
Abstract
In arterial myocytes, the canonical function of voltage-gated CaV1.2 and KV2.1 channels is to induce myocyte contraction and relaxation through their responses to membrane depolarization, respectively. Paradoxically, KV2.1 also plays a sex-specific role by promoting the clustering and activity of CaV1.2 channels. However, the impact of KV2.1 protein organization on CaV1.2 function remains poorly understood. We discovered that KV2.1 forms micro-clusters, which can transform into large macro-clusters when a critical clustering site (S590) in the channel is phosphorylated in arterial myocytes. Notably, female myocytes exhibit greater phosphorylation of S590, and macro-cluster formation compared to males. Contrary to current models, the activity of KV2.1 channels seems unrelated to density or macro-clustering in arterial myocytes. Disrupting the KV2.1 clustering site (KV2.1S590A) eliminated KV2.1 macro-clustering and sex-specific differences in CaV1.2 cluster size and activity. We propose that the degree of KV2.1 clustering tunes CaV1.2 channel function in a sex-specific manner in arterial myocytes.
Similar content being viewed by others


Introduction
Activation of dihydropyridine-sensitive, voltage-gated L-type CaV1.2 channels plays a crucial role in the development of myogenic tone1, a process of autoregulation that enables arteries to regulate their diameter in response to changes in intravascular pressure2. This mechanism, independent of neural or hormonal influences, is critical to maintain constant blood flow despite changes in blood pressure.
The current model of the myogenic response proposes that mechanical stretch of the membrane leads to the activation of TRPC63, TRPM44, and TRPP1 (PKD2)5 channels, which depolarizes arterial myocytes, activating CaV1.2 channels1. Activation of a single or a small cluster of CaV1.2 channels results in a local increase in intracellular Ca2+ concentration ([Ca2+]i) termed a “CaV1.2 sparklet”6,7,8. Summation of multiple CaV1.2 sparklets leads to a global increase in [Ca2+]i that triggers muscle contraction.
CaV1.2 channels form clusters in the plasma membrane via a stochastic self-assembly mechanism9. CaV1.2 channels within these clusters can gate cooperatively in response to Ca2+-driven physical interactions of adjacent channels10,11. CaV1.2 channels in this configuration allow for larger Ca2+ influx compared to random, independent openings of individual channels. In vascular smooth muscle, cooperative gating of CaV1.2 channels has been estimated to contribute up to ~50% of Ca2+ influx during the development of myogenic tone12.
One route of negative feedback regulation of membrane depolarization and Ca2+ influx via CaV1.2 channels occurs through the depolarization-mediated activation of delayed rectifier voltage-gated KV2.1 channels13,14. In its canonical role, KV2.1 proteins in arterial smooth muscle cells form ion conducting voltage-gated K+ channels whose activation results in membrane potential hyperpolarization, thereby affecting myocyte [Ca2+]i and myogenic tone13. Until recently, the accepted role of KV2.1 protein in arterial myocytes was to form K+ conducting channels. However, our recent work suggests that only about 1% of the KV2.1 channels in the plasma membrane of arterial smooth muscle are conductive15. Indeed, a growing body of evidence suggests that KV2.1 proteins have dual conductive and structural roles in the surface membrane of smooth muscle cells and neurons15,16,17.
In neurons and HEK293T cells, KV2.1 is expressed in large macro-clusters16,18,19,20,21,22,23. A 26 amino acid region within the C-terminus of the channel called the proximal restriction and clustering (PRC) domain was determined to be responsible for this expression pattern24. The high-density clustering of KV2.1 channels is influenced by phosphorylation of serine residues within the PRC domain25,26,27. Additionally, in heterologous expression systems, the majority of KV2.1 channels within macro-clusters are considered non-conductive18,19,28. Little to no channel activity was detectable within KV2.1 clusters, whereas currents could be recorded in areas with diffuse KV2.1 expression19. One of the structural roles of KV2.1 is to promote clustering of CaV1.2 channels, thus increasing the probability of CaV1.2-to-CaV1.2 interactions within these clusters16,17.
Both, the conductive and structural roles of KV2.1, depend on the level of expression of this protein, which in arterial myocytes varies with sex15. In female myocytes, where expression of KV2.1 protein is higher than in male myocytes, KV2.1 has both conductive and structural roles. Female myocytes have larger CaV1.2 clusters, [Ca2+]i, and myogenic tone than male myocytes. In contrast, in male myocytes, KV2.1 channels regulate membrane potential, but not CaV1.2 channel clustering.
Based on these data, a model was proposed in which KV2.1 function varies with sex15. In males, KV2.1 channels primarily control membrane potential, but in female myocytes KV2.1 plays dual electrical and CaV1.2 clustering roles. Currently, it is unclear whether the conductive and structural functions of KV2.1 in native arterial myocytes rely on its clustering ability, and if this relationship is sex-dependent.
In this study, we tested the hypothesis that conductive and structural roles of KV2.1 channels in male and female arterial myocytes depend on their capacity to form clusters in studies of wild-type (WT) and S586A mutant rat KV2.1 channels expressed in heterologous cells, and in arterial myocytes from a novel gene edited knock-in mouse expressing the S590A mutation. We focused on serine 586 within the PRC domain (amino acids 573-598) of rat KV2.1 because a point mutation changing this amino acid to a non-phosphorylatable alanine decreases the KV2.1 clustering phenotype24. This corresponds to a S590A point mutation in the mouse KV2.1 channel. Our data show that KV2.1 is expressed into large macro-clusters composed of micro-clusters that can only be resolved with super-resolution microscopy. The KV2.1S586A point mutation nearly eliminated KV2.1 macro-clusters but had a minimal impact on micro-clusters. Notably, we find that KV2.1 channel function is not dependent on its ability to form macro-clusters in arterial myocytes of either sex. Rather, KV2.1 macro-clustering enhances CaV1.2 channel clusters and activity. Based on these results, we propose a new model in which macro-clustering of KV2.1 in arterial myocytes alters CaV1.2 channel organization and function in a sex-specific manner but has no impact on its conductive function.
Results
KV2.1 macro-clusters are composed of micro-clusters of KV2.1 and are declustered by the KV2.1S586A mutation
We began our study by determining the spatial distribution in heterologous cells of exogenously expressed wild-type rat KV2.1 (KV2.1WT) channels and KV2.1 channels in which serine at position 586 was mutated to an alanine (KV2.1S586A) using confocal and super-resolution ground state depletion (GSD) microscopy (Fig. 1). Both channels were tagged at their N-terminus with the red-shifted fluorescent protein mScarlet.

a Confocal maximum intensity projection images of HEK293T cells transfected with KV2.1WT or KV2.1S586A tagged with mScarlet. Insets show single plane images of the bottom or center of each cell. b Number of KV2.1 cluster volumes of KV2.1WT (black) and KV2.1S586A (blue) in transfected HEK293T cells. Insets quantify relative frequency histograms as a percentage of KV2.1 volumes. Plots representing (c) number of clusters per cell, (d) total cluster volume per cell, and (e) mean cluster volumes (n = 11 KV2.1WT and 11 KV2.1S586A cells). f Representative super-resolution GSD images of immunolabeled KV2.1 channels in transfected HEK293T cells. Insets show 4 μm2 regions of interest. Red overlay depicts Gaussian blur. Cyan lines indicate distances of greater than 200 nm while yellow arrows represent distances less than 200 nm. g Number of clusters of KV2.1WT (black) and KV2.1S586A (blue) by cluster area in transfected HEK293T cells. Insets quantify relative frequency histograms as a percentage of KV2.1 areas. h Summary plot of mean KV2.1 cluster areas from super-resolution images (n = 9 KV2.1WT and 8 KV2.1S586A cells). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars indicate mean ± SEM.
Figure 1a shows confocal maximum intensity projection images of 3D reconstructions of representative HEK293T cells expressing KV2.1WT and KV2.1S586A. To the right of each image, we show single plane images from the bottom and center of each cell. Figure 1b–e shows a quantitative analysis of the number and volume of KV2.1 clusters from these 3D confocal images. The frequency distributions of KV2.1WT (black) and KV2.1S586A (blue) cluster volumes could be fit with a single exponential function. Of note, the number of clusters were larger in most volume bins for the KV2.1S586A mutation compared to KV2.1WT. For example, in the same number of cells (n = 11), we detected a total of 25,174 KV2.1WT clusters and 31,893 KV2.1S586A clusters. The number of clusters per cell was 2286 ± 188.4 in KV2.1WT (median = 2227 clusters) and 2899 ± 259.9 clusters in KV2.1S586A (median = 2889 clusters) (P = 0.04) (Fig. 1c). The total cluster volume per cell of KV2.1WT was 254.5 ± 20.8 μm3 (median = 246.0 μm3) compared to 192.5 ± 23.5 μm3 in cells expressing KV2.1S586A (median = 149.0 μm3) (P = 0.03) (Fig. 1d). Interestingly, the mean cluster volumes were larger between KV2.1WT channels at 0.12 ± 0.01 μm3 (median = 0.12 μm3) and KV2.1S586A at 0.06 ± 0.01 μm3 (median = 0.06 μm3) (P = < 0.0001) (Fig. 1e). These data suggest that KV2.1WT channels are expressed into clusters and that KV2.1S586A expression is more diffuse and exhibits a more uniform distribution.
Analysis of our confocal microscopy data showed that in HEK293T cells, KV2.1WT is expressed in clusters of different sizes but do in fact form large macro-clusters. Using this confocal microscopy analysis, we sought to define the size of a KV2.1 macro-cluster. We began with the mean cluster volume of KV2.1WT generated in Fig. 1e. Our analysis provided a mean cluster volume of 0.12 μm3. Assuming the volume of a cluster is spherical, we extrapolated the diameter of the macro-cluster to be 278 nanometers (nm). The standard deviation of these measurements was 0.03. Thus, the mean minus 2 standard deviations provides a lower end limit with a 95% confidence and aligned with the lateral resolution of our confocal microscopy. Accordingly, we define the lower limit of a macro-cluster as a cluster that is larger than 0.06 μm3 or 193 nm in diameter, with clusters smaller than this classified as micro-clusters.
Figure 1f provides representative super-resolution ground-state depletion (GSD) TIRF images (lateral resolution ≈ 40 nm) from representative cells expressing KV2.1WT or KV2.1S586A. Note that data are provided on an area basis since we are imaging a single plane footprint of the cell in contrast to capturing multiple Z-slices. We also provide two regions of interest by each image. To increase our confidence in this observation, we utilized a Gaussian blur (shown in red) to decrease the resolution of the GSD signal to match that of confocal microscopy. Cyan arrows indicate distances greater than 200 nm between signals produced by GSD, while yellow arrows indicate distances less than 200 nm. Notably, our GSD image with a Gaussian blur accurately reproduced the clustering phenotype of KV2.1WT at the confocal level, providing further evidence that macro-clusters are composed of micro-clusters. Although we do not observe the large macro-clusters in KV2.1S586A expressing cells, we can still resolve KV2.1 micro-clusters.
KV2.1WT and KV2.1S586A channels are organized into clusters of varied sizes (Fig. 1g), and consistent with the lower resolution confocal data, the distribution of KV2.1S586A channel clusters was more diffuse than that of KV2.1WT. It should be noted that the regions of interest (ROI) of our GSD images reveal that the macro-clusters observed at the confocal level are, in fact, made up of numerous micro-clusters. The frequency distribution of the cluster areas of KV2.1WT and KV2.1S586A obtained from GSD imaging could both be fit with an exponential function (Fig. 1g). The mean cluster area of KV2.1WT was 2634 ± 143 nm2 (median = 2568 nm2), larger than that of KV2.1S586A channels which was 1860 ± 104 nm2 (median = 1907 nm2) (P = 0.0003) (Fig. 1h). This is likely due to the absence of larger clusters in cells expressing KV2.1S586A. Notably, cluster density was 41 ± 5 clusters per micron and 29 ± 4 clusters per micron for KV2.1 and KV2.1S586A, respectively.
Our finding that the size distributions of KV2.1WT and KV2.1S586A clusters could be described by exponential functions, the hallmark of a Poisson process, suggests that these clusters are formed stochastically29. To test this hypothesis, we implemented the stochastic modeling approach employed by Sato et al.9 to determine whether we could reproduce our cluster distributions and make testable predictions regarding plasma membrane protein organization. As shown in Supplementary Fig. 1a, b, our stochastic self-assembly model effectively reproduced the steady-state size distributions that we measured for KV2.1WT and KV2.1S586A proteins embedded in the surface membrane of HEK293T cells. The parameters used in the model are summarized in Supplementary Fig. 1c. These in silico data suggest that in HEK293T cells, KV2.1WT has a higher probability of nucleation and cluster growth than KV2.1S586A channels. The probability of channel removal was similar.
KV2.1S590A mutation does not affect overall KV2.1 channel expression but declusters smooth muscle KV2.1 macro-clusters in a sex-specific manner
Next, we investigated whether, as in heterologous expression system (i.e., Fig. 1), KV2.1 channels cluster in arterial myocytes and whether this clustering could be disrupted via mutation of critical amino acids in the PRC domain. To do this, we used CRISPR/Cas gene editing to generate a knock-in mouse line expressing the S590A point mutation, corresponding to the S586A mutation in rat KV2.1, at the KCNB1 locus of a C57BL/6 J mouse (see Methods section for a full description of how these mice were generated).
We isolated, fixed, permeabilized, and labeled arterial myocytes from both male and female KV2.1WT and KV2.1S590A knock-in mice. We double labeled with wheat germ agglutinin-488 (WGA488) to identify the sarcolemma and KV2.1-specific antibodies in male and female KV2.1WT and KV2.1S590A myocytes using confocal microscopy.
Figure 2a shows representative 3D images of fixed male mesenteric smooth muscle cells labeled with WGA488 and immunolabeled for KV2.1. We investigated whether the KV2.1S590A mutation leads to altered expression levels of the channel in arterial myocytes. Our analysis showed that total, cell-wide KV2.1-associated fluorescence normalized to cell membrane area was similar in sex-matched KV2.1WT and KV2.1S590A myocytes (Fig. 2b), suggesting that the KV2.1 expression is similar in these cells.

a Representative maximum projection images of KV2.1 channel clusters at the surface membrane of KV2.1WT and KV2.1S590A male myocytes. b Quantification of immunofluorescence labeled KV2.1 normalized to cell membrane area in male myocytes (n = 24 KV2.1WT and 15 KV2.1S590A male myocytes). c Relative frequency as a percentage of KV2.1 cluster volumes from KV2.1WT male and KV2.1S590A male myocytes. Summary data of KV2.1 clusters in myocytes showing (d) mean cluster volumes, (e) clusters per cell, and (f) percent of the surface membrane occupied by KV2.1 channels in male myocytes (n = 12 KV2.1WT male cells and 9 KV2.1S590A male cells). g Representative maximum projection images of KV2.1 channel clusters at the surface membrane of KV2.1WT and KV2.1S590A female myocytes. h Quantification of immunofluorescence labeled KV2.1 normalized to cell membrane area in female myocytes (n = 18 KV2.1WT and 11 KV2.1S590A female myocytes). i Relative frequency as a percentage of KV2.1 cluster volumes from KV2.1WT female and KV2.1S590A female myocytes. Summary data of KV2.1 clusters in myocytes showing (j) mean cluster volumes, (k) clusters per cell, and (l) percent of the surface membrane occupied by KV2.1 channels in female myocytes (n = 11 KV2.1WT and 13 KV2.1S590A female cells). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars indicate mean ± SEM.
Further analysis of KV2.1 clusters was restricted to those that overlapped with the WGA-mapped plasma membrane. In both, the KV2.1WT and KV2.1S590A males, the frequency distribution of KV2.1 cluster sizes were similar in terms of relative values in all volume bins and could be fit with an exponential decay function (Fig. 2c). The mean cluster volume in KV2.1WT males was 0.07 ± 0.01 μm3 (median = 0.07 μm3) compared to a mean of 0.07 ± 0.01 μm3 (median = 0.06 μm3) in KV2.1S590A male myocytes. (P = 0.34) (Fig. 2d). Additionally, total clusters per cell of 551.6 ± 71.1 clusters, (median = 505 clusters) in KV2.1WT male myocytes were not significantly different from total clusters per cell of 444.2 ± 33.4 clusters (median = 419 clusters) in KV2.1S590A males (P = 0.10) (Fig. 2e). The percentage of the membrane occupied by clusters in KV2.1WT male myocytes was on average 5.5 ± 0.9% (median = 5.1%), similar to the average in KV2.1S590A males of 5.0 ± 1.0% (median = 4.6%) (P = 0.34) (Fig. 2f).
We next investigated whether the KV2.1S590A mutation leads to altered expression levels of the channel in female arterial myocytes. Figure 2g shows representative 3D images of fixed female mesenteric smooth muscle cells labeled with WGA488 and immunolabeled for KV2.1. Similar to male myocytes, our analysis revealed that the total KV2.1-associated fluorescence, normalized to the cell membrane area, exhibited no significant difference between sex-matched KV2.1WT and KV2.1S590A myocytes (Fig. 2h).
In sharp contrast to male myocytes, KV2.1S590A female myocytes exhibited an increased proportion of smaller clusters as compared to those from KV2.1WT females (Fig. 2i). Accordingly, mean cluster size of KV2.1WT in female myocytes was 0.14 ± 0.10 μm3 (median = 0.14 μm3), significantly larger than mean cluster size of 0.10 ± 0.01 μm3 (median = 0.09 μm3) (P = 0.007) in KV2.1S590A females (Fig. 2j). KV2.1WT female myocytes had 796 ± 67 clusters per cell, (median = 815 clusters) compared to only 422 ± 34 clusters per cell (median = 415 clusters) in KV2.1S590A female myocytes (P < 0.0001) (Fig. 2k). Similarly, the percentage of the plasma membrane occupied by KV2.1 was higher in KV2.1WT female myocytes at 16.7 ± 1.8% (median = 17.7%) in contrast to KV2.1S590A female myocytes in which KV2.1 clusters occupied on average 7.2 ± 1.0% of the plasma membrane (median = 6.0) (P < 0.0001) (Fig. 2l). The significantly lower KV2.1 clustering profile in all metrics measured indicates that unlike in males, the S590A mutation decreases channel clustering in female myocytes.
Using the threshold set from our confocal imaging (i.e., macro-clusters are >0.06 μm3), we quantified the number of macro-clusters expressed in myocytes from KV2.1WT and KV2.1S590A mice. Around 41% of clusters in KV2.1WT male myocytes were identified as macro-clusters, with a similar percentage of 35% observed in samples from males with the KV2.1S590A mutation. Approximately 57% of KV2.1 clusters in KV2.1WT female myocytes were classified as macro-clusters. Remarkably, in KV2.1S590A female myocytes, macro-clusters accounted for approximately 50% of the total KV2.1 clusters.
We also quantified KV2.1 micro-clusters. Although the proportion of micro-clusters was similar in male KV2.1WT and KV2.1S590A myocytes (59% and 65%, respectively), female KV2.1S590A myocytes exhibited a much larger proportion of micro-clusters (50%) compared to myocytes from KV2.1WT females (42%). Hence, it can be reasoned that the S590A mutation has a sex-specific effect of reducing the extent of KV2.1 macro-clustering in female but not male arterial myocytes without impacting channel expression.
KV2.1 S590 phosphorylation is higher in myocytes from female versus male KV2.1WT mice
To investigate the potential role of the S590 phosphorylation site in the sex-specific differences in KV2.1 clustering, we conducted immunocytochemistry analyses on arterial myocytes (Fig. 3). We tested the hypothesis that KV2.1WT female myocytes exhibit a higher degree of KV2.1 S590 phosphorylation compared to males, which could contribute to the observed sex-specific variations in KV2.1 clustering. Accordingly, we utilized a monoclonal antibody (mAb L100/130) specific for KV2.1 that is phosphorylated at serine 590 (pS590).

a Representative maximum projection images of immunolabeling for pS590 KV2.1 and total KV2.1channel clusters at the surface membrane of KV2.1WT female (red), KV2.1S590A female (blue), KV2.1WT male (black), and KV2.1S590A male (purple) cells. b Summary data of pS590 KV2.1 clusters per cell (n = 37 KV2.1WT male, 15 KV2.1S590A male, 34 KV2.1WT female and 15 KV2.1S590A myocytes). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars indicate mean ± SEM.
Figure 3a shows cells double immunolabeled for pS590 and total KV2.1 using confocal microscopy. One advantage of our study is that our KV2.1S590A mice serve as an ideal negative control. As expected, KV2.1S590A males exhibited 2.3 ± 0.6 clusters (median = 2.0 clusters) per cell and KV2.1S590A females exhibited 1.2 ± 0.4 clusters (median = 1.0 clusters) per cell confirming the specificity of this mAb for KV2.1 phosphorylated at S590. Remarkably, phosphorylated KV2.1 clusters were observed in KV2.1WT females (Fig. 3b) exhibiting on average 12.2 ± 1.7 of phosphorylated clusters (median = 10.0 clusters) per cell, whereas KV2.1WT males exhibited 3.4 ± 0.6 clusters (median = 2.5 clusters) per cell. Collectively, these findings suggest that basal levels of KV2.1 pS590 phosphorylation are higher in KV2.1WT females, which could account for their increased clustering of KV2.1. Furthermore, these data support the notion that KV2.1 phosphorylation in KV2.1WT male myocytes is constitutively low, making the S590A mutation functionally indistinguishable from non-phosphorylated Kv2.1WT and thus ineffective in altering KV2.1 clustering in male myocytes.
Expression of clustering impaired KV2.1S590A does not affect channel activity in arterial myocytes
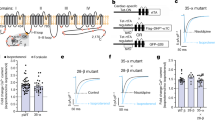
Three studies, one using Xenopus oocytes28, one using HEK293T cells19 and another from our group using arterial myocytes15 have suggested that the vast majority of KV2.1 channels (i.e., 98–99%) expressed in the plasma membrane of these cells are non-conductive. O’Connell et al.19 suggested that, at least in HEK293T cells, KV2.1 channel activity depends on their density and that channels within large, dense macro-clusters are non-conductive. A testable hypothesis raised by these data is that a larger fraction of KV2.1S590A should be conductive and hence the amplitude of KV2.1 currents in native arterial myocytes should differ between cells from KV2.1WT and KV2.1S590A mice (Fig. 4).

Computationally modeled IKv2.1 in male (a) and female (b) myocytes assuming 100% (red), 50% (blue) or 10% (green) of KV2.1 channels present in the plasma membrane are functional. Computationally modeled IKv2.1 in male (c) and female (d) myocytes assuming 1% (gray) and 0.1% (black) KV2.1 channels are functional. e Representative IKv2.1 traces at +50 mV from KV2.1WT male (black) and KV2.1S590A male (purple) arterial myocytes and voltage dependence of IKv2.1 in KV2.1WT (black) and KV2.1S590A (purple) male myocytes from −50 to +50 mV. f Representative IKv2.1 traces at +50 mV from KV2.1WT female (red) and KV2.1S590A (blue) arterial myocytes and voltage dependence of IKv2.1 in KV2.1WT (red) and KV2.1S590A (blue) female myocytes from −50 to +50 mV. IKV2.1 traces were obtained by subtracting currents after the application of RY785 from control IK traces. n = 18 KV2.1WT male, 6 KV2.1S590A male, 19 KV2.1WT female and 13 KV2.1S590A myocytes. Error bars indicate mean ± SEM.
We tested this hypothesis using a multipronged approach. First, we used a mathematical modeling approach31 to determine the predicted changes in macroscopic KV2.1 currents with varied levels of functional channels (i.e., 0.1, 1, 10, 50, or 100%) in male and female arterial myocytes (Fig. 4a–d). The rationale for this analysis is that it provides a set of potential outcomes that can provide insights into the degree of KV2.1 declustering in KV2.1S590A myocytes. This model incorporated data (e.g., voltage-dependencies and number of channels in the sarcolemma) from O’Dwyer et al.15.
As shown in Fig. 4a, the model predicts that with 100%, 50%, or 10% functional KV2.1 channels in male myocytes would produce current densities at +50 mV of 7,006, 3,503, and 701 pA/pF, respectively. By contrast, at the same voltage, the in silico female arterial myocytes produce current densities of 17,293, 8,646, and 1,729 pA/pF with 100%, 50%, or 10% functional KV2.1 channels (Fig. 4b). We also simulated the current-voltage relationships in male (Fig. 4c) and female (Fig. 4d) myocytes assuming 1% and 0.1% of KV2.1 channels are conductive, which are more within the range with previous experimental results in heterologous systems19,28 and native cells15. The magnitude of in silico KV2.1 current densities with 1% or 0.1% functional channels was 70.1 and 5.57 pA/pF in male myocytes and 173 and 16.7 pA/pF in female myocytes.
Next, we recorded voltage-gated K+ (KV) currents in male and female KV2.1WT and KV2.1S590A arterial myocytes in response to 500 ms depolarizations to voltages between −50 and +50 mV before and after applying the KV2.1 blocker RY785 (1 μM)32,33. This compound decreases KV2.1 currents by blocking the pore of these channels32 rather than by immobilizing their voltage sensor, as stromatoxin does34. As a first step in these experiments, we tested the specificity of the RY785 by recording KV currents before and after the application of this molecule in male and female KV2.1WT and KV2.1 null (KV2.1−/−) myocytes (Supplementary Fig. 2a–d). Notably, application of 1 μM RY785 decreased the amplitude of K+ currents in KV2.1WT but not in KV2.1−/− myocytes of either sex. This indicates that RY785 is a specific blocker of KV2.1 channels in arterial myocytes.
Having completed these critical control experiments, we recorded KV currents from KV2.1WT and KV2.1S590A myocytes. We noted that the amplitude of the composite K currents were similar in myocytes from KV2.1S590A mice compared to myocytes from sex-matched KV2.1WT littermates (Supplementary Figs. 2e, f).
Importantly, for both sexes, RY785-sensitive KV2.1 currents were also similar in male (Fig. 4e) and female (Fig. 4f) KV2.1WT and KV2.1S590A myocytes. Indeed, a comparison of the experimental and in silico amplitudes of the macroscopic KV2.1 currents suggests that less than 1% of the channels are functional in myocytes from both male and female KV2.1WT and KV2.1S590A mice. When taken together with our analyses of Kv2.1 clustering detailed above, these findings suggest that in arterial myocytes KV2.1 channel activity is not determined by the extent and nature of its clustering.
The KV2.1S590A mutation diminishes KV2.1 and CaV1.2 proximity in female myocytes
We used the proximity ligation assay (PLA) to interrogate the impact of the KV2.1S590A mutation on protein-protein proximity, at a resolution of approximately 50 nm35,36 (Supplementary Fig. 3). We first evaluated KV2.1-KV2.1 proximity within isolated mesenteric smooth muscle cells by using two different antibodies directed against different epitopes in the KV2.1 cytoplasmic C-terminus. In this case both intra- and inter-molecular proximity of the two epitopes would yield a PLA signal. Inter-molecular proximity could be visualized as “puncta”, and we hypothesized that more puncta would be exhibited in cells where KV2.1 was more clustered since there would be increased intermolecular proximity, allowing for more PLA reactions to occur. Confocal images of KV2.1WT and KV2.1S590A myocytes subjected to PLA show that puncta of KV2.1-KV2.1 PLA signals were randomly distributed throughout the cell, and that PLA signal could be detected in all cells consistent with our confocal data above showing that KV2.1 micro-clustering still occurs in mutant myocytes of both sexes (Supplementary Fig. 3a). The density of PLA puncta of 0.035 ± 0.003 puncta/μm2 (median = 0.035 μm2) in KV2.1WT males was similar to 0.030 ± 0.003 puncta/μm2 (median = 0.028 μm2) in KV2.1S590A male mice (P = 0.148) (Supplementary Fig. 3b). Total puncta area per cell was similar between KV2.1WT males at 8.494 ± 1.143 um2 (median = 7.376 um2) and KV2.1S590A males at 6.236 ± 2.166 um2 (median = 2.3838 um2) (P = 0.1578) (Supplementary Fig. 3c). Consistent with the confocal imaging analysis, the density of KV2.1-KV2.1 PLA puncta was greater in KV2.1WT females, with an average of 0.153 ± 0.011 puncta/μm2 (median = 0.162 μm2), compared to 0.046 ± 0.003 puncta/μm2 (median = 0.037 μm2) in KV2.1S590A females (P < 0.0001) (Supplementary Fig. 3d). Similarly, total puncta area per cell was larger in KV2.1WT females at 24.33 ± 2.289 um2 (median = 23.90 um2) and KV2.1S590A females at 6.484 ± 0.968 um2 (median = 5.338 um2) (P = < 0.0001) (Supplementary Fig. 3e). Together, this data suggests that the KV2.1S590A mutation reduces the level of KV2.1 clustering in female myocytes.
Previous work from our group has shown that KV2.1 expression promotes CaV1.2 clustering and activity in neurons16 and arterial myocytes15. Following from this and the data above, we hypothesize that in arterial myocytes KV2.1 plays a sex-specific structural role as an organizer to bring CaV1.2 channels together in female but not male myocytes. We again used PLA to test the hypothesis that the declustering of KV2.1 channels in female but not male myocytes from KV2.1S590A mice would decrease KV2.1-CaV1.2 channel proximity in a sex-specific manner. Representative images of KV2.1-CaV1.2 PLA puncta show randomly distributed interactions across the cell (Supplementary Fig. 3f).
Quantification showed that KV2.1-CaV1.2 puncta density was unchanged between KV2.1WT male myocytes with a mean of 0.022 ± 0.002 puncta/μm2 (median = 0.016 puncta/μm2) and KV2.1S590A male myocytes with a mean of 0.018 ± 0.002 puncta/μm2 (median = 0.016 puncta/μm2; P = 0.217) (Supplementary Fig. 3g).
Total puncta area per cell was similar between KV2.1WT males at 4.443 ± 0.5655 um2 (median = 2.700 um2) and KV2.1S590A males at 4.398 ± 0.4332 um2 (median = 3.476 um2) (P = 0.4746) (Supplementary Fig. 3h). However, KV2.1-CaV1.2 proximity decreased in female KV2.1S590A myocytes with a mean of 0.030 ± 0.004 puncta/μm2 (median = 0.026 puncta/μm2) compared to KV2.1WT female myocytes with a mean of 0.044 ± 0.004 puncta/μm2 (median = 0.026 puncta/μm2; P = 0.013) (Supplementary Fig. 3i). Total puncta area per cell was larger in KV2.1WT females at 6.328 ± 0.7606 um2 (median = 4.171 um2) than KV2.1S590A females at 4.268 ± 0.5517 um2 (median = 3.504 um2) (P = 0.0217) (Supplementary Fig. 3j). These data further support a sex-specific structural role for KV2.1 channels, facilitating CaV1.2-CaV1.2 clustering.
Female myocytes expressing KV2.1S590A have reduced macroscopic CaV1.2 currents
We recorded macroscopic CaV1.2 currents (ICa) from male and female KV2.1WT and KV2.1S590A arterial myocytes (Fig. 5a–d). ICa was activated by applying 300 ms voltage step depolarizations from a holding potential of −80 to +60 mV. We show ICa traces recorded during a depolarization to 0 mV from representative male (Fig. 5a) and female (Fig. 5b) KV2.1WT and KV2.1S590A arterial myocytes. Note that the amplitude and kinetics of ICa in these male KV2.1WT and KV2.1S590A arterial myocytes were similar. By contrast, we found that peak ICa was smaller in female Kv2.1S590A myocytes compared to those in KV2.1WT cells. In Fig. 5c and d, we show the voltage dependence of the amplitude of ICa from all the cells examined over a wider range of membrane potentials. This analysis shows that the amplitude of ICa is similar in KV2.1WT and KV2.1S590A male myocytes at all voltages examined. However, in female myocytes, ICa was smaller in KV2.1S590A than in KV2.1WT at all voltages examined. Indeed, at 0 mV, ICa amplitude in KV2.1S590A cells was approximately 50% of that of WT females.

ICa records (0 mV) from representative KV2.1WT and KV2.1S590A male (a) and KV2.1WT and KV2.1S590A female (b) myocytes. Voltage dependence of ICa from male (c) and female (d) myocytes at membrane potentials ranging from −50 to +50 mV (n = 6 KV2.1WT male, 7 KV2.1S590A male, 20 KV2.1WT female and 20 KV2.1S590A myocytes). Representative images of immunolabeled CaV1.2 in myocytes from KV2.1WT male (e, black), KV2.1S590A male (e, purple), KV2.1WT female (f, red), and KV2.1S590A female (f, blue) mice. g Summary data from real-time quantitative PCR experiments of CaV1.2 mRNA expression relative to β-actin in male (n = 2 from 4 KV2.1WT and 4 KV2.1S590A males where 2 artery beds were pooled for each sample). h Quantification of immunofluorescence of labeled CaV1.2ɑ subunits in male myocytes (n = 20 KV2.1WT and 19 KV2.1S590A male myocytes). i Summary data from real-time quantitative PCR experiments of CaV1.2 mRNA expression relative to β-actin in female (n = 2 from 4 KV2.1WT and 4 KV2.1S590A females where 2 artery beds were pooled for each sample). j Quantification of immunofluorescence of labeled CaV1.2ɑ subunits in female myocytes (n = 16 KV2.1WT and 20 KV2.1S590A myocytes). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars indicate mean ± SEM.
Next, we determined the level of expression of CaV1.2 protein in male and female KV2.1S590A and KV2.1WT vessels using RT-PCR and immunocytochemistry (Fig. 5e, f) approaches. Our analysis suggests that mRNA expression (Fig. 5g) and total CaV1.2 protein (Fig. 5h) is similar in KV2.1S590A and KV2.1WT males. Likewise, mRNA expression (Fig. 5i) and total CaV1.2 protein (Fig. 5j) is similar in female KV2.1S590A and KV2.1WT vessels. This suggests that the smaller ICa in female KV2.1S590A than KV2.1WT myocytes is not likely due to lower CaV1.2 expression in these cells.
KV2.1S590A mutation decreases CaV1.2 cluster sizes in female but not male arterial myocytes
In a previous study15, we suggested a model that differences in ICa amplitude between female and male arterial myocytes were due to sex-specific differences in KV2.1-mediated CaV1.2 clustering that impacted the probability of cooperative gating of these channels. Our data above show differences in ICa amplitude between female KV2.1WT and KV2.1S590A myocytes in the absence of differences in CaV1.2 expression levels. Thus, we investigated whether KV2.1S590A expression altered CaV1.2 channel clustering in a sex-specific manner using GSD super-resolution microscopy (Fig. 6).

a Representative super-resolution GSD images of immunolabeled CaV1.2 labeled channels in KV2.1WT and KV2.1S590A male myocytes. Insets show 4 μm2 regions of interest. b Relative frequency as a percentage of CaV1.2 cluster areas of KV2.1WT (black) and KV2.1S590A (purple) in male myocytes. c Summary plot of mean CaV1.2 cluster areas in male myocytes (n = 13 KV2.1WT and 14 KV2.1S590A male myocytes). d Representative super-resolution GSD microscopy images of immunolabeled CaV1.2 labeled channels in KV2.1WT and KV2.1S590A female myocytes. e Relative frequency as a percentage of CaV1.2 cluster areas of KV2.1WT (red) and KV2.1S590A (blue) female myocytes. f Summary plot of mean CaV1.2 cluster areas in female myocytes (n = 12 KV2.1WT and 9 KV2.1S590A female myocytes). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars indicate mean ± SEM.
We show GSD run in TIRF mode super-resolution images from representative male (Fig. 6a) myocytes from KV2.1WT and KV2.1S590A mice. The insets show expanded views of two regions of interest (1 µm2) within each cell image. Our TIRF images show that CaV1.2 clusters of various sizes are expressed throughout these cells. The frequency distribution of CaV1.2 cluster areas in KV2.1WT and KV2.1S586A male could both be fit with an exponential function (Fig. 6b). The mean area of CaV1.2 clusters in male KV2.1WT of 2259 ± 55 nm2 (median = 2219 nm2) was similar to the KV2.1S590A male mean of 2345 ± 82 nm2 (median = 2354 nm2) (P = 0.173) (Fig. 6c), suggesting that S590A mutation in male myocytes does not affect CaV1.2 channel clustering.
Figure 6d shows representative super-resolution images from female myocytes of KV2.1WT and KV2.1S590A mice. Similar to males, the frequency distribution of CaV1.2 cluster areas in KV2.1WT and KV2.1S586A females could both be fit with an exponential function (Fig. 6e). However, CaV1.2 cluster sizes were significantly smaller in KV2.1S590A female myocytes with a mean area of 2381 ± 91 nm2 (median = 2251 nm2) compared to KV2.1WT female myocytes whose mean area was 3098 ± 164 nm2 (median = 3117 nm2) (P = 0.0001) (Fig. 6f). Taken together with our electrophysiological data, our findings suggest that the clustering and activity of CaV1.2 channels is modulated by the degree of KV2.1 clustering.
As shown in Supplementary Fig. 4a, b, our stochastic self-assembly model effectively reproduced the steady-state size distributions that we measured for CaV1.2 clustering in KV2.1WT and KV2.1S586A arterial myocytes. The parameters used in the model are summarized in Supplementary Fig. 4c. These in silico data suggest that CaV1.2 clusters in KV2.1S590A female myocytes have a higher probability of growth than those in female KV2.1WT arterial myocytes.
KV2.1S586A reduces CaV1.2-CaV1.2 channel interactions
Having previously shown that KV2.1 enhances CaV1.2-CaV1.2 channel interactions in arterial myocytes15, we used a split-Venus fluorescent protein system to determine if a decrease in KV2.1 macro-clustering would lead to a reduction in CaV1.2-CaV1.2 interactions and channel activity. This system involves fusing CaV1.2 channels with either the N-terminal fragment (CaV1.2-VN) or the C-terminal fragment (CaV1.2-VC) of Venus protein. Individually, neither CaV1.2-VN nor CaV1.2-VC emits fluorescence. However, when brought into close enough proximity for interaction to occur, they can reconstitute a full fluorescence emitting Venus protein. Thus, the split-Venus fluorescence can be utilized to indicate interactions between neighboring CaV1.2 channels. Accordingly, we compared the split-Venus fluorescence in HEK293T cells expressing CaV1.2-VN and CaV1.2-VC and co-expressing either KV2.1WT or KV2.1S586A (Fig. 7). The voltage protocols used for these experiments to promote Venus reconstitution via the complementary interaction of clustered CaV1.2-VN and CaV1.2-VC are similar to those used in two recent studies10,15 and are described in detail in the Methods section of this paper. Briefly, we recorded ICa in response to short 200 ms depolarization steps to voltages ranging from −50 to +50 mV before and after the application of a conditioning 9 s step depolarization to 0 mV, to allow for Venus reconstitution between adjacent CaV1.2-VN and CaV1.2-VC channels.

a Representative currents measured at 0 mV from pre- (purple) and post- (red) conditioning protocol from HEK293T cells expressing CaV1.2-VN, CaV1.2-VC, and DsRed-KV2.1P404W. Normalized (to peak current in pre-conditioning protocol) pre- and post- IV relationships from HEK293T cells expressing CaV1.2-VN, CaV1.2-VC, and DsRed-KV2.1P404W. b Representative currents measured at 0 mV from pre- (green) and post- (blue) conditioning protocol from HEK293T cells expressing CaV1.2-VN, CaV1.2-VC, and DsRed-KV2.1P404W,S586A. Normalized pre- and post- IV relationships from HEK293T cells expressing CaV1.2-VN, CaV1.2-VC, and DsRed-KV2.1P404W,S586A (n = 13 DsRed-KV2.1P404W and 10 DsRed-KV2.1P404W,S586A cells). c Representative TIRF images of Venus fluorescence reconstitution in HEK293T cells from cells transfected with CaV1.2-VN, CaV1.2-VC and DsRed-KV2.1P404W or CaV1.2-VN, CaV1.2-VC and DsRed-KV2.1P404W,S586A. Pre- and post-conditioning CaV1.2-Venus (green), KV2.1P404W or KV2.1P404W,S856A (red), and the merge of the two channels are presented. d Summary of CaV1.2-Venus fluorescence (F/F0) (n = 22 DsRed-KV2.1P404W and 12 DsRed-KV2.1P404W,S586A cells). *P < 0.05. Error bars indicate mean ± SEM.
We first transfected HEK293T cells with CaV1.2-VN, CaV1.2-VC, and the non-conducting but macro-clustering-competent rat KV2.1P404W channel37 tagged with red-shifted fluorescent protein DsRed. The P404W mutation confers a non-conductive KV2.1 phenotype, allowing us to study the structural clustering role of KV2.1 without masking of the Ca2+ currents by K+. Our data show that submitting these cells to the conditioning protocol increased ICa at most membrane potentials, with the peak ICa at 0 mV being about 51% larger compared to control (i.e., before application of conditioning pulse) currents (Fig. 7a).
We next tested the hypothesis that reduction of KV2.1 macro-clusters with the S586A point mutation would decrease CaV1.2 channel activity. We co-expressed CaV1.2-VN, CaV1.2-VC and DsRed-KV2.1P404W,S586A in HEK293T cells and repeated the above protocol. We found that ICa exhibited an increase of about 9% between pre- and post- conditional protocols at 0 mV (Fig. 7b).
Analysis of the associated TIRF images show that Venus fluorescence increased by approximately 21% with stimulation from control to post-conditioning steps in cells cotransfected with CaV1.2-VN, CaV1.2-VC and DsRed-KV2.1P404W, suggesting an increase in KV2.1 macro-cluster dependent CaV1.2-CaV1.2 interactions (Fig. 7c, d). Furthermore, Venus fluorescence with KV2.1P404W,S586A expression increased by about 9%, a level similar to what was previously published15,17 with CaV1.2-VN and CaV1.2 VC alone (Fig. 7d). We propose this increase is due to the intrinsic ability of CaV1.2 channels to interact with one another. Together these data further support a model for the structural role KV2.1 macro-clusters play in enhancing CaV1.2-CaV1.2 interactions and activity.
The activity of CaV1.2 channels is reduced in KV2.1S590A female but not male arterial myocytes
We next examined whether variations in the activity of CaV1.2 channels could explain the differences in ICa observed in myocytes from KV2.1WT and KV2.1S590A male and female mice. CaV1.2 channel activity was determined by recording CaV1.2 sparklets using TIRF microscopy as previously described6,7,12,17,38,39,40 (Fig. 8). TIRF microscopy of near-plasma membrane intracellular Ca2+ levels provides a powerful tool for recording Ca2+ entry via individual or small clusters of CaV1.2 channels, as it enables the activity of individual channels to be recorded from a relatively large membrane area allowing for the identification of discrete sarcolemma signaling domains. In this analysis, CaV1.2 sparklet activity is expressed as nPs, where n is the number of quantal levels reached by the sparklet site and Ps is the probability of sparklet occurrence. Detailed analysis of CaV1.2 sparklets sites38 revealed heterogeneity in activity at different sites. Therefore, CaV1.2 sparklets sites were separated into low and high activity sites, using an nPs cutoff of 0.2.

Representative sparklet traces from KV2.1WT male (a) and KV2.1S590A male (b) myocytes. κ values are shown above each trace. c nPs values from low and high activity sites in male myocytes. d Coupling coefficient values (κ) from male myocytes. e Sparklet sites per cell from male myocytes (n = 16 KV2.1WT and 9 KV2.1S590A male myocytes). Representative sparklet traces from KV2.1WT female (f) and KV2.1S590A female (g) myocytes. h nPs values from low and high activity sites in female myocytes. i Coupling coefficient values (κ) from female myocytes. j Sparklet sites per cell from female myocytes (n = 16 KV2.1WT and 10 KV2.1S590A female myocytes. *P < 0.05, **P < 0.01. Error bars indicate mean ± SEM.
Representative CaV1.2 sparklet traces are provided from low activity sparklet sites. Of note, the majority of the sparklet activity that occurs in male myocytes is produced by a signal that corresponds to a single channel opening (one quantal unit) (Fig. 8a, b). The strength of the coupled gating is denoted by the κ value, and in these traces, the κ values are close to or equal to 0, indicating no or weak coupling between the channels.
We found that in low activity sparklet sites, the average nPs was not different between KV2.1WT and KV2.1S590A male myocytes (Fig. 8c). KV2.1WT sparklet sites had an average nPs of 0.07 ± 0.01 (median = 0.07) compared to KV2.1S590A where the nPs average was 0.07 ± 0.02 (median = 0.06) (P = 0.46). In KV2.1WT male myocytes, we detected 7 high activity sparklet sites with a mean nPs of 0.25 ± 0.02 (median = 0.24) (Fig. 8c). Interestingly, we could not detect any high activity sparklets sites from KV2.1S590A male myocytes. We therefore included cells from where we could not record any sparklet activity from. Including this data, the average nPs of sparklets recorded from KV2.1WT males were 0.13 ± 0.04 compared to an nPs of 0 from KV2.1S590A males. Furthermore, we did not observe a difference in the number of CaV1.2 sparklet sites with an average of 1.50 ± 0.17 sites (median = 1.50 sites) in KV2.1WT males compared with 1.2 ± 0.20 sites (median = 1.00 sites) in KV2.1S590A male myocytes (P = 0.15) (Fig. 8d).
Previous work40 showed that CaV1.2 sparklet sites appeared to arise from the simultaneous opening and/or closing of multiple channels suggesting that small groups of channels may be functioning cooperatively. To examine such coupling, we employed a coupled Markov chain model to determine the coupling coefficient (κ) among CaV1.2 channels at Ca2+ sparklet sites. The κ value ranges from 0 for channels that gate independently to 1 for channels that are tightly coupled and open and close simultaneously. A detailed description of this model is provided in the expanded Methods section. Using this analysis, we found that the average κ value of 0.28 ± 0.09 (median = 0.30) in KV2.1WT male myocytes was not significantly different from 0.20 ± 0.09 (median = 0.22) in KV2.1S590A male myocytes (P = 0.27) (Fig. 8e).
In contrast, the KV2.1WT female trace (Fig. 8f) from a low activity site exhibited coordinated multi-channel openings, of up to 3 channels with a κ value of 0.466. Interestingly, the activity of sparklet sites from KV2.1S590A female myocytes (Fig. 8g) were similar to those of KV2.1WT and KV2.1S590A male myocytes, exhibiting mostly single channel openings and few coupled gating events.
When we compared nPs in low activity sites in KV2.1WT and KV2.1S590A female myocytes, we could not discern a difference in their average nPs values (Fig. 8h). KV2.1WT female mean nPs was 0.10 ± 0.01 (median = 0.09), which was similar to 0.06 ± 0.03 (median = 0.04) in KV2.1S590A female myocytes (P = 0.08). High activity sites averaged an nPs of 0.31 ± 0.02 (median = 0.30) in KV2.1WT cells. We only recorded a single high activity sparklet site with an nPs of 0.29 in KV2.1S590A female cells, similar to that of KV2.1WT female myoctytes (P = 0.50) (Fig. 8h). As with the male myocytes, we included cells with no sparklet activity where the average nPs in KV2.1WT females was 0.20 ± 0.04 (median = 0.27) compared to 0.05 ± 0.05 (median = 0.0) in KV2.1S590A female myocytes. Additionally, KV2.1WT female myocytes exhibited 2.73 ± 0.27 (median = 3.0) CaV1.2 sparklet sites per cell, higher than 1.40 ± 0.25 (median = 1.0) CaV1.2 sparklet sites in KV2.1S590A female myocytes (P = 0.008) suggesting decreased CaV1.2 channel activity with S590A mutation (Fig. 8i). Together this data suggests a cumulative higher activity in KV2.1WT female myocytes.
However, the average κ value of 0.36 ± 0.05 (median = 0.38) in female KV2.1WT myocytes, was significantly higher than 0.14 ± 0.07 (median = 0) (P = 0.0082) in female KV2.1S590A myocytes, suggesting more coupled events (Fig. 8j). Taken together, these data indicate increased CaV1.2 channel activity and coupled gating in myocytes from KV2.1WT females compared to those with the KV2.1S590A mutation, suggesting that clustering of KV2.1 modulates CaV1.2 channel activity.
Discussion
In this study, we show that arterial smooth muscle cells from mice expressing a gene-edited point mutation of the KV2.1 channel that selectively eliminates its characteristic macro-clustered localization have properties remarkably like those from KV2.1 knock-out mice. This leads us to formulate a new model in which KV2.1 expression, by itself, is not sufficient for this channel to exert its structural functions on modulating CaV1.2 clustering and activity, but rather depends on KV2.1 channel’s capacity to form macro-clusters. Notably, the presence of KV2.1 macro-clusters in female, but not male myocytes underlie sex-specific differences in Ca2+ influx via CaV1.2 channels in arterial smooth muscle (Fig. 9). Our data suggest a new paradigm whereby the clustering of ion channels underlies their physiological functions, independent of their ability to conduct ions.

Enhanced S590A phosphorylation (yellow circles labeled with a “P”) of KV2.1 channels (red) in KV2.1WT female myocytes increases KV2.1 macro-clustering concomitant with larger CaV1.2 channel clusters (blue) and increased CaV1.2 channel activity (red trace). KV2.1WT male and S590A mutant myocytes exhibit decreased or no S590 phosphorylation compared to KV2.1WT female myocytes, disrupting the formation of KV2.1 macro-clusters. CaV1.2 channel clusters are smaller and exhibit less activity.
Analysis of super-resolution images indicates that clustering of KV2.1 and CaV1.2 channels is random and hence does not involve an active process. This stochastic self-assembly mechanism leads to micro- and macro-clusters of varying sizes that represent the default organization of KV2.1 and CaV1 channels expressed endogenously in neurons and smooth muscle cells or exogenously in heterologous cells9. Furthermore, we found that KV2.1 macro-clusters are composed of groups of micro-clusters. This is consistent with a recent study showing that in developing neurons KV2.1 macro-clusters formed from the coalescence of numerous micro-clusters41 and suggests that the organization of KV2.1 clusters is hierarchical.
An important finding in this study is that KV2.1 clustering is more prominent in female than in male arterial myocytes, with female myocytes expressing a larger proportion of macro-clusters. In this context, the development of the KV2.1S590A mouse allowed us to investigate the separatable structural clustering and ion conducting roles of this channel. We found that expression KV2.1S590A nearly eliminated macro-clustering in female myocytes but had no impact on KV2.1 micro-clusters in cells from male or female myocytes. Because the S590A mutation eliminated a phosphorylation site in the PRC domain that causes macro-clustering, these findings suggested that the potential mechanism of these sex-specific differences in KV2.1 clustering was differential phosphorylation of this specific serine in male and female myocytes.
Indeed, KV2.1 phosphorylation and macro-clustering is regulated by a myriad of protein kinases such as CDK5 and protein phosphatases such as calcineurin42. These kinases and phosphatases work as a rheostatic mechanism to regulate the phosphorylation status of KV2.1 based on physiological demands26,27,42. Accordingly, we found that that the phosphorylation state of KV2.1 in arterial myocytes differs between the two sexes, specifically, that KV2.1 in male myocytes is phosphorylated to a much lower degree.
It is intriguing to consider the potential clustering mechanisms that are impacted by inhibiting phosphorylation at the 590/586 specific site of the PRC domain by the serine to alanine point mutation. One hypothesis is that VAP proteins act to modulate the probability of macro-cluster formation. Studies show that KV2.1 clusters are expressed at sites where the endo/sarcoplasmic reticulum is brought into close juxtaposition to the plasma membrane41 and this interaction and accumulation of channels relies on the tethering of KV2.1 to VAP proteins21,43. The transmembrane endo/sarcoplasmic reticulum VAP proteins (VAPA and VAPB) interact with the phosphorylated KV2.1 PRC domain and have been proposed to function to increase the local concentration of KV2.1 channels at endo/sarcoplasmic reticulum-plasma membrane junctions resulting in KV2.1 macro-clustering.
Consistent with this, Kirmiz et al. 21, found that knock-out of VAPA in RAW664.7 macrophage cells resulted in a decrease in KV2.1 channel clustering. Knockdown of endogenous VAP proteins similarly impaired clustering of KV2.1 heterologously expressed in HEK293T cells43. Interestingly, the model proposed in these prior papers21,43 suggested that the phosphorylated PRC domain is necessary and sufficient for macro-clustering of KV2 channels. This is consistent with prior studies showing that mutations disrupting or eliminating the PRC domain22,37,43 or treatments that impact KV2.1 phosphorylation26,27,42 impact KV2.1 clustering. It is presumed that the phosphorylation of multiple serine residues, including S590, within the PRC domain provide the negative charges needed to generate a functional VAP-binding FFAT — two phenylalanines in an acidic tract — motif, as has been shown for numerous other proteins that exhibit phosphorylation-dependent binding to VAPs44.Therefore, one possible mechanism for the decrease in macro-clustering in the S590A mutant is the inability of VAP proteins to recognize the PRC domain of mutated channels preventing cluster growth. The similarity in the patterns of cluster sizes and densities between HEK293T cells and arterial myocytes of both WT and S590A channels is noteworthy, indicating the possibility of a shared set of mechanisms. Further research will be necessary to uncover the underlying factors that govern these clustering patterns.
Prior studies have suggested that the bulk of KV2.1 channels heterologously expressed in Xenopus oocytes28 or HEK293T cells19 as well as endogenous KV2.1 in hippocampal neurons45 and arterial myocytes15 are in a nonconducting state. The prevailing view is that aggregation of KV2.1 channels into high density clusters is what renders most of these channels incapable of conducting K+18. Although our study does not address this issue comprehensively, at a minimum, our data suggest that KV2.1 conduction is not dependent on macro-clustering formation. Future studies should investigate whether the formation of KV2.1 micro-clusters may be sufficient to electrically silence these channels.
We demonstrate the structural role of KV2.1 clustering in regulating CaV1.2 channel clustering and activity that occurs in native cells. This is important because the generally accepted view is that the functional impact of ion channel clustering is to exclusively concentrate ion conducting roles at specific sites. For example, Na+ channel clustering at nodes of Ranvier46, neuronal Ca2+ channel clustering at active zones in presynaptic terminals47, and skeletal muscle Ca2+ channels at SR Ca2+ release units48. In the case of ventricular myocytes, it is concentrating voltage sensors at specific sites in the junctional dyad49,50. We propose that KV2.1 clustering is distinct in playing a role in modulating the localization and activity of an otherwise seemingly unrelated ion channel: CaV1.2 channels. This functional impact of KV2.1 is due to the density-dependent cooperative gating that is an intrinsic property of CaV1.2 channels51.
The observation that the number of KV2.1-KV2.1 PLA puncta decreased with the S590A mutation suggests that the distance between individual channels increased above the PLA limits (i.e., ∼50 nm35), which we propose contributes to a large loss of macro-clusters in cells expressing these clustering-deficient channels. However, the observation of a significant, albeit more modest decrease in CaV1.2-KV2.1 puncta (i.e., compared to KV2.1-KV2.1 puncta) suggests to us that, on average, at least some KV2.1S590A channels are still expressed within ∼50 nm of a CaV1.2 cluster. The increase in the variance of the number of CaV1.2-KV2.1 PLA puncta per cell due to the S590A mutation likely indicates increased independence, or randomness, in the spatial arrangement of the stochastically self-assembled CaV1.2 and KV2.1 clusters.
Remarkably, the overall impact of KV2.1S590A expression is that the differences between the ICa amplitude of wild-type male and female myocytes were eliminated in myocytes expressing the KV2.1S590A mutation, similar to what we observed in homozygous KV2.1 knockout mice15. Thus, declustering KV2.1 macro-clusters appears to have the same impact as fully eliminating KV2.1 expression on CaV1.2 clustering and activity in male and female myocytes. As our work also suggests that in arterial myocytes the conductive function of KV2.1 channels is independent of the degree of its clustering, in our model it is the extent of KV2.1 clustering that is the key determinant of the sex-specific differences in Ca2+ influx observed in these cells.
Our previous work established that KV2.1 serves dual roles, encompassing both conductive and structural functions, which yield opposing functional consequences in arterial myocytes, with the former predominating in males and the latter in females15. In female myocytes, which exhibit greater expression of KV2.1 protein than males, KV2.1 has a larger structural role, serving as sites for larger clusters of CaV1.2, ultimately leading to higher [Ca2+]i, and greater myogenic tone than male myocytes. Together this would suggest that KV2.1-CaV1.2 interactions contribute to the sex-specific regulation of vascular smooth muscle excitability, Ca2+ dynamics and myogenic tone. Additional questions raised in this study regard the physiological consequences of the KV2.1S590A mutation. We would hypothesize that KV2.1S590A females would have decreased myogenic due to decreased CaV1.2 cluster size and activity. Future studies will aim to further elucidate the functional consequences of this mutation and whether targeting this interaction could provide a therapeutic option for blood pressure regulation.
The findings in this study raise an important question: Are hormonal differences between male and female underlying sex-specific differences in KV2.1 and CaV1.2 organization and function? Although our data do not provide an answer to this question, an intriguing approach would be to investigate the impact of ovariectomy and castration in mice and the role of CaV1.2 and KV2.1 channel clustering and gating in arterial smooth muscle function. These experiments would help to understand whether estrogen or testosterone are responsible for the sex-specific variations in CaV1.2 and KV2.1 channels reported here. For instance, would ovariectomy result in more “male-like” myocytes with smaller KV2.1 and CaV1.2 clusters or the opposite for castrated males?
The work contained here hints at several broad implications in vascular physiology. From a general point of view, our data suggest that modulation of the phosphorylation state of KV2.1 channels influence their capacity to form macro-clusters. Increased phosphorylation of KV2.1 channels at serine 590 can lead to KV2.1 macro-clustering promoting the formation of larger CaV1.2 channel clusters. This, in turn, increases the probability of cooperative gating of these channels and hence increases Ca2+ influx, [Ca2+]i, and myogenic tone altering arterial diameter and arterial blood flow. In principle, this could happen in male and female myocytes and represents a novel mode of regulation of Ca2+ influx in these cells.
A recent paper by Hernandez-Hernandez et al.31 describes a computational model of mesenteric smooth muscle which incorporated electrophysiology and Ca2+ signaling data to study sex-specific differences in CaV1.2 and KV2.1 channel function. This study yielded results that suggests female myocytes may be more sensitive to Ca2+ channel blockers compared to male. Interestingly, several clinical studies52,53,54 have observed increased responses to Ca2+ channel blockers in hypertensive women compared to men. For example, Kloner et al.52, demonstrated a significantly larger proportion of women (91.4%) reached the goal diastolic blood pressure response compared to men (83%) when subjected to dihydropyridine-type channel blockers like amlodipine or nifedipine. Notably, following adjustments for potential confounding variables such as baseline blood pressure, age, weight, and dosage per kilogram, the differences remained significant. A more recent study by van Luik et al.54, corroborated these findings, showing that treatment with calcium channel blockers led to a substantial reduction in systolic blood pressure, diastolic blood pressure, and mean arterial pressure in both sexes, with a notably more significant decrease observed in females compared to males. While many factors could be at play, including whether our mouse data fully translates to humans, our data, when considered alongside modeling and clinical research, prompt an interesting hypothesis. This data suggests the hypothesis that higher KV2.1 macro-clustering, and hence CaV1.2 channel clustering and Ca2+ influx, in arterial myocytes could be responsible for the increased responsiveness to these drugs in women than in men. However, future studies are warranted.
To conclude, we propose a model by which KV2.1 serves a structural role in promoting CaV1.2 channel clustering and activity in a sex-dependent manner. Of note, KV2.1S590A mutation reduced CaV1.2 clustering and function in female myocytes but had no effect on male myocytes. KV2.1 clustering is not necessary for KV2.1 channel function however, KV2.1 macro-clusters alter CaV1.2 channel organization. Together, our data suggest that the interactions between KV2.1 and CaV1.2 are crucial for sex-based differences in arterial smooth muscle physiology.
Methods
Generation of the CRISPR/Cas9-edited KV2.1S590A (KCNB1 S590A) knock-in mouse
The KCNB1 S590A mutation changes a AGC codon to GCC in Exon 2, thus converting a serine to an alanine (S590A) in the KV2.1 polypeptide. The knock-in mouse was generated in collaboration with the UC Davis Mouse Biology Program by using CRISPR/Cas mediated homology directed repair. KCNB1 S590A mice were generated by introducing a mixture of gRNA (15 ng/L), single-stranded oligodeoxynucleotide (ssODN) repair template and Cas9 protein (30 ng/μL) by pronuclear microinjection into C57BL/6 J (RRID: IMSR_JAX000664) mouse zygotes. Twenty zygotes were injected and implanted into the oviducts of one surrogate dam. A total of 6 pups were born, and genomic DNA was extracted from tail biopsies followed by PCR amplification using a specific primer set to identify a single male founder (F0). DNA-Seq analysis was used to confirm the mouse genotype. The correctly integrated single mutant F0 male mouse was further backcrossed with WT C57BL/6 J female mice to produce offspring (F1) followed by intercrossing for two additional generations to obtain KCNB1 S590A heterozygotes which were used for breeding. Heterozygous and homozygous mutants were identified by a PCR genotyping protocol.
Animals
All experiments were conducted in accordance with the University of California Institutional Animal Care and Use Committee guidelines. Animals were housed under standard light-dark cycles and allowed to feed and drink ad libitum. Mice were euthanized with a single, lethal dose of sodium pentobarbital (250 mg/kg) intraperitoneally.
Arterial myocyte isolation
Third- and fourth-order mesenteric arteries were carefully cleaned of surrounding adipose and connective tissue, dissected out, and placed in ice-cold dissecting solution containing (in mM) 5 KCl, 140 NaCl, 2 MgCl2, 10 glucose, and 10 HEPES adjusted to pH 7.4 with NaOH. Arteries were first placed in dissecting solution supplemented with 1.23 mg/ml papain (Worthington Biochemical, LS003119) and 1 mg/ml DTT at 37 °C for 14 min. This was immediately followed by a five-min incubation in dissecting solution supplemented with 1.6 mg/ml collagenase H, 0.5 mg/ml elastase (Worthington Biochemical, LS002292), and 1 mg/ml trypsin inhibitor from Glycine max at 37 °C. Arteries were rinsed three times with dissection solution and single cells obtained by gentle trituration with a wide-bore glass pipette. Myocytes were maintained at 4 °C until used.
HEK293T cell culture and transfection
HEK293T (AATC, CRL-3216) cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco, 11955) supplemented with 10% fetal bovine serum (Gibco, 26140) and 1% penicillin/streptomycin (Gibco, 15140122) and maintained at 37 °C in a humidified 5% CO2 atmosphere. Cells were transiently transfected using JetPEI (Polyplus Transfection, 101000053) according to manufacturer’s protocol and passaged 24 h later onto 25 mm square #1.5 coverslips or 18 mm square collagen coated #1.5 coverslips (Neuvitro Corporation, GG-18-15-Collagen) for GSD experiments. Plasmids encoding DsRed-KV2.1WT, DsRed-KV2.1S586A, DsRed-KV2.1P404W, and DsRed-KV2.1P404W, S586A were used for this study17,21. mScarlet-tagged versions of these plasmids were generated by GenScript, replacing the sequence encoding DsRed with sequence encoding mScarlet55. For the bimolecular fluorescence experiments, cells were transfected with the pore-forming subunit of the rabbit CaV1.2 (α1c, kindly provided by Dr. Diane Lipscombe; Brown University, Providence, RI) with the carboxy tail fused to either the N-fragment (VN) or the C-fragment (VC) of the Venus protein (Addgene, Cambridge, MA, 27097, 22011), auxiliary subunits CaVα2δ, CaVβ3 (kindly provided by Dr. Diane Lipscombe, Brown University, Providence, RI) and either DsRed-KV2.1P404W or DsRed-KV2.1P404W, S586A-DsRed. HEK293T cells were transfected with CaV1.2-VN, CaV1.2-VC, CaVα2δ, CaVβ3 and DsRed-KV2.1-dsRed in a 1.0:1.0:1.0:1.5:0.4 ratio.
Live cell confocal imaging
HEK293T cells transfected with 200 ng of mScarlet-KV2.1WT or mScarlet-KV2.1S586A and seeded onto 25-mm square 1.5 coverslips approximately 16 hours before experiments. Imaging was performed in Tyrode III solution consisting of (in mM) 140 NaCl, 5.4 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, and 5.5 glucose, pH 7.4 with NaOH. Cells were imaged with an Olympus Fluoview 3000 confocal laser-scanning microscope equipped with an Olympus Plan-Apochromat 60x oil immersion lens (NA = 1.40).
Stacks of images were analyzed using Imaris 10 (Andor, Belfast). Briefly, KV2.1-associated mScarlet signal was mapped to x/y/z centroid co-ordinates in each image stack using the Spots tool. Spots were assigned to all signal surpassing a fixed signal threshold and restricted to puncta greater than 100 nm (x/y) and 150 nm diameter (z), such that any bright signal with a volume greater than two voxels was identified as a KV2.1 cluster. ‘Region Growing’ was utilized with a fixed manual threshold to apply variable sizing to KV2.1 Spots, in line with the volume and brightness of mScarlet puncta. Finally, the Cell segmentation function was used to estimate cell boundaries based on low-intensity mScarlet signal and obtain an approximate cell volume.
KV2.1 immunofluorescence immunocytochemistry
Immunofluorescence labeling was performed on freshly dissociated arterial myocytes. Cells were left to adhere for one hour at room temperature prior to fixation, fixed with 4% formaldehyde (Electron Microscopy Sciences, 50980487) diluted in phosphate-buffered saline (PBS) (Fisher Scientific, Hampton, NH) for 15 min at room temperature, washed, and incubated with 50 mM glycine (BioRad, 1610718) for 10 min to reduce aldehydes. The surface membrane was stained with wheat germ agglutinin (WGA) Alexa Fluor 488 (1 μM, ThermoFisher, W11261) for 10 minutes at room temperature followed by washing. Cells were then incubated in blocking buffer made of 3% w/v bovine serum albumin and 0.25% Triton X-100 in PBS, followed by incubation with mouse anti-Kv2.1 (mAb K89/34; RRID: AB_2877280; NeuroMab, Davis, CA, 1:200) diluted in blocking buffer for one hour at room temperature or overnight at 4 °C. Myocytes were washed, incubated at room temperature for one hour with Alexa Fluor 647-conjugated donkey anti-mouse IgG diluted in blocking buffer (2 μg/ml, Molecular Probes, A31571) followed by washes in PBS. For experiments investigating KV2.1 phosphorylation state, double labeling was performed with the mouse anti-KV2.1 pS590 phosphospecific mAb L100/130 together with rabbit anti-KV2.1 (KC23; Trimmer laboratory, RRID:AB_2315767; 1:100). Myocytes were washed, incubated at room temperature for one hour with Alexa Fluor 568-conjugated goat anti-mouse IgG (2 μg/ml, Molecular Probes, A11004) and Alexa Fluor 647-conjugated donkey anti-rabbit IgG (2 μg/ml, Molecular Probes, A31571) diluted in blocking buffer followed by washes in PBS. All washes were performed with PBS three times for 10 minutes. Coverslips were mounted onto microscope slides in Vectashield mounting medium (Vector Labs) and sealed with clear nail polish. Images were collected on a Dragonfly 200 spinning disk confocal (Andor), coupled to a DMi* Leica microscope (Leica, Wetzlar, Germany) equipped with a 60x oil immersion objective (NA = 1.40) and acquired using an Andor iXon EMCCD camera. Images were collected via Fusion software, in optical planes with a z-axis of 0.13 μm/step.
Image stacks were segmented and analyzed in Imaris 10. WGA-488 signal was background-subtracted and a fixed threshold applied to consistently map the plasma membrane, using the Surfaces tool. Alexa Fluor-647 signal (denoting KV2.1 puncta) was assessed using the Spots tool, as described above. Spots marking KV2.1 clusters were categorized into internal and plasma membrane-restricted components, with the latter utilized for analysis.
Super-resolution microscopy
HEK293T cells transfected with 200 ng mScarlet-Kv2.1WT or mScarlet-Kv2.1S586A-mScarlet and arterial myocytes were plated onto collagen coated glass coverslips (Neuvitro Corporation, GG-18-1.5-Collagen) followed by fixation with 3% formaldehyde and 0.1% glutaraldehyde diluted in PBS for 15 min at room temperature. After washing with PBS, cells were incubated with 50 mM glycine for 10 min to quench aldehydes. Cells were washed and incubated for one hour at room temperature with a blocking buffer made with 3% w/v BSA and 0.25% Triton X-100 in PBS. Cells were then incubated with either mouse anti-Kv2.1 (HEK293T experiments, mAb K89/34; RRID: AB_2877280; UC Davis/NIH Neuromab Facility, Davis, CA; 1:20) or mouse anti-CaV1.2 (arterial myocytes experiments, mAb L57/23; RRID: AB_2802123; 1:5). After extensive washings with PBS (three quick washes followed by three 30-min washes), cells were incubated at room temperature for one hour with Alexa Fluor 647-conjugated goat anti-mouse diluted in blocking buffer to a concentration of 2 µg/ml and afterwards extensively washed with PBS.
The imaging buffer contained 10 mM MEA, 0.56 mg/ml glucose oxidase, 34 µg/ml catalase, and 10% w/v glucose in TN buffer (200 mM Tris-HCl pH 8, 10 mM NaCl). A super resolution ground state deletion system (SR-GSD, Leica, Wetzlar, Germany) based on stochastic single-molecule localization was used to generate super-resolution images of CaV1.2 and KV2.1 labeling. The Leica SR-GSD is a Leica DMI6000B TIRF microscope system equipped with a 160x HCX Plan-Apochromat (NA 1.43) oil-immersion lens and an EMCCD camera (iXon3 897, Andor Technology, Belfast, United Kingdom). Fluorophores were excited with a 642 nm laser (used for both pumping to the dark state and image acquisition). For all experiments, the camera was running in frame-transfer mode at a frame rate of 100 Hz (11 ms exposure time). Fluorescence was detected through Leica high power TIRF filter cubes (488 HP-T, 532 HP-T, 642 HP-T) with emission band-pass filters of 505–605 nm, 550–650 nm, and 660–760 nm. A total of 35,000 images were collected per cell and used to construct the super resolution localization images. Fluorescence signals in each image were fit with a 2D Gaussian function which localized the coordinates of centroids of single molecule fluorescence within the LASAF software (Leica). Images were rendered at 20 nm/pixel (normalized Gaussian mode), threshold (# photons/event) using the GSD software and exported as binary TIF images. Particle analyses were determined in ImageJ. Representative images were rendered down to 2 nm for visualization purposes.
To accomplish the Gaussian blur, the GSD generated pixel in an image was replaced by a weighted average of 200 nm of its neighboring pixels. The amount of blur applied to the image was controlled by the size of the kernel, which determines the radius of the neighboring pixels used in the calculation, such that the larger the kernel, the more pixels are included in the calculation, and the stronger the blur effect.
Quantitative PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen) as per manufacturer’s instructions. Isolated mRNA was then reverse transcribed using the AffinityScript qPCR cDNA Synthesis Kit (Agilent 600559) following manufacturer’s protocol. Quantitative PCR (qPCR) analysis was performed using a QuantStudio 7 Pro Real-time PCR System (Applied Biosystems), using PowerUP SYBR Green Master Mix (Thermo Fisher Scientific, A25742) as the fluorescence probe. The cycling conditions were 50 °C for 2 minutes and 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 56 °C for 1 min. A dissociation curve protocol (ramping temperatures between 60 °C and 95 °C) was added at the end to verify amplification specificity of each qPCR reaction.
Specific primers were designed in this experiment, including β-actin (NM_007393.5): sense nt (895-914): CCAGCCTTCCTTCTTGGGTA, antisense nt (989-967): AGAGGTCTTTACGGATGTCAACG; and CaV1.2 (NM_009781.4): sense nt (5-23): CTGAAAGCAGAAGCTCGGA, antisense nt (181-163): CATTGTGGCTTCCAGTTGG. Primer efficiencies were tested to be in between 90% and 110%. The relative abundance of CaV1.2 transcript was normalized to β-actin transcript expression.
Proximity ligation assay
A Duolink In Situ PLA kit (Sigma DUO92007) was used to detected KV2.1-KV2.1 and KV2.1-CaV1.2 complexes in freshly isolated mesenteric arterial myocytes. All protocols post incubation of primary antibodies were followed in accordance with the manufacturer’s instructions. Briefly, cells were plated on glass coverslips and allowed to adhere for 1 h at room temperature. Cells were fixed with 4% paraformaldehyde for 20 min, quenched in 10 mM glycine for 15 min, washed in PBS two times for three minutes, and permeabilized 20 min in 0.1% Triton X-100. After blocking for 1 h at 37 °C in Duolink Blocking Solution, cells were incubated overnight at 4 °C using the following primary antibodies: mouse anti-KV2.1 (mAb K89/34; RRID: AB_2877280; UC Davis/NIH Neuromab Facility, Davis, CA; 1:200), rabbit anti-KV2.1 (KC23; RRID:AB_2315767; 1:100) and rabbit anti-CaV1.2. The anti-CaV1.2 rabbit polyclonal antibody “CaV1.2 II-III” was generated by immunizing two New Zealand white rabbits with a His-tagged recombinant protein fragment corresponding to a.a. 785-900 of mouse CaV1.2 (accession number Q01815). Antibodies were affinity purified from serum on nitrocellulose strips containing the CaV1.2 II-III His-tagged recombinant protein fragment following the method of Olmsted56. Cells incubated with only one primary antibody served as negative controls. KV2.1−/− null mice also served as negative biological controls as previously reported15. Secondary oligonucleotide-conjugated antibodies (PLA probes: anti-mouse MINUS and anti-rabbit PLUS) were used to detect KV2.1 and CaV1.2 interactions. Fluorescent signal was detected using an Olympus FV3000 confocal microscope equipped with a 60x oil immersion lens (NA = 1.40). Images were collected with a z-axis of 0.5 µm/step optical planes. Stacks of images were combined in ImageJ and used for analysis of puncta/µm2 per cell restricting the PLA signal to the edge of the cell.
Patch-clamp electrophysiology
All electrophysiological recordings were acquired at room temperature using an Axopatch 200B amplifier and Digidata 1440 digitizer (Molecular Devices, Sunnyvale, CA). Borosilicate patch pipettes were pulled and polished to resistances of 3–6 MΩ using a micropipette puller (model P-97, Sutter Instruments, Novato, CA).
IKv2.1 was measured in arterial myocytes using conventional whole-cell voltage-clamp electrophysiology at a frequency of 50 kHz and low-pass filter of 2 kHz. Cells were continuously perfused with an external solution containing (in mM) 130 NaCl, 5 KCl, 3 MgCl2, 10 glucose, and 10 HEPES adjusted to pH 7.4 with NaOH. Micropipettes were filled with an internal solution containing (in mM) 87 K-aspartate, 20 KCl, 1 CaCl2, 1 MgCl2, 5 MgATP, 10 EGTA, and 10 HEPES pH 7.2 with KOH. A liquid junction potential of 13 mV was corrected for offline. To measure current-voltage relationships, cells were subjected to a series of 500 ms test pulses increasing from −70 mV to +70 mV. In order to isolate the RY785-sensitive Kv2 current, cells were first bathed and recorded in external solution. Cells were then exposed to 1 μM RY785 (MedChemExpress, HY-114608) to inhibit Kv2.1 activity. RY785-sensitive currents were calculated by subtracted the RY785 exposed traces from the composite IK traces.
ICa was measured in isolated arterial myocytes using conventional whole-cell electrophysiology. Currents were measured at a frequency of 50 kHz and low-pass filtered at 2 kHz. Myocytes were continuously bathed in an external solution with (in mM) 115 NaCl, 10 TEA-Cl, 0.5 MgCl2, 5.5 glucose, 5 CsCl, 20 BaCl2, and 10 HEPES adjusted to a pH of 7.4 using CsOH. Micropipettes were filled with (in mM) 20 CsCl, 87 aspartic acid, 1 MgCl2, 10 HEPES, 5 MgATP, and 10 EGTA adjusted to pH 7.2 via CsOH. A voltage error of 9.4 attributed to the liquid junction potential of the recording solutions was corrected for offline. Cells were exposed to a series of 300 ms depolarizing pulses from a holding potential of −70 mV to test potentials ranging from −70 mV to +60 mV to attain current-voltage relationships.
Bimolecular fluorescence complementation
Spontaneous interactions of CaV1.2 channels were assayed using biomolecular fluorescence complementation. HEK293T cells were transfected with CaV1.2 channels tagged at their C-terminus to either a non-fluorescent N-(VN(1-154, I152L)) or C-terminal (VC(155-238, A206K)) halves of a ‘split’ Venus fluorescent protein. When CaV1.2-VN and CaV1.2-VC are brought close enough together to interact, the full Venus protein can fold into its functional, fluorescent conformation. The scale of Venus fluorescence emission can therefore be an indicator of CaV1.2-CaV1.2 interactions. Venus fluorescence was monitored using TIRF microscopy.
For whole-cell current recordings from HEK293T cells, pipettes were filled with a solution containing (mM) 84 Cs-aspartate, 20 CsCl, 1 MgCl2, 10 HEPES, 1 EGTA, and 5 MgATP adjusted to pH 7.2 using CsOH. HEK293T cells were continuously perfused with an external solution comprising of (in mM) 5 CsCl, 10 HEPES, 10 glucose, 140 NMDG, 1 MgCl2, and 20 CaCl2 with a pH of 7.4 (HCl).
CaV1.2 sparklets
Ca2+ sparklets were recorded using a through-the-lens TIRF microscope built around an inverted microscope (IX-70; Olympus) equipped with a Plan-Apochromat (60x; NA 1.49) objective (Olympus) and an electron-multiplying charge-coupled device camera (iXON; Andor Technology, UK). Myocytes were loaded via the patch pipette with a solution containing (in mM) 0.2 Fluo-5F (Invitrogen, F14221), 87 Cs-aspartate, 20 CsCl, 1 MgCl2, 5 MgATP, 10 HEPES, 10 EGTA, pH 7.2 with CsOH. Cells were perfused in an external solution containing 140 NMDG, 5 CsCl, 51 MgCl2, 10 glucose, 10 HEPES, 2 CaCl2, pH 7.4 with HCl. After obtaining a GΩ seal in 2 mM Ca2+ external solution, the cell was broken into and allowed to dialyze for 3 min. The external solution was exchanged with a solution containing (in mM) 120 NMDG, 5 CsCl, 1 MgCl2, 10 glucose, 10 HEPES, 20 CaCl2, pH 7.4 with HCl. Images for the detection of sparklets were recorded at a frequency of 100 Hz using TILL Image software. Cells were held to a membrane potential of −70 mV using the whole-cell configuration of the patch-clamp technique. Sparklets were automatically detected and later analyzed using custom software (Source code 2) written in MATLAB (RRID:SCR_001622)10.
In silico modeling
Simulations were performed using the Sato et al.9 and Hernandez-Hernandez et al.31 mathematical models for cluster formation and smooth muscle electrophysiology, respectively.
Statistics and reproducibility
Statistical testing was performed using GraphPad Prism (GraphPad Software, Inc.). All data were tested for normality using Shapiro-Wilk test. Data are expressed as mean ± standard error of the mean (SEM). Data were compared using one tailed unpaired t-tests when comparing two groups or one-way analysis of variance (ANOVA) with a Tukey post-hoc test for more than two groups. P < 0.05, denoted by * in the figures, was considered statistically significant and further classified as P < 0.01 (**), P < 0.001 (***), P < 0.0001 (****).
Chemicals
All chemical reagents were acquired from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All source data are available in the Supplementary Data file.
References
Knot, H. J. & Nelson, M. T. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J. Physiol. 508, 199–209 (1998).
Bayliss, W. M. On the local reactions of the arterial wall to changes of internal pressure. J. Physiol. 28, 220–231 (1902).
Spassova, M. A., Hewavitharana, T., Xu, W., Soboloff, J. & Gill, D. L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl Acad. Sci. USA 103, 16586–16591 (2006).
Gonzales, A. L. et al. A PLCgamma1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci. Signal 7, ra49 (2014).
Bulley, S. et al. Arterial smooth muscle cell PKD2 (TRPP1) channels regulate systemic blood pressure. Elife 7, https://doi.org/10.7554/eLife.42628 (2018).
Navedo, M. F., Amberg, G. C., Votaw, V. S. & Santana, L. F. Constitutively active L-type Ca2+ channels. Proc. Natl Acad. Sci. USA 102, 11112–11117 (2005).
Navedo, M. F. & Santana, L. F. CaV1.2 sparklets in heart and vascular smooth muscle. J. Mol. Cell Cardiol. 58, 67–76 (2013).
Wang, S. Q., Song, L. S., Lakatta, E. G. & Cheng, H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature 410, 592–596 (2001).
Sato, D. et al. A stochastic model of ion channel cluster formation in the plasma membrane. J. Gen. Physiol. 151, 1116–1134 (2019).
Dixon, R. E. et al. Graded Ca(2)(+)/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. Elife 4, https://doi.org/10.7554/eLife.05608 (2015).
Dixon, R. E., Yuan, C., Cheng, E. P., Navedo, M. F. & Santana, L. F. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc. Natl Acad. Sci. USA 109, 1749–1754 (2012).
Amberg, G. C., Navedo, M. F., Nieves-Cintron, M., Molkentin, J. D. & Santana, L. F. Calcium sparklets regulate local and global calcium in murine arterial smooth muscle. J. Physiol. 579, 187–201 (2007).
Amberg, G. C. & Santana, L. F. Kv2 channels oppose myogenic constriction of rat cerebral arteries. Am. J. Physiol. Cell Physiol. 291, C348–C356 (2006).
Nelson, M. T. et al. Relaxation of arterial smooth muscle by calcium sparks. Science 270, 633–637 (1995).
O’Dwyer, S. C. et al. Kv2.1 channels play opposing roles in regulating membrane potential, Ca(2+) channel function, and myogenic tone in arterial smooth muscle. Proc. Natl Acad. Sci. USA 117, 3858–3866 (2020).
Vierra, N. C., Kirmiz, M., van der List, D., Santana, L. F. & Trimmer, J. S. Kv2.1 mediates spatial and functional coupling of L-type calcium channels and ryanodine receptors in mammalian neurons. Elife 8, https://doi.org/10.7554/eLife.49953 (2019).
Vierra, N. C., O’Dwyer, S. C., Matsumoto, C., Santana, L. F. & Trimmer, J. S. Regulation of neuronal excitation-transcription coupling by Kv2.1-induced clustering of somatic L-type Ca(2+) channels at ER-PM junctions. Proc. Natl Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2110094118 (2021).
Maverick, E. E. & Tamkun, M. M. High spatial density is associated with non-conducting Kv channels from two families. Biophys. J. 121, 755–768 (2022).
O’Connell, K. M., Loftus, R. & Tamkun, M. M. Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc. Natl Acad. Sci. USA 107, 12351–12356 (2010).
Du, J., Tao-Cheng, J. H., Zerfas, P. & McBain, C. J. The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 84, 37–48 (1998).
Kirmiz, M., Vierra, N. C., Palacio, S. & Trimmer, J. S. Identification of VAPA and VAPB as Kv2 channel-interacting proteins defining endoplasmic reticulum-plasma membrane junctions in mammalian brain neurons. J. Neurosci. 38, 7562–7584 (2018).
Scannevin, R. H., Murakoshi, H., Rhodes, K. J. & Trimmer, J. S. Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2.1 K+ channel. J. Cell Biol. 135, 1619–1632 (1996).
Trimmer, J. S. Immunological identification and characterization of a delayed rectifier K+ channel polypeptide in rat brain. Proc. Natl Acad. Sci. USA 88, 10764–10768 (1991).
Lim, S. T., Antonucci, D. E., Scannevin, R. H. & Trimmer, J. S. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 25, 385–397 (2000).
Park, K. S., Mohapatra, D. P., Misonou, H. & Trimmer, J. S. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science 313, 976–979 (2006).
Misonou, H. et al. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 7, 711–718 (2004).
Misonou, H. et al. Bidirectional activity-dependent regulation of neuronal ion channel phosphorylation. J. Neurosci. 26, 13505–13514 (2006).
Benndorf, K., Koopmann, R., Lorra, C. & Pongs, O. Gating and conductance properties of a human delayed rectifier K+ channel expressed in frog oocytes. J. Physiol. 477, 1–14 (1994).
Guarina, L. et al. Biological noise is a key determinant of the reproducibility and adaptability of cardiac pacemaking and EC coupling. J. Gen. Physiol. 154, https://doi.org/10.1085/jgp.202012613 (2022).
Cobb, M. M., Austin, D. C., Sack, J. T. & Trimmer, J. S. Cell cycle-dependent changes in localization and phosphorylation of the plasma membrane Kv2.1 K+ channel impact endoplasmic reticulum membrane contact sites in COS-1 cells. J. Biol. Chem. 291, 5527 (2016).
Hernandez, G. H. et al. A computational model predicts sex-specific responses to calcium channel blockers in mesenteric vascular smooth muscle. eLife 12, RP90604 (2023).
Marquis, M. J. & Sack, J. T. Mechanism of use-dependent Kv2 channel inhibition by RY785. J. Gen. Physiol. 154, https://doi.org/10.1085/jgp.202112981 (2022).
Herrington, J. et al. Identification of novel and selective Kv2 channel inhibitors. Mol. Pharmacol. 80, 959–964 (2011).
Tilley, D. C. et al. The tarantula toxin GxTx detains K(+) channel gating charges in their resting conformation. J. Gen. Physiol. 151, 292–315 (2019).
Fredriksson, S. et al. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 20, 473–477 (2002).
Koos, B. et al. Analysis of protein interactions in situ by proximity ligation assays. Curr. Top Microbiol. Immunol. 377, 111–126 (2014).
Kirmiz, M. et al. Remodeling neuronal ER-PM junctions is a conserved nonconducting function of Kv2 plasma membrane ion channels. Mol. Biol. Cell 29, 2410–2432 (2018).
Navedo, M. F., Amberg, G. C., Nieves, M., Molkentin, J. D. & Santana, L. F. Mechanisms underlying heterogeneous Ca2+ sparklet activity in arterial smooth muscle. J. Gen. Physiol. 127, 611–622 (2006).
Navedo, M. F. et al. Ca(v)1.3 channels produce persistent calcium sparklets, but Ca(v)1.2 channels are responsible for sparklets in mouse arterial smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 293, H1359–H1370 (2007).
Navedo, M. F. et al. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ. Res. 106, 748–756 (2010).
Antonucci, D. E., Lim, S. T., Vassanelli, S. & Trimmer, J. S. Dynamic localization and clustering of dendritic Kv2.1 voltage-dependent potassium channels in developing hippocampal neurons. Neuroscience 108, 69–81 (2001).
Cerda, O. & Trimmer, J. S. Activity-dependent phosphorylation of neuronal Kv2.1 potassium channels by CDK5. J. Biol. Chem. 286, 28738–28748 (2011).
Johnson, B. et al. Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc. Natl Acad. Sci. USA 115, E7331–e7340 (2018).
Di Mattia, T. et al. FFAT motif phosphorylation controls formation and lipid transfer function of inter-organelle contacts. EMBO J. 39, e104369 (2020).
Fox, P. D., Loftus, R. J. & Tamkun, M. M. Regulation of Kv2.1 K(+) conductance by cell surface channel density. J. Neurosci. 33, 1259–1270 (2013).
Stuart, G. & Sakmann, B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron 15, 1065–1076 (1995).
Issa, N. P. & Hudspeth, A. J. Clustering of Ca2+ channels and Ca(2+)-activated K+ channels at fluorescently labeled presynaptic active zones of hair cells. Proc. Natl Acad. Sci. USA 91, 7578–7582 (1994).
Takekura, H., Sun, X. & Franzini-Armstrong, C. Development of the excitation-contraction coupling apparatus in skeletal muscle: peripheral and internal calcium release units are formed sequentially. J. Muscle Res. Cell Motil. 15, 102–118 (1994).
Dixon, R. E. & Trimmer, J. S. Endoplasmic reticulum-plasma membrane junctions as sites of depolarization-induced Ca(2+) signaling in excitable cells. Annu. Rev. Physiol. 85, 217–243 (2023).
Nirenberg, V. A. & Yifrach, O. Bridging the molecular-cellular gap in understanding ion channel clustering. Front. Pharmacol. 10, 1644 (2019).
Dixon, R. E., Navedo, M. F., Binder, M. D. & Santana, L. F. Mechanisms and physiological implications of cooperative gating of clustered ion channels. Physiol. Rev. 102, 1159–1210 (2022).
Kloner, R. A., Sowers, J. R., DiBona, G. F., Gaffney, M. & Wein, M. Sex- and age-related antihypertensive effects of amlodipine. Amlodipine Cardiovasc. Community Trial Study Group Am. J. Cardiol. 77, 713–722 (1996).
Zanchetti, A. et al. Outcomes in subgroups of hypertensive patients treated with regimens based on valsartan and amlodipine: An analysis of findings from the VALUE trial. J. Hypertens 24, 2163–2168 (2006).
van Luik, E. M. et al. Sex differences in the anti-hypertensive effect of calcium-channel blockers: a systematic review and meta-analysis. Biomedicines 11, 1622 (2023).
Bindels, D. S. et al. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14, 53–56 (2017).
Olmsted, J. B. Affinity purification of antibodies from diazotized paper blots of heterogeneous protein samples. J. Biol. Chem. 256, 11955–11957 (1981).
Acknowledgements
We thank Dellaney Rudolph-Gandy and Ernesto Javier Rivera for technical assistance. We thank Dr. Joshua Tulman for assistance with the diagram. This work was supported by grants from US National Institutes of Health HL085686 (L.F.S.), US National Institutes of Health HL128537 (L.F.S., C.E.C.), US National Institutes of Health HL144071 (L.F.S., J.S.T.), US National Institutes of Health NS114210 (L.F.S., J.S.T.), US National Institutes of Health 1OT2OD026580 (L.F.S., C.E.C.), Amazon AWS Cloud Credits for Research (G.H.H.).
Author information
Authors and Affiliations
Contributions
Conceptualization: C.M., L.F.S., J.S.T. Methodology: C.M., L.F.S., N.C.V., J.S.T. Investigation: C.M., S.C.O., D.M., G.H.H., P.R., Z.F., D.S. Formal Analysis: C.M., S.C.O., D.M., P.R., Z.F., L.F.S. Visualization: C.M. Data Curation: G.H.H., D.S. Software: G.H.H., D.S. Supervision: L.F.S., C.E.C., J.S.T. Writing—original draft: C.M., L.F.S. Writing—review & editing: C.M., S.C.O., D.M., G.H.H., P.R., Z.F., N.C.V., J.S.T., L.F.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Mark Nelson, Osama Harraz and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Mark Collins and Manuel Breuer. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matsumoto, C., O’Dwyer, S.C., Manning, D. et al. The formation of KV2.1 macro-clusters is required for sex-specific differences in L-type CaV1.2 clustering and function in arterial myocytes. Commun Biol 6, 1165 (2023). https://doi.org/10.1038/s42003-023-05527-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05527-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
