 157–168 (2001).
Yabuuchi, E. et al. Proposals of Sphingomonas paucimobilisgen. nov. and comb. nov., Sphingomonas parapaucimobilis sp. nov., Sphingomonas yanoikuyae sp. nov., Sphingomonas adhaesiva sp. nov., Sphingomonas capsulata comb. nov., and two genospecies of the genus Sphingomonas. Microbiol. Immunol. 34, 99–119 (1990).
Ophel, K. & Kerr, A. N. D. A. Agrobacterium vitis sp. nov. for strains of Agrobacterium biovar 3 from grapevines. Int. J. Syst. Bacteriol. 40, 236–241 (1990).
Sheu, S. Y., Cho, N. T., Arun, A. B. & Chen, W. M. Terrimonas aquatica sp. nov., isolated from a freshwater spring. Int. J. Syst. Evol. Microbiol. 60, 2705–2709 (2010).
Jiao, Y. et al. Changpingibacter yushuensis gen. nov., sp. nov., isolated from fluvial sediment in Qinghai Tibet Plateau of China. J. Microbiol. 60, 147–155 (2022).
Herbold, C. W. et al. A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes. Front. Microbiol. 6, 731 (2015).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016).
Walters, W. et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1, e0009–e0015 (2015).
Apprill, A., Mcnally, S., Parsons, R. & Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137 (2015).
Sequencing microbial communities in the ant-plant Azteca-Cecropia association. NCBI BioProject PRJNA777006. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA777006.
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
R Core Team. R: a language and environment for statistical computing. https://www.r-project.org/ (2021).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596 (2013).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217 (2013).
Oksanen, J. et al. vegan: community ecology package. https://cran.r-project.org/package=vegan (2020).
Wickham, H. ggplot2: elegant graphics for data analysis. (Springer-Verlag New York, 2016). https://ggplot2.tidyverse.org.
McArdle, B. H. & Anderson, M. J. Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82, 290–297 (2001).
Fernandes, A. D. et al. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2, 15 (2014).
Lahti, L. & Shetty, S. microbiome R package. https://microbiome.github.io/tutorials/ (2019).
Hamilton, N. E. & Ferry, M. ggtern: ternary diagrams using ggplot2. J. Stat. Softw. 87, 1–17 (2018).
Community analysis scripts in mnepel/bacteria_in_arboreal_ant_nests. GitHub. https://github.com/mnepel/bacteria_in_arboreal_ant_nests (2023).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Acknowledgements
This work was funded by a project grant (P-31990-B to VEM) from the Austrian Science Fund (FWF). MN was supported by a DOC fellowship (24388) from the Austrian Academy of Sciences (ÖAW) and a fellowship by the Society for the promotion of the La Gamba Field Station. We thank the staff at the Estación Tropical La Gamba for great working conditions and we thank Costa Rican authorities for their permissions to conduct this research (R-046-2015-OT-CONAGEBIO, SINAC SE-GUS-PI-113-2015, INV-ACOSA-001-2016, INV ACOSA-013-2018, DGVS-481-2015, DGVS-061-2016, DGVS-088-2018). We also thank Adrián Pinto-Tomás for the warm welcome and interesting discussions every time we visited San José, and for facilitating to hold a joint Fluorescence in situ Hybridization (FISH) workshop at the Universidad de Costa Rica.
Author information
Authors and Affiliations
Contributions
D.W., V.E.M. and M.N. designed this study. V.E.M., M.N. and D.W. conducted fieldwork and collected samples. M.N. performed lab work, analyzed the data and wrote the original draft of the manuscript. All authors, including V.B.S., contributed substantially to revisions. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Marilia B. Bitar and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Tobias Goris. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nepel, M., Mayer, V.E., Barrajon-Santos, V. et al. Bacterial diversity in arboreal ant nesting spaces is linked to colony developmental stage. Commun Biol 6, 1217 (2023). https://doi.org/10.1038/s42003-023-05577-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05577-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
