
Abstract
Adult T-cell leukemia/lymphoma (ATL) is caused by human T-cell leukemia virus type 1 (HTLV-1). In addition to HTLV-1 bZIP factor (HBZ), a leukemogenic antisense transcript of HTLV-1, abnormalities of genes involved in TCR-NF-κB signaling, such as CARD11, are detected in about 90% of patients. Utilizing mice expressing CD4+ T cell-specific CARD11(E626K) and/or CD4+ T cell-specific HBZ, namely CARD11(E626K)CD4-Cre mice, HBZ transgenic (Tg) mice, and CARD11(E626K)CD4-Cre;HBZ Tg double transgenic mice, we clarify these genes’ pathogenetic effects. CARD11(E626K)CD4-Cre and HBZ Tg mice exhibit lymphocytic invasion to many organs, including the lungs, and double transgenic mice develop lymphoproliferative disease and increase CD4+ T cells in vivo. CARD11(E626K) and HBZ cooperatively activate the non-canonical NF-κB pathway, IRF4 targets, BATF3/IRF4/HBZ transcriptional network, MYC targets, and E2F targets. Most KEGG and HALLMARK gene sets enriched in acute-type ATL are also enriched in double transgenic mice, indicating that these genes cooperatively contribute to ATL development.
Similar content being viewed by others


Introduction
Adult T-cell leukemia/lymphoma (ATL) is an aggressive peripheral T-cell lymphoma that develops in about 5% of people infected with human T-cell leukemia virus type 1 (HTLV-1), usually after decades of clinical latency1,2,3,4,5. Viral oncogenes encoded by HTLV-1, namely tax and HTLV-1 bZIP factor (HBZ), are thought to play a major role in ATL development6,7. Tax possesses transforming activity in vitro, and Tax transgenic (Tg) mice develop cancer in vivo8,9. However, Tax is highly immunogenic, and its expression is suppressed and disrupted in many ATL cells10,11. In contrast, HBZ, which is encoded in the minus strand of the HTLV-1 genome, is transcribed in all ATL cases, and HBZ promotes ATL cell proliferation in vivo and induces T-cell lymphoma and inflammation in vivo12,13,14,15.
The development of ATL involves not only viral oncogenes, but also presumably the accumulation of gene mutations in T cells during the long latency period from HTLV-1 infection in infancy to ATL onset later in life4. We previously reported that about 90% of ATL patients possessed mutations in genes involved in T-cell receptor (TCR)–NF-κB signaling and downstream or associated pathways16. Among them, mutations in CARD11, a cytoplasmic scaffolding protein required for antigen receptor-induced NF-κB activation in both T and B cells, were detected in 24% of ATL patients16. The most frequently observed CARD11 mutation, comprising 17% of the total, was the E626K gain-of-function mutation16. Gain-of-function mutations in CARD11 were also reported in 9.6% of patients with diffuse large B-cell lymphoma (DLBCL) of the activated B-cell (ABC) subtype, and mice harboring active CARD11 mutations in B cells developed aggressive B-cell lymphoproliferation, leading to early postnatal lethality17,18,19.
Here we generated a mouse model involving the conditional expression of a CARD11(E626K) gain-of-function mutant, which is the most common mutation observed in ATL patients16, and demonstrated that CARD11 activation in CD4+ T cells induced lymphadenopathy and T-cell infiltration of many organs. We also showed that the combination of HBZ expression and CARD11 mutation cooperatively accelerated the onset of lymphoproliferative disease, affected the expression of many genes, and served as the molecular basis of acute-type ATL.
Results
Generation of CARD11(E626K)CD4-Cre and HBZ Tg mice
To investigate constitutively active CARD11 signaling in CD4+ T cells in vivo, we generated CARD11(E626K)CD4-Cre mice; this mutation is the homologue of human CARD11(E626K), the most commonly observed mutation in ATL patients (Supplementary Fig. 1a). Compared to WT CD4+ T cells, CD4+ T cells from CARD11(E626K)CD4-Cre mice demonstrated a 1.2-fold increase in the ratio of phosphorylated CARD11 Ser652 to CARD11 (Supplementary Fig. 2a). HBZ Tg mice were generated by HBZ cDNA expression under a mouse CD4 promoter (Supplementary Fig. 1b). Crossing CARD11(E626K)CD4-Cre and HBZ Tg mice resulted in CARD11(E626K)CD4-Cre;HBZ Tg mice.
Western blotting showed that compared with WT mice, the level of CARD11 in CD4+ T cells was increased by 3-fold and 2.5-fold in CARD11(E626K)CD4-Cre mice and CARD11(E626K)CD4-Cre;HBZ Tg mice, respectively (Supplementary Fig. 2b). There was no difference between these mouse types regarding the CARD11 protein level. HBZ was detected in CD4+ T cells from HBZ Tg mice, but not in those from WT mice (Supplementary Fig. 2b). The protein level of HBZ in CARD11(E626K)CD4-Cre;HBZ Tg mice was comparable to that in HBZ Tg mice.
The median survival time of CARD11(E626K)CD4-Cre, HBZ Tg, and CARD11(E626K)CD4-Cre;HBZ Tg mice was 14, 8, and 6 months, respectively (Fig. 1a).

a Kaplan–Meier survival curves of mutant mice (wild type (WT), n = 38; CARD11(E626K)CD4-Cre, n = 37; HBZ Tg, n = 88; CARD11(E626K)CD4-Cre;HBZ Tg, n = 31). The median survival times of WT, CARD11(E626K)CD4-Cre, HBZ Tg, and CARD11(E626K)CD4-Cre;HBZ Tg mice, respectively, are as follows: not reached, 14, 8, and 6 months after birth. b The number of white blood cells and CD4+ T cells in peripheral blood in WT (n = 23), CARD11(E626K)CD4-Cre (n = 6), HBZ Tg (n = 10), and CARD11(E626K)CD4-Cre;HBZ Tg (n = 4) mice at 3 months after birth, and those in WT (n = 27), CARD11(E626K)CD4-Cre (n = 12), HBZ Tg (n = 13), and CARD11(E626K)CD4-Cre;HBZ Tg (n = 13) mice at 6 months after birth. c The differential white blood cell count was performed manually, and a peripheral blood slide revealed that about 5% of lymphocytes in CARD11(E626K)CD4-Cre;HBZ Tg mice demonstrated pleomorphic nuclear features (WT, n = 5; CARD11(E626K)CD4-Cre, n = 3; HBZ Tg, n = 3; CARD11(E626K)CD4-Cre;HBZ Tg, n = 3). The blood smear was stained with Wright-Giemsa. p values were calculated by the Tukey test after one-way ANOVA, and *, **, ***, and **** represent p values less than 0.05, 0.01, 0.001, and 0.0001, respectively.
Lymphocyte invasion in many organs in CARD11(E626K)CD4-Cre and HBZ Tg mice
In CARD11(E626K)CD4-Cre mice, leukocytosis and an increased number of CD4+ cells were observed in peripheral blood (Fig. 1b). Morphologically abnormal/atypical cells were rarely observed (Fig. 1c). CARD11(E626K)CD4-Cre mice did not show an increase in splenic nucleated cells (Fig. 2a). Normal murine splenic histological architecture contains clearly bordered white and red pulp. White pulp consists of aggregates of nucleated cells, mainly lymphocytes, while red pulp consists of a vascularized sinus meshwork. At 6 months after birth, the splenic histological architecture in CARD11(E626K)CD4-Cre mice was almost intact, but the interface between white and red pulp was uneven and obscure (Fig. 2b). This was attributed to focal infiltration of CD3+ T cells from the white pulp periarteriolar region into the red pulp. At 12 months after birth, areas of white pulp were decreased in number and size, while those of red pulp were larger, and the interface had become disrupted. CD3+ T cells had diffusely infiltrated both the shrunken white pulp and the expanding red pulp. The size and cellularity of the thymus was unchanged in CARD11(E626K)CD4-Cre mice (Supplementary Fig. 3).

a Absolute numbers of nucleated cells in the spleen at 6 months after birth (wild type (WT), n = 21; CARD11(E626K)CD4-Cre, n = 15; HBZ Tg, n = 20; CARD11(E626K)CD4-Cre;HBZ Tg, n = 13) and 12 months after birth (WT, n = 11; CARD11(E626K)CD4-Cre, n = 5; HBZ Tg, n = 12). b Histologic analysis of the spleen at 6 and 12 months after birth (n = 5 for each mouse type). Spleens were stained with hematoxylin and eosin (HE) and anti-CD3 and anti-B220 antibodies. The interface between white and red pulp is shown at high magnification (HE 400×). CD3+ T cells in red pulp are shown at high magnification (CD3 400×).
Most CARD11(E626K)CD4-Cre mice developed lymphadenopathy. However, the swollen lymph nodes (LNs) were not big, with a mean diameter of about 3 mm. Lymphadenopathy was observed in 12 of 18 mice (67%) at 6 months after birth, and in 12 of 14 mice (86%) at 12 months after birth (Fig. 3a). The LN histological structure in WT mice consists of the cortex, paracortex, and medulla. Lymph follicles are located within the cortex, and the paracortex contains veins, with a high proportion of endothelial cells, into which circulating lymphocytes enter. At 6 months after birth, CARD11(E626K)CD4-Cre mice exhibited slightly shrunken follicles, a thickened cortex, and an expanded paracortex with CD3+ T-cell infiltration (Fig. 3b). The interface between the cortex and paracortex was obscured by infiltrating T cells. At 12 months, the normal LN architecture was barely present in CARD11(E626K)CD4-Cre mice. The cortex had become thickened and the follicles had disappeared. The paracortex was expanded by CD3+ T cells, and the interface was disrupted.

a The frequency of lymphadenopathy at 6 and 12 months after birth. Swollen lymph nodes (LNs) are not large, with a mean diameter of about 3 mm. Differences between frequencies of lymphadenopathy were assessed by Fisher’s exact test with Benjamini–Hochberg correction. b Histologic analysis of LNs at 6 and 12 months after birth (n = 5 for each mouse type). LNs were stained with hematoxylin and eosin (HE), and anti-CD3 and anti-B220 antibodies. The interface between the cortex and paracortex is shown with high magnification (CD3 400×).
The most affected organ in CARD11(E626K)CD4-Cre mice was the lung. In WT mice, alveolar ducts, alveolar sacs, and alveoli have very thin walls invested with fine close-meshed networks of large thin-walled capillaries. In CARD11(E626K)CD4-Cre mice at 6 months after birth, focal lymphocyte infiltration was observed; both T and B cells had infiltrated the perivascular interstitium and occasionally the alveolar septa. Over time this infiltration increasingly consisted of CD3+ T cells, which formed diffuse lung lesions with thickening of the interstitium and alveolar septa at 12 months (Fig. 4).

Histologic analysis of the lung at 6 and 12 months after birth (n = 5 for each mouse type). Lungs were stained with hematoxylin and eosin (HE), as well as anti-CD3 and anti-B220 antibodies. Lymphocyte infiltration into the lung perivascular interstitium and alveolar septa is observed in mutant mice at 6 and 12 months.
HBZ Tg mice exhibited leukopenia and a decreased number of CD4+ T cells in peripheral blood (Fig. 1b). Morphologically abnormal/atypical cells were rarely observed (Fig. 1c). The number of nucleated cells in the spleen was comparable between HBZ Tg and WT mice (Fig. 2a). Histological examination at 6 months after birth revealed that the spleen was almost normal; however, at 12 months after birth, the white pulp exhibited expansion (Fig. 2b). CD3+ T cells had caused expansion of both white and red pulp, which disrupted their interface.
The thymus was shrunken in size in HBZ Tg mice at 6 months after birth. In one lobe of the thymus, the nucleated cell number in HBZ Tg mice was comparable to that in WT mice, and the number of CD4+ T cells was lower than that in WT mice (Supplementary Fig. 3).
Lymphadenopathy was obvious at 12 months after birth in HBZ Tg mice (Fig. 3a). The frequency of lymphadenopathy at 12 months after birth was 67%, which was similar to that in a previous report15. The normal LN architecture was destroyed. The follicles were shrunken, the cortex was thickened, and the paracortex was expanded (Fig. 3b). CD3+ T cells were mainly observed in the paracortex, and they infiltrated the cortex.
As in CARD11(E626K)CD4-Cre mice, the most affected organ in HBZ Tg mice was the lung. Diffuse infiltration of lymphocytes was observed at 6 months, and lymphocytes comprising mainly T cells invaded the perivascular interstitium and alveolar septa. The alveolar space was filled with exudate at 6 months after birth (Fig. 4). These abnormalities were much more pronounced at 12 months after birth. Severe lung findings were observed, with exudate and thickening of the interstitium and alveolar septa. These features, except for the exudate, resembled those in CARD11(E626K)CD4-Cre mice.
The combination of CARD11 mutation and HBZ expression induced the development of lymphoproliferative disease
HBZ is thought to play a major role in ATL development6,7 and is detected in CD4+ T cells from almost all patients with acute-type ATL, while CARD mutations are found in about one-fourth of patients with ATL. We therefore analyzed how HBZ expression and CARD mutation affected the development of lymphoproliferative neoplasms in vivo. The subsequent analysis was performed at 6 months after birth since most CARD11(E626K)CD4-Cre;HBZ Tg mice died within 9 months after birth. CARD11(E626K)CD4-Cre;HBZ Tg mice exhibited leukopenia and a decreased number of CD4+ T cells in peripheral blood (Fig. 1b). About 5% of lymphocytes demonstrated pleomorphic nuclear features, whereas other lymphocytes had nearly normal morphology (Fig. 1c).
In the spleen, neither splenomegaly nor an increased number of nucleated cells was observed (Fig. 2a, b). The splenic architecture was destroyed and the interface between white and red pulp was disrupted. CD3+ T cells were diffusely present throughout the spleen. The thymus was shrunken in size in CARD11(E626K)CD4-Cre;HBZ Tg mice at 6 months after birth compared with WT mice (Supplementary Fig. 3). Accordingly, the numbers of nucleated cells and CD4+CD8− T cells in one lobe of the thymus were lower in CARD11(E626K)CD4-Cre;HBZ Tg mice compared with WT mice.
Lymphadenopathy was detected in 17 of 20 (85%) CARD11(E626K)CD4-Cre;HBZ Tg mice (Fig. 3). The normal LN architecture was destroyed; follicles had almost disappeared, the cortex was thickened, and the paracortex was expanded. CD3+ T cells were observed mainly in the paracortex and had massively infiltrated the cortex, leading to the highly disrupted interface.
The most affected organ in CARD11(E626K)CD4-Cre;HBZ Tg mice was the lung. The lung interstitium and alveolar septa were thickened with infiltrating cells consisting mainly of lymphocytes (Fig. 4). CD3+ T cells had accumulated around capillary blood vessels. The alveolar space had filled with exudate, and subsequently with macrophages, and consequently almost no air spaces were observed. Mice with both CARD11 mutation and HBZ expression developed the aggressive lymphoproliferative disease.
Increment of CD4+ T cells, effector/memory T cells (Tem), and regulatory T cells (Treg) in CARD11(E626K)CD4-Cre;HBZ Tg mice
Since ATL is a neoplasm of CD4+ T lymphocytes, and both CARD11(E626K) and HBZ were expressed in CD4+ T cells in our mice, we next evaluated the absolute numbers of CD4+ T cells and their subsets, namely CD4+CD44+CD62L− Tem and CD4+CD25+ Treg, together with CD8+ T cells, in the body of each mouse as the sum of their numbers in the whole bone marrow and whole spleen20,21 (Fig. 5, Supplementary Fig. 4). In CARD11(E626K)CD4-Cre mice, the numbers of CD4+CD8− T cells, Tem, Treg, and CD4−CD8+ T cells per mouse were not increased compared with WT mice. In HBZ Tg mice, there was a decrease in the number of CD4−CD8+ T cells per mouse at 6 months after birth, and an increase of CD4+CD8− T cells and Tem per mouse at 12 months after birth. HBZ expression had no effect on the number of Treg. In CARD11(E626K)CD4-Cre;HBZ Tg mice, the numbers of CD4+CD8− T cells, Tem, and Treg were increased compared with WT mice. Consistent with this, the numbers of CD44+ and FOXP3+ cells were increased in LNs and lung in CARD11(E626K)CD4-Cre;HBZ Tg mice, and the ratio of Ki-67 positive cells among Tem and Treg in LNs were increased compared with WT mice (Supplementary Figs. 5, 6); this indicated a cooperative positive effect of CARD11 mutation and HBZ expression on the proliferation of each cell type. The number of CD4−CD8+ T cells in CARD11(E626K)CD4-Cre;HBZ Tg mice was comparable to that in WT mice.

Absolute numbers of CD4+CD8- T cells, CD4-CD8+ T cells, CD4+CD44+CD62L− effector/memory T cells (Tem), and CD4+CD25+ regulatory T cells (Treg) per mouse in wild type (WT) (n = 11), CARD11(E626K)CD4-Cre (n = 10), HBZ Tg (n = 12), and CARD11(E626K)CD4-Cre;HBZ Tg (n = 14) mice at 6 months after birth (a), and those in WT (n = 11), CARD11(E626K)CD4-Cre (n = 6), and HBZ Tg (n = 11) mice at 12 months after birth (b). p values were calculated by the Tukey test after one-way ANOVA, and *, **, and *** represent p values less than 0.05, 0.01, and 0.001, respectively.
A hallmark of malignant transformation is the ability of a transplanted tumor to proliferate in another host. We transplanted splenic CD4+ T cells from WT, CARD11(E626K)CD4-Cre, HBZ Tg, or CARD11(E626K)CD4-Cre;HBZ Tg mice into immunocompromised NOD/Shi-scid/IL2rγnull (NOG) mice. At 18 weeks after transplantation, no overt tumors were visible in recipient mice. In recipient mice transplanted with CD4+ T cells from CARD11(E626K)CD4-Cre;HBZ Tg mice, CD3+ T cells were detected in the spleen, whereas CD3+ T cells were rare in the spleens of recipient mice transplanted with CD4+ T cells from WT, CARD11(E626K)CD4-Cre, or HBZ Tg mice (Supplementary Fig. 7). Some small LNs were detected in recipient mice transplanted with CD4+ T cells from CARD11(E626K)CD4-Cre;HBZ Tg mice. These LNs contained mostly CD3+ T cells, and about 20% of the CD3+ T cells expressed Ki-67, suggesting that CARD11(E626K)CD4-Cre;HBZ Tg mice developed lymphoproliferative disease. On the other hand, the absence of CD3+ T cells in the spleen and the absence of LNs containing CD3+ T cells in the recipient mice transplanted with CARD11(E626K)CD4-Cre and HBZ Tg CD4+ T cells indicate that they developed inflammation, but not neoplasm.
Gene expression changes due to the CARD11 mutant and HBZ expression
To understand the basis for the phenotypes induced by the CARD11 mutant and HBZ expression in vivo, we performed gene expression profiling of Tem derived from WT, CARD11(E626K)CD4-Cre, HBZ Tg, and CARD11(E626K)CD4-Cre;HBZ Tg mice. Unsupervised hierarchical clustering of the global gene expression signatures revealed three major branches in the gene expression hierarchy (Supplementary Fig. 8a). WT and CARD11(E626K)CD4-Cre samples comprised the first branch, HBZ Tg samples comprised the second, and CARD11(E626K)CD4-Cre;HBZ Tg samples comprised the third. The principal component analysis also showed that the CARD11 mutant and HBZ expression induced distinct transcriptomes (Supplementary Fig. 8b).
Compared with WT mice, the CARD11(E626K)CD4-Cre, HBZ Tg, and CARD11(E626K)CD4-Cre;HBZ Tg mice showed upregulation of 17, 877, and 508 genes, respectively, and downregulation of 23, 546, and 297 genes, respectively (Fig. 6a). KEGG pathway analysis showed that TGF-beta signaling, mTORC1 signaling, inflammatory response, and apical junction were uniquely activated in CARD11(E626K)CD4-Cre mice; mitotic spindle signaling was uniquely activated in HBZ Tg mice; and PI3K/AKT/mTOR signaling, MYC target V1, G2M checkpoint, E2F target, and interferon alpha response were uniquely activated in CARD11(E626K)CD4-Cre;HBZ Tg mice (Fig. 6b). In contrast, TNF alpha signaling via NF-kappa B, IL-2/STAT5 signaling and interferon-gamma response were activated in CARD11(E626K)CD4-Cre, HBZ Tg, and CARD11(E626K)CD4-Cre;HBZ Tg mice. The p53 pathway and IL-6/JAK/STAT3 signaling were activated in both HBZ Tg and CARD11(E626K)CD4-Cre;HBZ Tg mice, but not in CARD11(E626K)CD4-Cre mice.

a Venn diagrams of the overlap between significantly up- and downregulated genes in CARD11(E626K)CD4-Cre (n = 3) vs. WT (n = 3) mice, HBZ-Tg (n = 4) vs. WT mice, and CARD11(E626K)CD4-Cre;HBZ Tg (n = 3) vs. WT mice. Splenic effector/memory T cells (Tem) from 4–6-month-old mice were sorted and underwent expressional analysis. Differentially expressed genes between sample groups were identified, using cut-offs of fold change (FC) > 1.2 and an FDR of <0.1. b Bar plots of significantly (FDR < 0.25) upregulated KEGG pathways. Some pathways are uniquely activated in CARD11(E626K)CD4-Cre mice, HBZ Tg mice, or CARD11(E626K)CD4-Cre;HBZ Tg mice. On the other hand, IL-2/STAT5 signaling and interferon-gamma response are commonly activated in all three types of mice.
CARD activates the NF-κB signaling pathway. GSEA showed heavy enrichment of canonical NF-κB pathway gene sets22 in CARD11(E626K)CD4-Cre mice, but not in HBZ Tg or CARD11(E626K)CD4-Cre;HBZ Tg mice (Supplementary Fig. 9). Non-canonical NF-κB pathway gene sets23,24 were significantly enriched in CARD11(E626K)CD4-Cre;HBZ Tg mice. Consistent with these results, high nuclear levels of RelA (p65) and p50 were detected in splenic CD4+ cells from CARD11(E626K)CD4-Cre mice, indicating that the canonical NF-κB pathway was activated by CARD11 mutation (Supplementary Fig. 10). In splenic CD4+ cells from CARD11(E626K)CD4-Cre;HBZ Tg mice, high nuclear levels of RelB were detected, indicating that the non-canonical NF-κB pathway was activated by the combined effects of CARD11 mutation and HBZ expression.
HBZ mRNA and its transcript directly bind to the promoter regions of many genes. The selective apoptosis- and proliferation-related genes directly regulated by HBZ mRNA25 were enriched in both HBZ Tg and CARD11(E626K)CD4-Cre;HBZ Tg mice, and the enrichment of HBZ target gene sets26, including indirect ones, were also observed in both mice (Supplementary Fig. 11).
The gene set enrichment pattern in Tem differed drastically between CARD11(E626K)CD4-Cre and HBZ Tg mice (Fig. 7). Gene sets related to cytokine signaling and inflammation were heavily enriched in CARD11(E626K)CD4-Cre mice. CARD activation and HBZ expression might cooperatively affect global gene expression. In CARD11(E626K)CD4-Cre;HBZ Tg mice, there was greater enrichment of gene sets related to the non-canonical NF-κB pathway, IRF4 targets, proliferation, T-cell differentiation, and cell adhesion than in WT mice. The gene sets whose expression was correlated with chromosomal instability were more enriched in CARD11(E626K)CD4-Cre;HBZ Tg mice (Supplementary Fig. 12), suggesting that the combination of HBZ expression and CARD11 mutation alter genes associated with genomic stability and contribute to the development of ATL27.

Comparisons of results are shown in the following order: CARD11(E626K)CD4-Cre vs. WT, HBZ Tg vs. WT, CARD11(E626K)CD4-Cre;HBZ Tg vs. WT, CARD11(E626K)CD4-Cre;HBZ Tg vs. HBZ Tg, and CARD11(E626K)CD4-Cre;HBZ Tg vs. CARD11(E626K)CD4-Cre.
Both HBZ Tg and CARD11(E626K)CD4-Cre;HBZ Tg mice exhibited similar phenotypes, with the major difference being disease severity or timing. HBZ Tg mice at 6 months after birth exhibited a milder phenotype than CARD11(E626K)CD4-Cre;HBZ Tg mice at 6 months after birth, but HBZ Tg mice at 12 months after birth exhibited similar severity as CARD11(E626K)CD4-Cre;HBZ Tg at 6 months after birth. We then compared the gene expression between HBZ Tg mice at 8–12 months and CARD11(E626K)CD4-Cre;HBZ Tg mice at 4–6 months (Supplementary Fig. 13a). A total of 430 genes and 227 genes were commonly up- and downregulated, respectively, and target gene sets of HBZ were highly enriched in both HBZ Tg mice at 8–12 months and CARD11(E626K)CD4-Cre;HBZ Tg mice at 4–6 months (Supplementary Figs. 11, 13b). Even though many genes were commonly up- or downregulated in both mouse types, there were genes that were uniquely up- or downregulated in only one type. Hierarchical clustering analysis also demonstrated that HBZ Tg mice at 8–12 months comprised a different gene expression hierarchy branch than CARD11(E626K)CD4-Cre;HBZ Tg mice at 4–6 months (Supplementary Fig. 13c). The uniquely upregulated gene sets in CARD11(E626K)CD4-Cre;HBZ Tg mice at 4–6 months (n = 78) were highly enriched in acute-type ATL patient samples compared with those from HTLV-1 carriers (Supplementary Fig. 13d). On the other hand, uniquely upregulated gene sets in HBZ Tg mice at 8–12 months (n = 2970) did not exhibit this enrichment. These observations suggested that CARD11(E626K)CD4-Cre;HBZ Tg mice and HBZ Tg mice had some oncogenic signals in common, but they did not completely overlap.
Finally, we compared the enriched KEGG and HALLMARK gene sets between human samples obtained from patients with acute-type ATL16 and samples from CARD11(E626K)CD4-Cre, HBZ Tg, and CARD11(E626K)CD4-Cre;HBZ Tg mice. As shown in Fig. 8a, about one-third of KEGG and HALLMARK gene sets were enriched in human acute-type ATL samples but not in healthy controls. About 10% of the enriched gene sets of human acute-type ATL samples from both the KEGG and HALLMARK databases were also enriched in CARD11(E626K)CD4-Cre, and none of them was enriched in HBZ Tg mice. In contrast, CARD11(E626K)CD4-Cre;HBZ Tg mice demonstrated enrichment of 74% and 94% of the enriched gene sets of human acute-type ATL samples from the two databases, respectively. The remaining gene sets that were only enriched in human acute-type ATL samples but not in CARD11(E626K)CD4-Cre;HBZ Tg mice included many involved in metabolism (Supplementary Table 1). In addition, the NOTCH signaling pathway was only enriched in human acute-type ATL samples and not in Tem from CARD11(E626K)CD4-Cre;HBZ Tg mice (Fig. 8b). Since there were many mutations besides those involving the TCR-NF-κB pathway in acute-type ATL patients, these other mutations might be responsible for the activation of the other signaling pathways that were enriched in human ATL samples but not in CARD11(E626K)CD4-Cre;HBZ Tg mice.

a Normalized enrichment scores (NESs) in gene set enrichment analysis (GSEA) of the entire KEGG and HALLMARK gene sets show that mice and humans share many of the same enriched gene sets. Each gene set is indicated by a dot and is highlighted in a different color according to the respective false discovery rate (FDR) values for mice and humans, with a common cutoff value 0.25. Each plot legend indicates the frequency (number) of significant/non-significant gene sets, based on the common FDR cutoff values in both mice and humans. In the table at the bottom of each plot, the frequency (number) of pathways is indicated, based on the positive/negative values for both mouse NES (NES_m) and human NES (NES_h). b Notch signaling is enriched in the acute-type ATL samples, but not in CARD11(E626K)CD4-Cre;HBZ Tg mice. Enrichment plots of the KEGG Notch signaling gene set in mice and humans are shown with their NESs and FDRs.
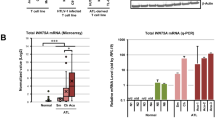
The BATF3/IRF4/HBZ transcriptional network was reported to be essential for the proliferation of cells from an ATL cell line28. As shown in Fig. 9a, this network was enriched in both CARD11(E626K)CD4-Cre;HBZ Tg mice and human acute-type ATL samples, but not in CARD11(E626K)CD4-Cre mice, HBZ Tg mice, or HTLV-1 carrier samples. BATF3 was upregulated in both HBZ Tg and CARD11(E626K)CD4-Cre;HBZ Tg mice (Fig. 9b). There was a 2.0-fold increase in IRF-4 protein levels in CD4+ T cells from CARD11(E626K)CD4-Cre;HBZ Tg mice compared with those from WT mice (Fig. 9c). Increased nuclear IRF4 protein was also observed in CD4+ T cells from CARD11(E626K)CD4-Cre;HBZ Tg mice, but not in those from HBZ Tg mice (Fig. 9d, e). In line with this, many IRF4 target gene sets were enriched in CARD11(E626K)CD4-Cre;HBZ Tg mice but not in HBZ Tg mice (Supplementary Fig. 14). In CARD11(E626K)CD4-Cre;HBZ Tg mice, the elevated BATF3 and IRF4 levels might induce the activation of the BATF3/IRF4/HBZ transcriptional network. Positive enrichment of proliferation-related gene sets such as MYC_TARGETS and E2F_TARGETS was also confirmed in CARD11(E626K)CD4-Cre;HBZ Tg mice, highlighting the cooperative effect between CARD11(E626K)CD4-Cre and HBZ (Supplementary Fig. 15).

a CARD11(E626K)CD4-Cre;HBZ Tg mice recapitulated a transcriptional network change that is characteristic of ATL. The BATF3/IRF4/HBZ transcriptional network28 is enriched in both CARD11(E626K)CD4-Cre;HBZ Tg mice and human acute-type ATL samples. Enrichment plots of each gene set in mice and humans are shown with their NESs and FDRs. b Quantitative PCR for Batf3 (WT, n = 4; CARD11(E626K)CD4-Cre, n = 3; HBZ Tg, n = 3; CARD11(E626K)CD4-Cre;HBZ Tg, n = 3). The expression level of Batf3 in each type of mutant mouse was normalized to the expression level of Gapdh, and is shown as the relative ratio to that in WT mice. Batf3 is transcriptionally upregulated in splenic CD4+ T cells from both HBZ Tg and CARD11(E626K)CD4-Cre;HBZ Tg mice. c Western blotting for IRF4 in CD4+ T cells. The protein level of IRF4 in each mouse type is normalized to the protein level of tubulin, and is shown as the relative ratio to that in WT mice. d Western blotting of cytoplasmic and nuclear fractions of IRF4 in CD4+ T cells from each mouse type. Expression levels of IRF4 were normalized to those of tubulin in cytosolic fractions, and to those of TATA-binding protein (TBP) in nuclear fractions. The expression level in each type of mutant mouse is shown as the relative ratio to that in WT mice. e Summary of the relative levels of IRF4 in the nuclear fraction. A bar graph depicts the results from 3 replicate of Western blotting. p values were calculated by the Tukey test after one-way ANOVA, and *** represent p values less than 0.001, respectively.
Discussion
We showed here that the combination of constitutive HBZ expression and a gain-of-function mutation in CARD11 in CD4+ T cells induced lethal lymphoproliferative disease in vivo, which includes LN swelling and lymphocyte invasion of many organs, especially the lungs. HBZ expression and CARD11 mutation cooperatively increased the numbers of CD4+ T cells, Tem, and Treg in vivo, and upregulated BATF3 and IRF4, MYC target genes, and E2F target genes in CD4+ T cells. These findings, and the fact that many of the KEGG and HALLMARK gene sets that were enriched in acute-type ATL patient samples were also enriched in CARD11(E626K)CD4-Cre;HBZ Tg mice, indicate that the combination of HBZ expression and CARD11 mutation forms the pathological basis for ATL development.
CARD11(E626K)CD4-Cre mice developed leukocytosis in peripheral blood, lymphadenopathy, and T-cell organ invasion, and exhibited shorter survival. The majority of infiltrating cells were Tem, which is consistent with previous reports that ATL clones are preserved in T memory stem cells (Tscm) and that the majority of ATL cells are conventional memory T cells29. However, the degree of leukocytosis was mild and the spleen was only minimally affected. This phenotype induced by the constitutive activation of CARD11 in T cells is very different from that in B cells. A mouse model that conditionally expressed the human DLBCL-derived CARD11(L225LI) mutant in B cells developed a rapidly lethal lymphoproliferative disorder with massive splenomegaly associated with a drastic increment in B-cell number19. Our observations might indicate that the in vivo signaling and other effects caused by constitutive activation of CARD11/BCL10/MALT1 (CBM) signaling differ between B and T cells, although the CARD11 mutation is frequently observed in both B-cell malignancies, as the DLBCL ABC type, and in T-cell malignancies, as ATL. These ideas are consistent with the clinical features of the B-cell expansion with NF-κB and T-cell anergy (BENTA) syndrome, which is caused by germline gain-of-function mutations in CARD1130. Patients with BENTA syndrome exhibit splenomegaly and an increased number of B cells in PB, but a normal number of T cells31,32. The absence of abnormal growth of transplanted CD4+ T cells with the CARD11 mutation and the lack of tumor development in recipient mice provide confirmation that CARD11(E626K)CD4-Cre mice developed inflammation rather than neoplasia.
HBZ was found to be constitutively expressed in many ATL cases, and HBZ Tg mice were reported to develop lymphoma and systemic inflammation15,33. The phenotype of our HBZ Tg mice resembled that in previous reports15,33. Lymphadenopathy was observed in 66.7% of HBZ Tg mice at 12 months after birth. In a report by Satou, 37.8% of HBZ Tg mice developed T-cell lymphoma after 16 months15. In our HBZ Tg mice, the increment of CD4+ T cells and Tem in the body was not observed at 6 months after birth, but became prominent at 12 months after birth. A long period of time might be required for the development of lymphoma in HBZ Tg mice.
Since HTLV-1-infected CD4+ T cells transformed into ATL cells after developing gene mutations, we next examined the effect of the CARD11(E626K) mutation on HBZ Tg. CARD11(E626K)CD4-Cre;HBZ Tg mice exhibited aggressive lymphoproliferative disease. The numbers of CD4+ T cells, Tem, and Treg per mouse increased due to the cooperative effect of CARD11 mutation and HBZ expression. There was increased infiltration of lymphocytes into the perivascular region and alveolar septum of the lung, causing almost complete obliteration of the alveolar space in CARD11(E626K)CD4-Cre;HBZ Tg mice. These mice demonstrated lymphadenopathy and complete destruction of the normal LN architecture. Further, T-cell invasion and growth in the spleen and LNs were observed in recipient mice transplanted with CD4+ T cells from CARD11(E626K)CD4-Cre;HBZ Tg mice. These observations support the theory that constitutive CARD11 activation and HBZ expression have a cooperative effect and cause the development of lymphoproliferative disease.
Gene set enrichment patterns differed drastically between CARD11(E626K)CD4-Cre and HBZ Tg mice. Canonical NF-κB pathway gene sets were enriched in CARD11(E626K)CD4-Cre mice. It was reported that activation of the canonical NF-κB pathway was selectively inhibited by HBZ expression34. In line with this, the gene set associated with this pathway was negatively enriched in HBZ Tg mice, but the finding was not statistically significant. The negative impact of HBZ on the canonical NF-κB pathway might negate the enrichment of these gene sets in CARD11(E626K)CD4-Cre;HBZ Tg mice. In addition to the NF-κB pathway, cytokine signaling cascades were heavily enriched in CARD11(E626K)CD4-Cre mice. On the other hand, HBZ target gene sets were highly enriched in HBZ Tg mice, but not in CARD11(E626K)CD4-Cre mice. In CARD11(E626K)CD4-Cre;HBZ Tg mice, CARD11 mutation and HBZ expression cooperatively activated many signaling cascades, including those associated with gene sets related to the non-canonical NF-κB pathway, IRF4 targets, T-cell differentiation, cell proliferation, and adhesion or migration, which was consistent with these mice having the most severe phenotype.
We previously reported that mutations in genes encoding components of TCR-NF-κB signaling pathways were observed in 90% of samples from patients with acute-type ATL, and the median number of driver gene mutations in these samples was 1016. In addition, constitutive HBZ expression was observed in acute-type ATL samples. We then compared the gene expressions in CARD11(E626K)CD4-Cre;HBZ Tg mice and acute-type ATL samples, and examined the combined effects of TCR-NF-κB signaling pathway activation and HBZ expression on the pathogenesis of ATL. Many signaling cascades that were activated in human acute-type ATL samples were also activated in CARD11(E626K)CD4-Cre;HBZ Tg mice, indicating that TCR-NF-κB signaling pathway activation and HBZ expression were together responsible for the basic molecular pathogenesis of acute-type ATL. In line with this, the BATF3/IRF4/HBZ transcriptional network28 was enriched in both CARD11(E626K)CD4-Cre;HBZ Tg mice and human acute-type ATL samples. The proliferation of an ATL cell line was reported to be dependent on BATF3 and IRF4, which cooperatively drive ATL-specific gene expression, and HBZ was reported to induce BATF3 expression28. In CARD11(E626K)CD4-Cre;HBZ Tg mice, upregulation of BATF3 and IRF4 was observed in CD4+ T cells. On the other hand, IRF4 was not upregulated in HBZ Tg mice. CARD activation and HBZ expression might be sufficient for the cooperative induction of proliferation/survival signals driven by BATF3 and IRF4 in ATL.
Patients with acute-type ATL often exhibit an extremely elevated lymphocyte count in PB, widespread lymphadenopathy, splenomegaly, and lymphocyte invasion of not only the lung but also the digestive tract, liver, adrenal grands, and brain. As this phenotype is much more severe than that in CARD11(E626K)CD4-Cre;HBZ Tg mice, additional mutations in T cells that already harbor NF-κB activation mutations and HBZ expression might be required for the explosive increase in T cell number observed in acute-type ATL patients. In our previous analysis, CARD11 mutation was an early event in ATL development16, suggesting that later mutations occur in cells that already harbor CARD11 mutations, thereby resulting in the full phenotype of aggressive ATL. The full development of acute-type ATL might be partially due to the activation of signaling cascade genes, for example the NOTCH signaling cascade gene set, that was activated in human acute-type ATL samples but not in CARD11(E626K)CD4-Cre;HBZ Tg mice.
Methods
Mouse models
Animal studies were approved and performed in accordance with the guidelines of University of Miyazaki Ethics Committee (#582). Targeting vector used to generate CARD11 mutant mice was constructed by recombination of the mRosa26 BAC (RP23-184A7) with cDNA containing the Card11E626K mutation, the homologue of CARD11(E626K), preceded by a loxP-flanked (FL) stop sequence (Supplementary Fig. 1a). The resulting vector was electroporated into C57BL/6 J embryonic stem (ES) cells. The recombinant ES cells were injected into ICR blastocysts, and subsequent chimera breeding resulted in CARD11(E626K)stopFL mice. Crossing these mice with CD4-Cre Tg mice35 resulted in CARD11(E626K)CD4-Cre mice. To generate HBZ Tg mice, HBZ cDNA was cloned from the ATL cell line Su9T36. The clones were inserted into a cassette vector containing a mouse CD4 promoter/enhancer (Supplementary Fig. 1b)37. Electroporation and subsequent chimera breeding resulted in HBZ Tg. Crossing CARD11(E626K)CD4-Cre and HBZ Tg mice resulted in CARD11(E626K)CD4-Cre;HBZ Tg mice. These strains were backcrossed at least eight times onto the C57BL/6 background for this study. Male and female mice between 3 months and 12 months after birth were used in the experiments. An experimental flowchart is shown in Supplementary Fig. 16. The materials used in the experiments are shown in Supplementary Table 2.
Gene expression profiling of mouse samples
Splenic CD4+CD44+CD62L− Tem (3 × 104 cells per mouse) were sorted from four types of mice: wild type (WT) (n = 3), CARD11(E626K)CD4-Cre (n = 3), HBZ Tg (n = 4), and CARD11(E626K)CD4-Cre;HBZ Tg (n = 3). Total RNA extraction, cDNA library preparation, and sequencing were performed as described in the Supplementary Methods. Gene set enrichment analysis (GSEA)22,23,38 was performed using a false discovery rate (FDR) cut-off of ≤0.25. Differentially expressed genes between sample groups were identified by Integrated Differential Expression and Pathway Analysis (iDEP) Tools, using cut-offs of fold change (FC) > 1.2 and an FDR of <0.139. Functional annotation of identified genes was carried out in Enrichr40.
Comparison of gene expression signatures enriched in mutant mice and acute-type ATL samples
To assess the extent to which the mouse models mimicked the molecular pathogenesis of ATL, we compared mouse and human GSEA results for the KEGG and HALLMARK gene sets, using their normalized enrichment score (NES) and FDR values. Human gene expression data were obtained from the healthy donors (n = 3), acute-type ATL patients (n = 21), and HTLV-1 carrier (n = 3) included in our previous report (EGAS00001001296)16. Using these expression data, GSEA of human samples was performed with the same protocol used with mouse samples, including the same gene sets and the same FDR cutoff value. To interpret the single and compound effects of the CARD mutant and HBZ expression on ATL pathogenesis, each gene set in the mouse and human GSEA results was linked by name, plotted according to the mouse and human NES values, and highlighted according to the mouse and human FDR values. This was done for each combination of mouse genotype (WT/CARD11(E626K)CD4-Cre/HBZ Tg/CARD11(E626K)CD4-Cre;HBZ Tg) and human condition (healthy/acute-type ATL/HTLV-1 carrier).
Statistics and reproducibility
Differences between groups in terms of numbers and frequencies of cells were analyzed by ANOVA complemented with the post-hoc Tukey–Kramer test. Differences in animal survival, assessed by the Kaplan–Meier method, were analyzed by the log-rank test. Differences between frequencies of lymphadenopathy were assessed by Fisher’s exact test with Benjamini–Hochberg correction. In vitro experiments were repeated three times individually and statistics of the measurements were calculated; in the case of only two replications were made, the measurements were directly displayed. p < 0.05 indicated statistical significance. Statistical analysis was performed using R v.4.0.3.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cDNA sequences used are listed in Supplementary Table 2. The newly generated plasmid has been deposited in the DNA Data Bank of Japan (LC739268). Gene expression datasets obtained from human samples have been deposited at the European Genome-phenome Archive (EGAD00001001411). Gene expression datasets obtained from mouse samples have been deposited in the DNA Data Bank of Japan (DRA015050). Raw data for graphs are uploaded as Supplementary Data. Uncropped blots are provided as Supplementary Figs. 17–19. For data-sharing requests, please contact Kazuya Shimoda (kshimoda@med.miyazaki-u.ac.jp).
References
Uchiyama, T., Yodoi, J., Sagawa, K., Takatsuki, K. & Uchino, H. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood 50, 481–492 (1977).
Vose, J., Armitage, J. & Weisenburger, D., International, T. C. L. P. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J. Clin. Oncol. 26, 4124–4130 (2008).
WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Adult T-cell leukaemia/lymphoma (ed. Swerdlow, S. H. et al.). 281–284 (IARC press Lyon, 2008).
Watanabe, T. Adult T-cell leukemia: molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood 129, 1071–1081 (2017).
Ishitsuka, K. & Tamura, K. Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol. 15, e517–e526 (2014).
Matsuoka, M. & Jeang, K. T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 7, 270–280 (2007).
Matsuoka, M. & Jeang, K. T. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene 30, 1379–1389 (2011).
Grossman, W. J. et al. Development of leukemia in mice transgenic for the tax gene of human T-cell leukemia virus type I. Proc. Natl Acad. Sci. USA 92, 1057–1061 (1995).
Hasegawa, H. et al. Thymus-derived leukemia-lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat. Med. 12, 466–472 (2006).
Kinoshita, T. et al. Detection of mRNA for the tax1/rex1 gene of human T-cell leukemia virus type I in fresh peripheral blood mononuclear cells of adult T-cell leukemia patients and viral carriers by using the polymerase chain reaction. Proc. Natl Acad. Sci. USA 86, 5620–5624 (1989).
Takeda, S. et al. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int. J. Cancer 109, 559–567 (2004).
Miyazaki, M. et al. Preferential selection of human T-cell leukemia virus type 1 provirus lacking the 5’ long terminal repeat during oncogenesis. J. Virol. 81, 5714–5723 (2007).
Satou, Y., Yasunaga, J., Yoshida, M. & Matsuoka, M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl Acad. Sci. USA 103, 720–725 (2006).
Arnold, J., Zimmerman, B., Li, M., Lairmore, M. D. & Green, P. L. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood 112, 3788–3797 (2008).
Satou, Y. et al. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 7, e1001274 (2011).
Kataoka, K. et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 47, 1304–1315 (2015).
Lenz, G. et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319, 1676–1679 (2008).
Young, R. M. & Staudt, L. M. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov. 12, 229–243 (2013).
Knies, N. et al. Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant B-cell proliferation via cooperative NF-kappaB and JNK activation. Proc. Natl Acad. Sci. USA 112, E7230–E7238 (2015).
Boggs, D. R. The total marrow mass of the mouse: a simplified method of measurement. Am. J. Hematol. 16, 277–286 (1984).
Hasan, S. et al. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNalpha. Blood 122, 1464–1477 (2013).
Staudt lab. Signature database. https://lymphochip.nih.gov/signaturedb/.
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Odqvist, L. et al. NIK controls classical and alternative NF-kappaB activation and is necessary for the survival of human T-cell lymphoma cells. Clin. Cancer Res. 19, 2319–2330 (2013).
Mitobe, Y., Yasunaga, J., Furuta, R. & Matsuoka, M. HTLV-1 bZIP Factor RNA and Protein Impart Distinct Functions on T-cell Proliferation and Survival. Cancer Res 75, 4143–4152 (2015).
Higuchi, Y. et al. HTLV-1 induces T cell malignancy and inflammation by viral antisense factor-mediated modulation of the cytokine signaling. Proc. Natl Acad. Sci. USA 117, 13740–13749 (2020).
Carter, S. L., Eklund, A. C., Kohane, I. S., Harris, L. N. & Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043–1048 (2006).
Nakagawa, M. et al. Targeting the HTLV-I-regulated BATF3/IRF4 transcriptional network in adult T cell leukemia/lymphoma. Cancer Cell 34, 286–297.e210 (2018).
Nagai, Y. et al. T memory stem cells are the hierarchical apex of adult T-cell leukemia. Blood 125, 3527–3535 (2015).
Snow, A. L. et al. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J. Exp. Med. 209, 2247–2261 (2012).
Brohl, A. S. et al. Germline CARD11 mutation in a patient with severe congenital B cell lymphocytosis. J. Clin. Immunol. 35, 32–46 (2015).
Buchbinder, D. et al. Mild B-cell lymphocytosis in patients with a CARD11 C49Y mutation. J. Allergy Clin. Immunol. 136, 819–821.e811 (2015).
Esser, A. K. et al. HTLV-1 viral oncogene HBZ induces osteolytic bone disease in transgenic mice. Oncotarget 8, 69250–69263 (2017).
Zhao, T. et al. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-kappaB. Blood 113, 2755–2764 (2009).
Lee, P. P. et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774 (2001).
Sarkar, B. et al. Degradation of p47 by autophagy contributes to CADM1 overexpression in ATLL cells through the activation of NF-kappaB. Sci. Rep. 9, 3491 (2019).
Yasuda, J., Miyao, T., Kamata, M., Aida, Y. & Iwakura, Y. T cell apoptosis causes peripheral T cell depletion in mice transgenic for the HIV-1 vpr gene. Virology 285, 181–192 (2001).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Ge, S. X., Son, E. W. & Yao, R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinforma. 19, 534 (2018).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 14, 128 (2013).
Acknowledgements
The authors would like to thank M. Takeyama and Y. Nakamura (Axcelead Drug Discovery Partners, Inc.) for their technical assistance with the generation of CARD11(E626K)stopFL mice, and S. Saito, M. Matsushita, and T. Shinmori for their technical assistance with the experiments. This work was supported by Grants-in-Aid for Scientific Research (C) (17K09931, 20K08715) (T.K.) from the Japan Society for the Promotion of Science, by Research Grants (H28) (T.K.) from the Shinnihon Foundation of Advanced Medical Treatment Research, by Research Grants (R1) (T.K.) from The Japanese Society of Hematology, and by Research Grants (K. Shimoda; 20ck0106538h0001: S.O; 20ck0106409h0003) from the Japan Agency for Medical Research and Development (AMED). This study was supported by the Frontier Science Research Center, University of Miyazaki. Cell sorting experiments were supported by Y. Kawagoe.
Author information
Authors and Affiliations
Contributions
Conceptualization, T.K. and K. Shimoda; data curation, T.K., Y. Kogure, J.K., K.K., and T.O; methodology, T.K., K. Shide, A.K., Y.T., D.M., M. Sugiyama, J.K., and Y. Kogure; formal analysis, T.K., Y. Kogure, T.Y.-N., S.K., G.S., Y. Kitai, T.M. and A.Y.; investigation, T.K., K. Shide, A.K., Y.T., D.M., M. Sugiyama, K.A., K.M., M. Sekine, T.H., and Y. Kubuki; writing, T.K. and K. Shimoda; funding acquisition, T.K., K. Shimoda, and S.O.; supervision, K. Shimoda.
Corresponding author
Ethics declarations
Competing interests
K. Shimoda has received consulting fees from Novartis Pharma, Takeda Pharmaceutical, Bristol-Myers, Shire Japan, and Celgene, all outside the submitted work, and has received research grants from Perseus Proteomics, Pharma Essentia Japan KK, AbbVie GK, Astellas Pharma, MSD, Chugai Pharmaceutical, Kyowa Kirin, Pfizer, Novartis Pharma, Otsuka Pharmaceutical, Asahi Kasei Medical, all outside the submitted work. K.K. holds stock in Asahi Genomics, has a patent for genetic alterations as a biomarker in T-cell lymphomas, and has received research funding from Chordia Therapeutics outside the submitted work. The remaining authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Lennart Lenk, Yaacov Ben-David and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Guideng Li and Eve Rogers. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kameda, T., Shide, K., Kamiunten, A. et al. CARD11 mutation and HBZ expression induce lymphoproliferative disease and adult T-cell leukemia/lymphoma. Commun Biol 5, 1309 (2022). https://doi.org/10.1038/s42003-022-04284-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-04284-x
This article is cited by
-
A case of T-cell acute lymphoblastic leukemia in retroviral gene therapy for ADA-SCID
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
