
Abstract
Oral lichen planus (OLP), a chronic inflammatory disorder, is characterized by the massive cell apoptosis in the keratinocytes of oral mucosa. However, the mechanism responsible for triggering oral keratinocyte apoptosis is not fully explained. Here, we identify that Gasdermin C (GSDMC) downregulation contributes to apoptosis in human oral keratinocytes. Mechanistically, we describe that activated nuclear factor kappa B (NF-κB) pathway induces overexpression of methyltransferase-like 14 (METTL14), which increases N6-adenosine methylation (m6A) levels in the epithelial layer of OLP. m6A modification is capable of regulating primary miR-6858 processing and alternative splicing, leading to miR-6858 increases. miR-6858 can bind and promote GSDMC mRNA degradation. Forced expression of GSDMC is able to rescue cell apoptosis in human oral keratinocyte models resembling OLP. Collectively, our data unveil that m6A modification regulates miR-6858 production to decrease GSDMC expression and to trigger keratinocyte apoptosis in the context of OLP.
Similar content being viewed by others


Introduction
Oral lichen planus (OLP) is considered to be a chronic inflammation-related disorder affecting oral mucosa1. The range of OLP prevalence is between 0.22% and 5% across the world2. OLP patients often suffer from these clinical symptoms such as burning, soreness and pain3,4. These symptoms become worsened amid chewing or tooth brushing, limiting patients’ daily activities3,4. Albeit the pathogenesis of OLP remains elusive, autoimmune response, genetic factors, infectious agents and psychological factors all contribute to the onset and development of OLP4. To date, OLP is identified to be initiated by basal keratinocytes apoptosis triggered by T lymphocytes in the oral stratified squamous epithelium5,6,7. Exploring the cell apoptosis process of oral keratinocytes might be an efficient strategy for OLP management.
Post-transcriptional modifications of RNAs are regarded as an indispensable mechanism regulating gene expression8. In mammalian cells, N6-adenosine methylation (m6A) of polyadenylated RNAs is considered to be the most prevalent internal modification in mRNAs at the post-transcriptional level9. The adenosine nucleotides regulated by m6A methylation in mammalian RNAs are approximately 0.4%, with 3–5 m6A modifications on average in each mRNA10,11. The deposition of m6A modification is organized in the mammalian cells’ transcriptome. The m6A sites have a consensus motif RRm6ACH [(G/A/U)(G > A)m6AC(U > A > C)], with enrichment in the coding sequence (CDS) and 3’untranslated region (3’UTR)12,13,14. RNA m6A modification is regulated by writers, erasers and readers dynamically to achieve a reversible balance. Writers are methyltransferases responsible for installing the methyl group on the N6 position of adenosine, while erasers are demethylases which remove methyl group from it reversibly. Readers are proteins accounting for interacting with and recognizing the m6A modification sites15. The methyltransferase of m6A modification is a complex consisting of Wilms tumor 1-associated protein (WTAP), methyltransferase-like 3 (METTL3) and METTL14. AlkB homolog 5 (ALKBH5) and fat mass and obesity-associated protein (FTO) are the two m6A erasers identified so far10,11. Three groups of m6A readers, YT521-B homology (YTH) domain-containing proteins (YTHDF1/2/3 and YTHDC1/2), heterogeneous nuclear ribonucleoproteins (HNRNPA2B1 and HNRNPC) and insulin-like growth factor 2 mRNA-binding proteins (IGF2BP1/2/3), utilizes RNA binding domains and RNA recognition motif to determine the fate of the modified mRNAs15,16. Although m6A modification is identified to regulate tumor growth, inflammatory responses and stem cell pluripotency17,18,19, its roles in OLP initiation and development remain unanswered.
Gasdermin (GSDM) family includes GSDMA/B/C/D/E and Pejvakin, which are highly expressed in keratinocytes and intestinal epithelial cells20,21. The main role of GSDM family is identified to enhance pyroptotic cell death22,23,24. However, recent studies have suggested that GSDMC in cancer cells is capable of switching apoptosis to pyroptosis, thereby leading to tumor necrosis25. Given that cell apoptosis is a typical feature of OLP, we set to investigate the functions of GSDMC in oral keratinocytes in the context of OLP. In this study, our results reveal that up-regulation of METTL14 in the setting of OLP promotes m6A methylation of primiR-6858, which results in miR-6858 overexpression. Increased miR-6858 targets and decreases GSDMC mRNA to induce cell apoptosis in oral keratinocytes.
Results
GSDMC expression is decreased in the keratinocytes of OLP
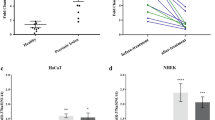
Since GSDMC expression in the epithelium of OLP is not clear, we detected the levels of it by real-time PCR and western blot. As shown in Fig. 1, both mRNA and protein levels of GSDMC were decreased largely in the epithelia derived from OLP biopsies compared to those from healthy controls (Fig. 1a–c). To establish cell models mimicking OLP in vitro, we used both lipopolysaccharides (LPS) and the supernatants of culture medium from activated CD4+ T cells to challenge human oral keratinocytes (HOKs)26,27. Consistently, GSDMC expression showed a remarkable decrease in the two cell models at both mRNA and protein levels (Fig. 1d–i). Importantly, GSDMC expression was down-regulated in a time course-dependent manner upon LPS or activated CD4+ T cells treatment (Supplementary Fig. 1), indicating that these stimulators decreased GSDMC levels in oral keratinocytes.

a GSDMC mRNA levels in oral epithelial layers from healthy or diseased tissues determined by real-time PCR, n = 14. Western blot (b) and densitometric analysis (c) showing GSDMC protein levels in oral epithelial layers from healthy or diseased tissues, n = 14. Data (b) are representative of 14 samples. GSDMC mRNA or protein expression detected by real-time PCR (d), western blot (e) or densitometric quantitation (f) in HOKs with or without 12-hour LPS treatment (100 ng/ml), n = 3. Data (e) are representative of 3 samples. GSDMC mRNA or protein levels measured by real-time PCR (g), western blot (h) or densitometric quantitation (i) in HOKs with or without 12-hour activated CD4+ T cells treatment, n = 3. Data (h) are representative of 3 samples. **P < 0.01, ***P < 0.001 vs corresponding health or control group; Ctrl, control. Data were expressed as means ± standard deviation. All experiments were carried out at least 3 times. Student’s t test (a, c, d, f, g and i) was performed for the statistical analysis.
Down-regulation of GSDMC induces cell apoptosis in oral keratinocytes
As previous studies suggested that GSDMC plays a critical role in cell apoptosis in tumor cells25, we investigated the effects of GSDMC on cell death in HOKs. The propidium iodide (PI) staining has been used for the assessment of cell apoptosis in different experimental models widely28. PI is able to bind to double stranded DNA in dead cells with defective plasma membranes, but is excluded from alive cells28. After binding with DNA, the quantum yield of PI is increased 20-30 fold and can be detected by fluorescence microscope28,29. As exhibited in Fig. 2, PI staining data demonstrated that most of oral keratinocytes were dead upon GSDMC knockdown (Fig. 2a). Cell death of HOKs was further confirmed by enzymatic assays (Fig. 2b). To explain the manner of cell death induced by GSDMC deficiency, we generated Caspase3/7 double knockout (dKO), Gasdermin D (GSDMD) KO, Mixed Lineage Kinase Domain Like Pseudokinase (MLKL) KO, BAX KO or BAK KO HOKs to investigate the roles of apoptosis, pyroptosis and necroptosis in HOKs’ death30 (Supplementary Fig. 2a). Of note, blockage of apoptosis pathway decreased HOKs’ death upon the loss of GSDMC (Fig. 2c, d). Moreover, caspase 3 activity, cleaved caspase 3 and cleaved caspase 9 levels were all elevated in HOKs after shGSDMC-lentivirus infection (Fig. 2e, f), noting that GSDMC knockdown induced cell apoptosis in HOKs. The apoptosis inhibitor, Q-VD-OPh, rescued deficient GSDMC-induced cell death (Fig. 2g, h). In comparison to our previous data27,31, we found that GSDMC expression showed positive correlations with the levels of antiapoptotic factors such as vitamin D receptor (VDR), miR-26a/b, B-cell lymphoma-2 (BCL-2) in the epithelial layers of human biopsies, but had negative correlations with miR-802 and cleaved caspase 3 which are known as pro-apoptotic factors (Supplementary Fig. 2b). Importantly, mitochondrial membrane potential was changed after GSDMC knockdown (Supplementary Fig. 3a, b), indicating the mitochondrial dysfunction. Overall, these findings uncover that GSDMC plays a protective role in oral keratinocytes in the setting of OLP. The excessive cell apoptosis in the epithelial layer of OLP mucosa has been identified in our previous studies31.

a PI staining and quantitative analysis in HOKs transduced with control- or shGSDMC-lentivirus. b Cell death detection of HOKs transduced with control- or shGSDMC-lentivirus. c PI staining analysis and cell death measurement of HOKs with GSDMC knockdown and caspase3/7, MLKL or GSDMD knockout. d PI staining analysis and cell death measurement of HOKs with GSDMC knockdown and BAX or BAK knockout. Caspase 3 activity and proteins levels in HOKs transduced with control- or shGSDMC-lentivirus evaluated by caspase 3 activity assay (e) or western blot (f). PI staining analysis (g) and cell death measurement (h) of HOKs transduced with control- or shGSDMC-lentivirus. HOKs were pretreated with or without Q-VD-OPh (100 nM) for 4 h. ***P < 0.001 vs corresponding control group; ##P < 0.01, ###P < 0.001 vs corresponding LPS or CD4 group; n = 3; Ctrl, control; KO, knockout; dKO, double knockout. Data were expressed as means ± standard deviation. All experiments were carried out at least 3 times. Student’s t test (a, b, e) and one-way ANOVA (c, d, g, h) were used for statistical analysis.
GSDMC protects HOKs from apoptosis in the cell models of OLP
To test whether GSDMC overexpression is able to block cell apoptosis, we treated HOKs with GSDMC-expressing lentivirus in the presence of LPS or activated CD4+ T cells. Accordantly, forced expression of GSDMC relieved cell death and pro-apoptotic pathway activation induced by challenges (Fig. 3a–h and Supplementary Fig. 3c, d). Interestingly, the overexpression of GSDMC in HOKs did not lead to cell death (Fig. 3a, b), indicating GSDMC, at least itself alone, could not trigger pyroptosis in HOKs.

PI staining analysis (a), cell death measurement (b), caspase 3 activity assay (c) or western blot (d) in control- or GSDMC-lentivirus-transduced HOKs with or without 12-hour LPS challenge (100 ng/ml). PI staining analysis (e), cell death measurement (f), caspase 3 activity assay (g) or western blot (h) in control- or GSDMC-lentivirus-transduced HOKs with or without 12-hour activated CD4+ T cells stimulation (30% final volumetric concentration). **P < 0.01, ***P < 0.001 vs corresponding control group; ##P < 0.01, ###P < 0.001 vs corresponding LPS or CD4 group; n = 3; Data were expressed as means ± standard deviation. All experiments were carried out at least 3 times. One-way ANOVA was used for statistical analysis.
m6A methylation levels are enhanced in the epithelial layer of OLP tissues due to the boosted METTL14 transcripts
m6A modification is reported to be a prevalent way to regulate gene expression10. To determine whether GSDMC expression was mediated by m6A methylation in oral keratinocytes, we detected m6A status in the epithelia of human specimens. Of interest, m6A modification levels showed a robust increase in the epithelial layer of OLP samples compared to that of controls (Fig. 4a), and also in the HOKs with treatments as indicated (Supplementary Fig. 4a, b). To explore the underpinning by which m6A is increased, we measured the writers and erasers expression in the oral keratinocytes of human samples. As shown, only METTL14 levels were changed considerably in the diseased samples among all of the methyltransferases and demethylases (Fig. 4b and Supplementary Fig. 4c–f). METTL14 protein levels were also increased in the diseased samples (Fig. 4c, d). In accordance, METTL14 expression was up-regulated in HOKs upon LPS or activated CD4+ T cells treatment (Supplementary Fig. 4g–j). Forced expression of METTL14 in HOKs was capable of elevating m6A levels (Fig. 4e, f). Next, to search the underpinning of METTL14’s upregulation, we screened the promoter region of human METTL14 and found a putative nuclear factor-κB (NF-κB) binding site (Fig. 4g). Chromatin immunoprecipitation (ChIP) data showed that NF-κB p65 selectively bound with this putative κB site in HOKs using a primer flanking the potential motif (Fig. 4h). Overexpression of IκB kinase β (IKKβ) which can activate NF-κB pathway improved METTL14 levels in HOKs (Fig. 4i, j). These results suggest that m6A methylation is increased in oral keratinocytes owing to METTL14 upregulation in the context of OLP. The activated NF-κB pathway in the epithelial layer of OLP biopsies has been confirmed in our previous studies32.

a Quantitation of m6A level in total RNAs isolated from oral keratinocytes of human mucosa, n = 14. METTL114 mRNA or protein expression assessed by real-time PCR (b), western blot (c) or densitometric quantitation (d) in oral keratinocytes of human samples, n = 14. METTL14 or m6A levels in HOKs transduced with control- or METTL14-expressing lentivirus determined by western blot (e) or m6A quantification assay (f), n = 3. g Schematic showing the potential NF-κB motif in the promoter of human METTL14 gene. h ChIP assays of HOKs against control or anti-p65 antibody, n = 3. METTL14 mRNA or protein expression in HOKs with control- or IKKβ-expressing lentivirus determined by real-time PCR (i) or western blot (j), n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 vs corresponding control or health group; Ctrl, control. Data were expressed as means ± standard deviation. All experiments were carried out at least 3 times. Student’s t test (a, b, d, e, f, h, i and j) was used for statistical analysis.
To interrogate whether the GSDMC mRNA in HOKs is regulated by RNA m6A methylation, we mined the online m6A-IP-sequencing database (GSE213714). After analysis, we found 4 putative m6A sites in the GSDMC mRNA from human oral keratinocytes (Supplementary Fig. 4k). However, the levels of m6A-related RNA fragments were comparable in IgG control and m6A groups (Supplementary Fig. 4l), indicating m6A methylation did not directly contribute to GSDMC mRNA decreases in HOKs.
miR-6858 targets and degrades GSDMC mRNA in HOKs
miRNAs are known to regulate gene expression in mammalian cells33. Here, targetscan database (http://www.targetscan.org/vert_72/) was used to predict the potential miRNAs responsible for GSDMC mRNA decreases. Based on the target score, we selected the top 5 miRNAs closely related to human GSDMC. Among them (miR-4795-3p, miR-548ax, miR-548ao-5p, miR-4689, miR-6858), only miR-6858 levels were boosted in the diseased human biopsies as well as the serum and saliva derived from OLP patients compared to those from healthy participants (Fig. 5a and Supplementary Fig. 5a–d). In line with these data relative to human samples, LPS or activated CD4+ T cells could induce miR-6858 levels in HOKs as well (Supplementary Fig. 5e, f). The miR-6858-targeted site in human GSDMC mRNA was exhibited in Fig. 5b. To verify the regulatory role of miR-6858 in GSDMC mRNA stability, we inserted the fragment containing the miR-6858-targeted site in the 3’UTR of GSDMC mRNA for luciferase assays. As shown, luciferase activity of HOKs transfected with pGL3-GSDMC carrying the miR-6858-targeted site, but not that with pGL3-GSDMCmut, was suppressed by forced expression of miR-6858 (Fig. 5c), confirming miR-6858 promoted GSDMC mRNA degradation. Next we applied gain-of-function and loss-of-function assays to detect the effects of miR-6858 on GSDMC expression. Accordantly, miR-6858 overexpression down-regulated GSDMC levels largely in HOKs at both mRNA and protein levels which were confirmed by real-time PCR and western blot assays (Fig. 5d, e and Supplementary Fig. 5g, h). On the contrary, miR-6858 inhibitors reversed LPS- or activated CD4+ T cells-induced GSDMC decreases in HOKs (Fig. 5f–i and Supplementary Fig. 5i–k).

a m6A quantitation assays showing miR-6858 levels in oral keratinocytes of biopsies, serum and saliva derived from human individuals, n = 14. b Schematic showing the predicted miR-6858 target site in the 3’UTR of human GSDMC mRNA. c Luciferase activities of HOKs transfected with control or miR-6858 mimics, pGL3-Promoter, pGL3-GSDMC or pGL3-GSDMCmut plasmids as indicated, n = 3. GSDMC levels in HOKs with or without miR-6858 mimics evaluated by real-time PCR (d) or western blot (e), n = 3. Real-time PCR (f) or western blot (g) analyses showing GSDMC levels in control or miR-6858 inhibitors-transfected HOKs in the presence or absence of LPS treatment, n = 3. Real-time PCR (h) or western blot (i) analyses showing GSDMC expression in control or miR-6858 inhibitors-transfected HOKs in the presence or absence of activated CD4+ T cells treatment, n = 3. The concentrations of microRNA oligonucleotides or plasmids are 200 nM or 500 ng, respectively. **P < 0.01, ***P < 0.001 vs corresponding control or health group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs corresponding LPS or CD4 group; Ctrl, control. Data were expressed as means ± standard deviation. All experiments were carried out at least 3 times. Student’s t test (a, d) and one-way ANOVA (c, f, h) were used for statistical analysis.
m6A methylation of primiR-6858 modulates the processing of miR-6858 in HOKs
To uncover the mechanism by which miR-6858 is upregulated in OLP, we screened the sequence of primiR-6858 using SRAMP database and found two m6A motifs, one is within premiR-6858 sequence and the other one is not (Supplementary Fig. 6a). we then designed primers flanking the m6A motif (GGACA) in the primiR-6858, but not in the premiR-6858, to perform m6A immunoprecipitation (IP)-quantitative PCR (qPCR). As shown in Fig. 5S, m6A antibody highly bound with this m6A site in HOKs (Supplementary Fig. 6b). Next, we used single-stranded methylated RNA bait (ss-m6A) or unmethylated control RNA (ss-A) to perform RNA pull-down assay (Supplementary Fi. 6c). m6A writers, METTL14 and METTL3, were confirmed to interact with the methylated RNA bait selectively with a higher affinity compared to the unmethylated control group (Supplementary Fig. 6d).
m6A methylation has an effect on primary miRNA processing and splicing34. We next overexpressed or knocked down METTL14 or METTL3 levels in HOKs to observe primiR-6858 and miR-6858 expression separately (Fig. 4e and Supplementary Fig. 6e–h). Forced expression of METTL14 or METTL3 enhanced miR-6858 level and inhibited primiR-6858 expression in HOKs (Fig. 6a, b), while knockdown of m6A writer played a reversed role in them (Fig. 6c, d). A R298P mutant METTL14 and a D394A/W397A METTL3 mutant are enzymatically inactive and defective in regulating RNA m6A modification35. In this investigation, mutant METTL14 or METTL3 did not change or rescue primiR-6858 and miR-6858 levels as wild type METTL14 or METTL3 did (Fig. 6a–d), suggesting the modulation of primiR-6858 was dependent on RNA m6A methylation. Overexpression of FTO, a demethylase, had the opposite functions in miRNAs compared to METTL14 or METTL3 (Fig. 6e and Supplementary Fig. 6i). HNRNPA2B1 is reported to be a reader protein in the m6A modification of primary miRNAs34. Similar to previous studies34, we found HNRNPA2B1 could bind with m6A site in the primiR-6858 in HOKs and regulate miR-6858 expression (Supplementary Fig. 6j, k and Fig. 6f).

Real-time PCR showing primiR-6858 or miR-6858 expression in HOKs infected with wild type/ mutant METTL14- (a) or METTL3- (b) lentivirus. c Real-time PCR showing primiR-6858 or miR-6858 expression in HOKs with shMETTL14-, wild type METTL14- or mutant METTL14-lentivirus as shown. d Real-time PCR analyses of primiR-6858 or miR-6858 expression in HOKs infected with shMETTL3-, wild type METTL3- or mutant METTL3-lentivirus as shown. e, f Primary miR-6858 or miR-6858 levels in HOKs with FTO- or shHNRNPA2B1-lentivirus determined by real-time PCR. n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 vs corresponding control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs corresponding shMETTL14 or shMETTL3 group. Data were expressed as means ± standard deviation. All experiments were carried out at least 3 times. Student’s t test (e and f) and one-way ANOVA (a, b, c and d) were used for statistical analysis.
Inhibition of miR-6858 or METTL14 rescues cell apoptosis in oral keratinocytes
As METTL14-induced miR-6858 overexpression can promote GSDMC mRNA degradation to trigger cell apoptosis in HOKs, we next sought to determine whether METTL14 or miR-6858 inhibition can attenuate cell apoptosis under OLP conditions. As shown, LPS or activated CD4+ T cells-induced cell apoptosis was ameliorated in METTL14-knockdown cells (Fig. 7a, b), and so was it in cells with miR-6858 inhibition (Fig. 7c, d).

Caspase 3 activity and protein expression in control- or shMETTL14-lentivirus-infected HOKs with 12-hour LPS (a) or activated CD4+ T cells (b) treatment. Caspase 3 activity and protein expression in 200 nM control- or miR-6858 inhibitors-transfected HOKs with 12-hour LPS (c) or activated CD4+ T cells (d) treatment. n = 3. **P < 0.01, ***P < 0.001 vs corresponding control group; ##P < 0.01, ###P < 0.001 vs corresponding LPS or CD4 group. Data were expressed as means ± standard deviation. All experiments were carried out at least 3 times. One-way ANOVA (a, b, c and d) was used for statistical analysis.
GSDMC overexpression rescues METTL14- or miR-6858-induced cell apoptosis
We further evaluated whether overexpression of METTL14 or miR-6858 could induce cell apoptosis in HOKs as LPS or activated CD4+ T cells did. Intriguingly, either METTL14- or miR-6858 overexpression triggered cell death and pro-apoptotic factors production in HOKs (Fig. 8a–e). However, forced expression of GSDMC rescued METTL14- or miR-6858-induced cell apoptosis (Fig. 8a–e), indicating METTL14- or miR-6858 initiated cell apoptosis in a GSDMC-dependent manner.

PI staining (a) and analysis (b), cell death measurement (c), caspase 3 activity assay (d) or western blot (e) in HOKs with METTL14-expressing lentivirus or miR-6858 mimics in the presence or absence of GSDMC-expressing lentivirus infection. f Schematic illustration of the m6A methylation-miR-6858-GSDMC axis in oral keratinocyte. The concentration of microRNA oligonucleotides is 200 nM. n = 3, **P < 0.01, ***P < 0.001 vs corresponding control group; ###P < 0.001 vs corresponding METTL14 or miR-6858 group. Data were expressed as means ± standard deviation. All experiments were carried out at least 3 times. One-way ANOVA was used for statistical analysis.
Discussion
Unlike other members of GSDM family (GSDMD and GSDME), the function of GSDMC in mammalian cells is still not fully explained36. In this study, we confirmed that GSDMC expression shows a decrease in the keratinocytes of OLP and the downregulation of GSDMC in oral keratinocytes results in cell apoptosis, consistent with other studies noting that GSDMC knockout is associated with apoptosis in tumor cell25. Moreover, GSDMC knockdown exhibits the ability to reduce colony formation in DLD-1 and LoVo cell lines37, indicating GSDMC deficiency impedes cell proliferation in colon cancer. Mechanistically, the loss of GSDMC in colonic epithelial cells disrupted mitochondrial membrane potential and triggered cytochrome c release to activate the pro-apoptotic pathway38. Therefore, GSDMC may regulate cell apoptosis in oral keratinocytes through affecting mitochondrial homeostasis. We hypothesize that GSDMC may influence the stability of apoptosis-related factors such as Bax and Bak, and this hypothesis needs to be confirmed by more extra data. However, overexpression of GSDMC is reported to initiate pyroptosis in intestine39, confirming the conclusion that GSDMC switches apoptosis to pyroptosis in cancer cells.
GsdmC is suggested to be directly fine-tuned by RNA m6A methylation in mouse colonic epithelial cells. On the contrary, we found that GSDMC is indirectly mediated by RNA m6A methylation in human oral keratinocytes. This discrepancy might be due to the differences on cell types or species. The reports concerning miR-6858 are rare, only one paper suggests that miR-6858 regulates the process of melatonin inhibition in glioma40. Herein, we indicate that GSDMC decreases in oral keratinocytes is due to the upregulation of miR-6858 levels in the context of OLP. miR-6858 targets the 3’UTR of GSDMC mRNA to promote its degradation, in alignment with other investigations demonstrating miRNAs play critical roles in OLP27,31. Furthermore, consistent with other studies claiming m6A modification regulates primary microRNA processing and alternative splicing34, we elucidated that elevated METTL14 in oral keratinocytes binds to m6A site of primiR-6858 to facilitate its processing, leading to miR-6858 upregulation. We also used METTL14 or METTL3 mutant to confirm that the regulation of primiR-6858 is dependent on m6A modification since the roles of METTL14 or METTL3 are not only limited in RNA methylation35.
However, overexpression of GSDMC or inhibition of miR-6858 can not completely rescue cell apoptosis in OLP cell models because other factors or pathways may be involved in the process of cell death. In contrast to intestinal tissues, overexpression of GSDMC fails to trigger pyroptosis in oral keratinocytes, implying the distinct functions of GSDMC in different tissues.
In this exploration, we identified a factor GSDMC which is related to oral keratinocyte apoptosis in the setting of OLP. Mechanistically, m6A methylation has a regulatory effect on primiR6858 to decrease GSDMC expression but not on GSDMC itself (Fig. 8f). Although we are unable to carry out experiments in vivo due to the lack of animal models mimicking OLP so far, the human and HOKs data we provided here are compelling to help us make conclusions. Given RNA methylation’s roles in cell apoptosis, FTO inhibitors might be helpful for OLP management.
Methods
Human samples
Human oral mucosa, blood and saliva were obtained from healthy and OLP participants in the Hospital of Stomatology of Shanxi Medical University31. OLP inclusion and exclusion criteria were according to the modified World Health Organization (WHO) diagnostic rules. This project was approved by the Institutional Ethical Committee of Shanxi Medical University (# 2016LL046). Informed consent from each individual was obtained and all ethical regulations relevant to human research participants were followed. Details of OLP patients’ information was listed in previous studies31.
Cell culture
HOKs were obtained from Chinese Shanghai Institution and cultured in plates using oral keratinocyte medium supplemented with fetal bovine serum (10%) and penicillin/streptomycin (1%). OLP is identified to be mainly triggered by T lymphocytes, the main lymphocytes involved are CD4+ and CD8 + T cells41. In addition, bacterial infection is considered to be a prominent contributing factor to OLP27. To this end, we used two methods to resemble OLP in vitro. First, lipopolysaccharides (LPS, 100 ng/ml, Sigma-Aldrich, St. Louis, MO) was added into the culture medium of HOKs for 12 h. Second, the supernatants from anti-CD3/28-activated CD4+ T cells were added into the culture medium of HOKs for 12 h at a 30% final volumetric concentration27. Isolation and activation of T cells from human were performed according to a protocol27. In brief, peripheral blood samples were collected and CD4 + T cells were enriched by anti-human CD4 magnetic particles (BD Biosciences, Franklin Lakes, NJ, USA). Anti-human CD3 and anti-human CD28 antibodies (BD Biosciences Pharmingen, SanDiego, CA, USA) were selected to activate CD4 + T cells. In another experiment, HOKs were challenged for 0, 4, 8, 12, hours by LPS or activated CD4+ T cells, respectively. For miR-6858 mimics or inhibitors (mybiosource, San Diego, CA) treatment, HOKs were transfected with 200 nM oligonucleotides for 36 h.
Oral mucosal epithelium isolation
Oral mucosal epithelium was separated from buccal tissues which were digested in 0.25% dispase II for 12 h by muscle forceps as reported27.
Western blotting
Proteins from human biopsies or HOKs were isolated using laemmli buffer. The same amounts of protein samples were separated by SDS–PAGE and subsequently transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore, Burlington, MA). After being blocked by 5% non-fat milk, membranes were treated with first and secondary antibodies. The information of primary antibodies was listed in Supplementary Table 1.
RT-PCR
Total RNAs from human tissues or HOKs were harvested using TRIzol (Invitrogen, Waltham, MA). The PrimeScript RT Reagent Kit (TaKaRa, Dalian, China) was used to synthesize the first strand cDNAs. The SYBR Premix Ex Kit (TaKaRa) was selected to perform qPCR in a real-time PCR system (Roche480). For miRNAs analyses, the miRNA isolation Kit (QIAGEN, Hilden, Germany) was chosen to extract miRNAs from human samples or cells. The specific miRNA First-strand cDNA Synthesis Kit (Aidlab Biotechnologies, Beijing, China) and a miRNA real-time PCR Assay Kit (Aidlab Biotechnologies) were applied to carry out the first strand cDNAs synthesis and qPCR, respectively. The relative amounts of transcripts were calculated using the 2-ΔΔCt formula. GAPDH or U6 were regarded as internal control for mRNA or miRNA examinations. The equal amount of exogenous cel-miR-39 was served as internal control for circulating miRNAs in human serum and saliva. The primers for qPCR were listed in Supplementary Table 2.
Lentiviral and plasmid constructs
The coding sequence of human METTL14, METTL3, FTO, GSDMC-FL, cDNA was amplified and subcloned into the pLV[Exp]-Neo-EF1A lentiviral vector (VectorBuilder, Chicago, IL). Site-specific mutation of METTL14 or METTL3 was generated using a QuickChange Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA)35. For luciferase reporter plasmids construction, DNA sequence (50 bp) containing the potential target site of miR-6858 in the 3′UTR of human GSDMC cDNA was synthesized and inserted in pGL3-Promoter vector (Promega, Madison, WI). The inserted sequence is: GAGTTTATATATTTGTCAAAACTCCTCAAATAGTATGTTAAAGACGTAAG. pGL3-GSDMCmut was constructed by mutating the target sequence 5’CUCCUCAA3’ to 5’CUGGGCAA3’ in pGL3-GSDMC plasmid by a Mutagenesis Kit. The METTL14, METTTL3 and HNRNPA2B1 shRNAs were obtained from Sigma and co-transfected with lentivirus packaging plasmids to generate lentivirus in HEK293T cells. A shRNA for HNRNPA2B1 (TRCN0000001058), a shRNA specific targeting METTL3 (TRCN0000034715), a shRNA specific for METTL14 (TRCN0000015933) and a control shRNA (SHC002) were used for experiments. The knockdown efficiencies of purchased shRNAs were all validated. Plasmids carrying GSDMC shRNA sequence were subcloned in pLV[shRNA]-EGFP-U6 lentiviral backbone (VectorBuilder)37. Scrambled shRNAs were served as controls for gene knockdown experiments (CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG). Empty vectors were used as controls for gene overexpression experiments. Negative miRNA mimic or inhibitor controls (Mybiosource) were selected for miRNA assays. All cloned DNA fragments and mutations were confirmed by DNA sequencing.
Transfection and transduction assays
MicroRNA oligonucleotides (200 nM) and plasmids were transfected transiently into HOK by Lipofectamin 3000 Reagent (Invitrogen). For transduction, lentivirus particles were added into HOKs at 10 MOI supplemented with 6 µg/ml polybrene for 36-hour culture.
Luciferase reporter assay
Luciferase reporter assays were carried out according to the protocol mentioned before with minor modifications18. In brief, HOKs plated onto 48-well plates were co-transfected with 200 nM miR-6858 mimics or 500 ng pGL3-Promoter, pGL3-GSDMC or pGL3-GSDMCmut plasmids by Lipofectamine 3000 reagent. After 36-hour incubation, HOKs were lysed and luciferase activities were measured using the Dual Luciferase Reporter Assay System (Promega) in a Lumet LB 9507 luminometer (Berthold Technologies, Bad Wildbad, Germany) and normalized to the activities of Renilla.
Cell death assays
Cell death was detected using the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega, Catalog #G1780) according to the manufactures’ instruction.
m6A quantitation
The m6A quantitation assays in total cellular RNAs were performed using an EpiQuik m6A RNA Methylation kit (Epigentek, Farmingdale, NY) in terms of the manufacturer’s instruction. Briefly, total RNAs extracted from cells were mixed with capture antibody in detection wells at 37 °C for 90 min. After several times washes, the absorbance of solutions was detected at 450 nm using a microplate reader.
m6A RNA-immunoprecipitation (RIP)-qPCR
The Magna MeRIP m6A Kit (Millipore) was used to perform m6A RIP assay. Briefly, total RNAs were fragmented by RNA Fragmentation Reagents. 5% samples were saved as input controls. The rest fragmented RNA samples were mixed with magnetic beads conjugated with m6A antibody for precipitation. The enriched RNA fragments were reversed-transcribed into cDNA and then quantified by real-time PCR. The enrichment of m6A was normalized to the input controls. Primers were listed in Supplementary Table 2.
Cross-linking and RNA immunoprecipitation (CLIP)
CLIP assays were performed according to previous studies18. HOKs were crosslinked by UV light at 0.15 J/cm2 and lysed in lysis buffer (150 mM KCl, 50 mM HEPES-KOH (pH 7.5), 1 mM NaF, 2 mM EDTA-NaOH (PH 8.0), 0.5% NP-40, 0.5 mM DTT, 1 μl/ml RNase inhibitor and 1x protease inhibitor cocktail) for nuclear extraction. After 15 min 1 U/ml RNase T1 treatment at 22 °C, 5% lysates were saved as input controls and the remaining lysates were rotated with control or anti-HNRNPA2B1 antibody-conjugated protein G magnetic beads for 1 h in cold room. Then the mixture was enriched with a magnet and washed by an IP wash buffer and a high-salt buffer, followed by complete resuspension in a proteinase K buffer. IP and input RNAs were extracted by TRIzol reagent and subjected to RT-qPCR. Primers for CLIP assays were listed in Supplementary Table 2.
RNA affinity chromatography
RNA affinity chromatography assay was carried out according to previous investigation16. Biotin-labelled ssRNA oligonucleotides harboring adenosine or m6A were from GE Dharmacon (Lafayette, CO). After being denatured at 99 °C for 10 min, the ssRNA baits were put on ice immediately. 0.4 pmol RNA oligonucleotide was mixed with 50 μl streptavidin magnetic beads (Thermo Fisher Scientific) in binding buffer (200 mM NaCl, 20 mM Tris, 5 mM potassium fluoride, 5 mM β-glycerophosphate, 6 mM EDTA, 2 μg/ml aprotinin at pH 7.5) for 4 h in cold room. The RNA bait-beads complexes were then incubated with 200 μg nuclear extract derived from HOKs in binding buffer in a final volume of 400 μl in cold room for 12 h. After a set of washes, RNA–protein mixtures were dissolved in laemmli buffer and subjected to western blot analysis.
Caspase 3 activity
Caspase-3 activities in oral keratinocytes were assessed according to a protocol27. In brief, 1 × 106 oral keratinocytes were dissolved in lysis buffer for 10 min. After 10-min centrifugation at 3000 × g, supernatants were harvested as cell lysate for analysis. Caspase-3 activities in lysate were evaluated by caspase substrate Ac-DEVD-AFC (Bio Vision, Milpitas, CA), and monitored using a plate reader under Ex360/Em530 condition. Data of caspase 3 activity were normalized to cell number of HOKs.
Image acquisition of cell death
Cell death in HOKs was determined by propidium iodide (1 μg/ml) staining and images were captured using a fluorescent microscope. Quantitative analysis was performed using Image J software.
ChIP assays
ChIP assays were performed according to a published protocol with some modifications42. Briefly, oral keratinocytes were fixed with formaldehyde (1%) and neutralized by glycine. The chromatins in cell lysates were then sheared to get 400–500 bp fragments by sonification. 10% sheared samples were collected as input controls. The remaining 90% lysates were purified and mixed with anti-p65 or control IgG antibodies (4 μg). After O/N incubation, mixture was precipitated by protein A agarose beads. Following elution and purification with a set of washes, the precipitated samples were quantified by qPCR. The primers for qPCR were listed in Supplementary Table 2.
CRISPR/Cas9-mediated knockout of Caspase3, Caspase7, GSDMD, MLKL, BAX and BAK
sgRNA sequences targeting the Caspase3 gene (5′- ATGTCGATGCAGCAAACCTC-3′), Caspase7 gene (5′- CGTTTGTACCGTCCCTCTTC-3′), GSDMD gene (5′- CGGCTCTCACCTGTCGCGGG-3′), MLKL gene (5′-TTGAAGCATATTATCACCCT-3′), BAX gene (5′- TTTCTGACGGCAACTTCAAC-3′) and BAK gene (5′- GCATGAAGTCGACCACGAAG-3′) were subcloned into lentiCRISPRv2 vector (Addgene, catalog 52961). Packaging plasmids and lenti-vector were co-transfected into HEK293T cells. Lentivirus was collected from the cell culture medium after 48 h and infected into HOKs with 4 μg/ml polybrenes.
Mitochondrial membrane potential measurement
Mitochondrial membrane potential was detected by flow cytometry with the MitoProbe JC-1 Assay Kit (ThermoFisher, Cat#:M34152) according to the manufacturers’ instruction using a BD LSRFortessa system (BD Biosciences).
Mitochondria isolation
Cytosolic and mitochondrial fractions were separated and harvested by a Mitochondria Isolation Kit (ThermoFisher, Cat#:89874) according to the manufacturer’s instruction.
Statistics and reproducibility
All experiments were carried out at least 3 times independently. The biological replicates of human samples or HOK experiments are n = 14 or n = 3, respectively. Data were expressed as means ± standard deviation. Student’s t test was performed for two groups statistical analysis and ANOVA was used for analyzing the significant difference among three or more than three groups. P < 0.05 was identified to be significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
References
Bermejo-Fenoll, A. & Lopez-Jornet, P. Familial oral lichen planus: presentation of six families. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodo. 102, e12–e15 (2006).
Gorouhi, F., Davari, P. & Fazel, N. Cutaneous and mucosal lichen planus: a comprehensive review of clinical subtypes, risk factors, diagnosis, and prognosis. ScientificWorldJournal 2014, 742826 (2014).
Gonzalez-Moles, M. A. et al. Worldwide prevalence of oral lichen planus: a systematic review and meta-analysis. Oral Dis. 27, 813–828 (2021).
Cheng, Y. S., Gould, A., Kurago, Z., Fantasia, J. & Muller, S. Diagnosis of oral lichen planus: a position paper of the American Academy of Oral and Maxillofacial Pathology. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 122, 332–354 (2016).
Wang, K. et al. Analysis of oral microbial community and Th17-associated cytokines in saliva of patients with oral lichen planus. Microbiol. Immunol. 59, 105–113 (2015).
Lavanya, N., Jayanthi, P., Rao, U. K. & Ranganathan, K. Oral lichen planus: an update on pathogenesis and treatment. J. Oral Maxillofac. Pathol. 15, 127–132 (2011).
Zhou, X. J., Sugerman, P. B., Savage, N. W., Walsh, L. J. & Seymour, G. J. Intra-epithelial CD8+ T cells and basement membrane disruption in oral lichen planus. J. Oral Pathol. Med. 31, 23–27 (2002).
Nachtergaele, S. & He, C. The emerging biology of RNA post-transcriptional modifications. RNA Biol. 14, 156–163 (2017).
Wei, C. M., Gershowitz, A. & Moss, B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 4, 379–386 (1975).
Shi, H., Wei, J. & He, C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 (2019).
Huang, H., Weng, H. & Chen, J. m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell 37, 270–288 (2020).
Narayan, P. & Rottman, F. M. An in vitro system for accurate methylation of internal adenosine residues in messenger RNA. Science 242, 1159–1162 (1988).
Kane, S. E. & Beemon, K. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol. Cell. Biol. 5, 2298–2306 (1985).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Zhao, B. S., Roundtree, I. A. & He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 (2017).
Huang, H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Lee, H. et al. Stage-specific requirement for Mettl3-dependent m(6)A mRNA methylation during haematopoietic stem cell differentiation. Nat. Cell Biol. 21, 700–709 (2019).
Du, J. et al. N(6)-adenosine methylation of Socs1 mRNA is required to sustain the negative feedback control of macrophage activation. Dev. Cell 55, 737–753.e737 (2020).
Han, D. et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274 (2019).
Feng, S., Fox, D. & Man, S. M. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J. Mol. Biol. 430, 3068–3080 (2018).
Tamura, M. et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 89, 618–629 (2007).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Hou, J. et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 22, 1264–1275 (2020).
Lu, R. et al. Overexpression and selectively regulatory roles of IL-23/IL-17 axis in the lesions of oral lichen planus. Mediators Inflamm. 2014, 701094 (2014).
Zhao, B. et al. Vitamin D/VDR signaling suppresses microRNA-802-induced apoptosis of keratinocytes in oral lichen planus. FASEB J. 33, 1042–1050 (2019).
Riccardi, C. & Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 1, 1458–1461 (2006).
Wang, R. et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 580, 386–390 (2020).
Anderson, C. J. et al. Microbes exploit death-induced nutrient release by gut epithelial cells. Nature 596, 262–267 (2021).
Du, J. et al. MicroRNA-26a/b have protective roles in oral lichen planus. Cell Death Dis. 11, 15 (2020).
Ge, X. et al. Renin promotes STAT4 phosphorylation to induce IL-17 production in keratinocytes of oral lichen planus. iScience 23, 100983 (2020).
Mendell, J. T. & Olson, E. N. MicroRNAs in stress signaling and human disease. Cell 148, 1172–1187 (2012).
Alarcon, C. R. et al. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell 162, 1299–1308 (2015).
Liu, P. et al. m(6)A-independent genome-wide METTL3 and METTL14 redistribution drives the senescence-associated secretory phenotype. Nat. Cell Biol. 23, 355–365 (2021).
Xia, S., Hollingsworth, L. R. & Wu, H. Mechanism and regulation of gasdermin-mediated cell death. Cold Spring Harb. Perspect. Biol. 12, a036400 (2020).
Miguchi, M. et al. Gasdermin C is upregulated by inactivation of transforming growth factor beta receptor type II in the presence of mutated Apc, promoting colorectal cancer proliferation. PloS One 11, e0166422 (2016).
Du, J. et al. N(6)-adenomethylation of GsdmC is essential for Lgr5(+) stem cell survival to maintain normal colonic epithelial morphogenesis. Dev. Cell 57, 1976–1994.e1978 (2022).
Xi, R. et al. Up-regulation of gasdermin C in mouse small intestine is associated with lytic cell death in enterocytes in worm-induced type 2 immunity. Proc. Natl Acad. Sci. USA 118, e2026307118 (2021).
Wang, C. et al. miR-6858 plays a key role in the process of melatonin inhibition of the malignant biological behavior of glioma. J. Clin. Neurosci. 87, 137–146 (2021).
El-Howati, A., Thornhill, M. H., Colley, H. E. & Murdoch, C. Immune mechanisms in oral lichen planus. Oral Dis. 29, 1400–1415 (2023).
Yuan, W. et al. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 282, 29821–29830 (2007).
Acknowledgements
This study was supported by National Natural Science Foundation of China grants (81800499, 81960565, 81560275), Fourth National Oral Health Epidemiology Survey Follow-up Research Project (201502002), Fundamental Research Program of Shanxi Province (202103021224231), Research Project of Shanxi Provincial Health Commission (2023049), Hainan Provincial Key Research and Development Program (ZDYF2020147), Hainan Natural Science Foundation Innovation Research Team Project (2018CXTD350) and a grant from Hainan Province Clinical Medical Center. We thank Jie Du (Shanxi Medical University) for his assistant work for this project.
Author information
Authors and Affiliations
Contributions
X.G. conceived and designed the research. X.W., S.L. ST, Y.D., R.G., H.S. and X.S. performed the experiments. R.L. collected human samples. X.W. analyzed data. X.G. and wrote the manuscript. X.W. and Y.D. acquired funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Kohsuke Tsuchiya and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Kaiwen Chen and Manuel Breuer. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Li, S., Song, H. et al. METTL14-upregulated miR-6858 triggers cell apoptosis in keratinocytes of oral lichen planus through decreasing GSDMC. Commun Biol 6, 976 (2023). https://doi.org/10.1038/s42003-023-05360-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05360-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
