
Abstract
Membrane proteins are essential for cellular growth, signalling and homeostasis, making up a large proportion of therapeutic targets. However, the necessity for a solubilising agent to extract them from the membrane creates challenges in their structural and functional study. Although amphipols have been very effective for single-particle electron cryo-microscopy (cryoEM) and mass spectrometry, they rely on initial detergent extraction before exchange into the amphipol environment. Therefore, circumventing this pre-requirement would be a big advantage. Here we use an alternative type of amphipol: a cycloalkane-modified amphiphile polymer (CyclAPol) to extract Escherichia coli AcrB directly from the membrane and demonstrate that the protein can be isolated in a one-step purification with the resultant cryoEM structure achieving 3.2 Å resolution. Together this work shows that cycloalkane amphipols provide a powerful approach for the study of membrane proteins, allowing native extraction and high-resolution structure determination by cryoEM.
Similar content being viewed by others

Introduction
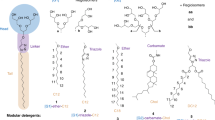
Membrane proteins represent ~30% of open reading frames in the human genome, and an important class of drug targets1 and yet make up only 3% of reported structures in the PDB. Despite their prevalence in the cell and importance for ion transport and cell signalling, amongst other functions, they remain challenging research targets due to problems of overexpression, extraction and stabilisation of their native structure2,3,4,5. Traditionally extraction and purification of a membrane protein involves the use of a detergent, from which the protein may then be transferred into other surfactants, be they detergents of different chemical composition, protein-based nanodiscs6, peptidiscs7 or amphipathic polymers8,9 (structures for which can be seen in Fig. 1a–e). Extraction of a membrane protein into a detergent micelle functions by disrupting the interaction between protein and its surrounding lipid molecules10. Detergent molecules replace the bulk of lipids at the hydrophobic surface of a membrane protein but poorly mimic the lipid bilayer in terms of lateral pressure and thickness which has been shown to cause perturbations in the structure11,12. Moreover, the closely associated lipids which can be important for gating, regulation and stability, maybe displaced by competition with the detergent13,14,15,16. In addition, detergent purification buffers must contain the detergent above its critical micelle concentration (CMC) in all downstream steps which can exacerbate reduction in activity, protein complex dissociation, unnatural oligomerisation and loss of lipid cofactors, amongst other problems17,18,19. Detergent micelles in single-particle cryoEM lead to reduced contrast and increased noise20,21 and must be disassembled in native mass spectrometry (MS)22. Due to the importance of membrane proteins and the problems associated with detergents, there exist several membrane mimetic alternatives developed to circumvent these. The predominant ones are protein-based nanodiscs6 and amphipathic polymers8,9.
Classical amphipols (APols) are short and flexible amphipathic polymers able to form complexes with membrane proteins and maintain the proteins in a water-soluble form8. They have been used for decades and are well-characterised in their applicability for stabilising membrane proteins8,23. The prototypical APol A8-35 is a poly(acrylic acid) (PAA) polymer randomly modified with octylamine and isopropylamine side chains24, and many different functionalities have been tethered to the polymer for specific purposes23,25. APol A8-35 facilitated the first high-resolution single-particle cryoEM structure of a membrane protein, that of TRPV126. Since then, the number of high-resolution cryoEM structures of membrane proteins using APols (mainly A8-35 and PMAL-C8)27 has increased28. Of those cryoEM structures deposited within the EMDB, the best resolution achieved using classical APols is 2.17 Å29. In addition, APols are amenable to native electrospray ionisation (ESI)-MS30. However, A8-35 and the other classical APols traditionally require initial detergent extraction of the protein31. Recently, this limitation has been overcome with the development of cycloalkane-modified APols (which contain cyclic rather than linear aliphatic groups) showing much greater efficiency at extracting proteins directly from the membrane than the common A8-35 APol32.
The advantage of using polymers for extraction and purification of membrane proteins emerged a decade ago with the use of styrene and maleic acid (SMA) co-polymers in the field of membrane protein research33. SMAs heralded the advent of ‘native’ nanodiscs containing a protein directly extracted from the membrane, with its endogenous lipids and without the requirement for conventional detergents9,34,35,36,37,38. The styrene-maleic acid-lipid particles (SMALPs) formed39 lend themselves to a plethora of biophysical techniques, including cryoEM40,41. However, SMALPs also have their limitations (sensitivity to pH extremes and divalent cations), as such there is a continuous interest in developing new SMA-like polymers such as the acrylic acid and styrene polymers (AASTY)42 which can be used to directly extract proteins from the membrane, but currently, their applicability to cryoEM has been limited to ~18 Å resolution. Moreover, to date SMA-derived polymers are generally not amenable to ESI MS, having only successfully and recently been applied to native ESI MS for bacteriorhodopsin43. Although it has recently been demonstrated that A8-35 can be utilised following protein extraction with SMA44, an APol-like polymer combining the extraction capability of SMA with the applications of A8-35 would be highly advantageous.
Here we demonstrate that the properties of A8-35 and SMA can be combined through cycloalkane-modified APols with SMALP-like properties for direct extraction32. Using Escherichia coli AcrB, we demonstrate that these APol derivatives (henceforth distinguished as CyclAPols) are capable of solubilising the protein of interest directly from the membrane. The CyclAPols can be utilised at exceptionally low concentrations (0.1–0.5%), minimising the risk of destabilisation due to high APol concentrations45,46. We present the first cryoEM structure of a protein in CyclAPols, at 3.2 Å resolution, demonstrating their applicability to high-resolution structure determination by cryoEM and making these APols an important new tool in the study of membrane proteins.
Results
Cycloalkane-modified amphiphile polymers can solubilise proteins directly from membranes
The CyclAPols (C6-C2-50 and C8-C0-50) in addition to A8-35, were compared for their capability for direct membrane solubilisation. E. coli membranes overexpressing the exporter AcrB were homogenised and incubated with each polymer before ultracentrifugation to remove insoluble material. Western blot analysis showed that all polymers are capable of solubilising membranes and extracting AcrB, with the amphipathic polymers CyclAPol C6-C2-50 and C8-C0-50 showing greater solubilisation efficacy than A8-35 (Fig. S1 and supplementary methods). It was determined that a 0.1% (w/v) concentration of CyclAPols was sufficient for downstream experiments, with yields only slightly lower relative to that obtained for the SMA polymer, despite the lower polymer concentration for CyclAPols (0.1 vs 1%).

SDS-PAGE of purification of AcrB in a DDM, b SMA, c A8-35, d C6-C2-50, and e C8-C0-50. Purification is shown with flow-through (FT) from affinity beads, and wash (W) steps, in addition to soluble (S) and insoluble (I) samples for the amphipol purifications (for experimental details, see the M&M section). All polymers showed the ability to directly extract and purify AcrB with different efficiencies and resulting in different purities. The CyclAPols C6-C2-50 and C8-C0-50 showed a reduced efficiency to DDM and SMA (mainly because their concentration used at the solubilisation step is ten times lower) but with much-improved efficiency than the A8-35 polymer.
A one-step purification with affinity resin was carried out of AcrB extracted with SMA, DDM, A8-35 or CyclAPol (Fig. 1a–e). Under the experimental conditions used, C8-C0-50 appeared to perform better than C6-C2-50. This one-step purification procedure with SMA has previously been observed to result in clean homogenous protein44,47, with increased purity of SMA-solubilised AcrB relative to detergent44. While minor modifications were made to optimise buffers for compatibility with the polymers, purification with CyclAPols resulted in clear elution fractions containing relatively pure AcrB protein consistent with a one-step purification (Fig. 1). The resultant elution fractions of each purification were pooled and dialysed to remove imidazole and protein concentrated to ~1 mg/mL.
Negative stain electron microscopy was used to assess the homogeneity and stability of AcrB trimer extracted and purified in CyclAPols C6-C2-50 and C8-C0-50 showing homogenous, monodisperse protein, with less background contamination than typically observed for detergent micelles and similar to SMA-purified AcrB (Fig. 2 a, c, d). The low level of aggregation, clear trimers and low background observed in the negative stain data for the C6-C2-50, C8-C0-50 and SMA samples were indicative of a sample suitable for cryoEM. However, images of A8-35-purified AcrB showed large aggregates which likely contain several copies of AcrB and only a small percentage of monodispersed AcrB (Fig. 2b). The large aggregates suggest that A8-35 at similar concentrations than the other polymers (0.5 vs 0.1% for CyclApols and 1% SMA) is not as efficient as CyclAPols at breaking apart the membrane and may produce larger lipid bilayer fragments containing multiple copies of AcrB. 2D classification of AcrB purified with C6-C2-50 (e) and C8-C0-50 (f) showed typical features to those seen with AcrB-SMA48 along with increased high angle views in addition to the typical side and top views, particularly for C8-C0-50 (f, green boxes).

Representative micrographs were collected at 49k magnification of AcrB following purification in (a) SMA, (b) A8-35, (c) C6-C2-50, and (d) C8-C0-50. Scale bar is 100 nm in all images. Negative stain 2D classes of AcrB in (e) C6-C2-50 and (f) C8-C0-50 are also shown with some representative high-angle classes highlighted in green. Images (a) and (b-d) were taken using Tecnai F20 and G2-spirit T12 transmission electron microscopes, respectively.
Single-particle cryoEM of AcrB in CyclAPol C8-C0-50
We next investigated if CyclAPols, like the classic APols such as A8-35 and PMAL-C8, were also capable of providing a suitable environment for high-resolution structure determination by cryoEM. Purified AcrB was vitrified on Quantifoil grids for single-particle cryoEM analysis. While AcrB extracted and purified in A8-35 was not suitable for cryoEM due to particle aggregation, in the screening of grids both CyclAPols exhibited sufficient particle distribution. AcrB in C8-C0-50 showed the best distribution and was taken forward for data collection. Consistently, the C8-C0-50 polymer marginally outperformed C6-C2-50, with slightly increased purity, yield (Fig. 1) and particle homogeneity as seen in negative stain (Fig. 2) and screening in cryoEM (Fig. S2).
Following data collection, particle picking was carried out with CrYOLO49, and extraction and further processing were carried out in RELION50. Approximately 400k particles were initially extracted from 1837 micrographs. Following two rounds of 2D classification, ~200k particles were selected for further 3D classification and processing. Initial 2D classes showed a clear AcrB trimer, with a good angular distribution within the data (Fig. 3a). This is important in reducing the anisotropy in the data that can arise through preferred orientations resulting in reduced map quality. It was noted that a small population of the 2D classes exhibited clear doublets of AcrB trimers (0.5–1% of particles) which had previously been seen in negative stain studies of AcrB in SMA48, but not reported in the published structures40,41.

a Classes of AcrB in C8-C0-50 following one round of 2D classification. The classes demonstrate a range of views such as a high angle view showing putative threefold symmetry (highlighted in green) with classes showing clear doublets highlighted in red. b Side view of the AcrB cryoEM map at 3.2 Å final resolution. c Representative map density of AcrB in C8-C0-50 around the top of the vestibule region. d Overlay of the cryoEM structure of AcrB in SMA40 (PDB accession code: 6baj; pink) with the AcrB in C8-C0-50 structure (PDB accession code 7B5P; grey) demonstrating the close similarity of structures.
The resultant 3D reconstruction, processed with C1 symmetry, achieved a final global resolution of 3.2 Å with clearly resolved density for the secondary structure and in most cases the side chains (Fig. 3, b, c). The local resolution is lower for those helices on the surface of AcrB within the membrane domain, where density for the side chains could not be unambiguously resolved. The previously derived EM structure of AcrB in SMA40 was used as a starting point for model building and refinement, with the resultant model being highly similar to previously published AcrB structures40,41,51 (Fig. 3d). The structure is asymmetric and exhibits a clear cavity at the interior of the trimer, which after model fitting was devoid of any density that could be assigned to lipids (Fig. 4a–d). This is especially apparent when viewed from the base of the structure, where the trimeric pseudo-symmetry and resolved helices are very clear. Particularly, the structure appears well resolved at the transmembrane region with clear density for the side chains, consistent with a resolution of ~3.5 Å.

Density map of C8-C0-50-purified AcrB seen around the transmembrane region from the side and base. The lipids from AcrB solved by cryoEM in SMA are superimposed and shown in purple, red and orange for carbon, oxygen and phosphate, respectively. The density contour level in (a) and (b) is comparable to that of Fig. 3c (0.025), a lower threshold of density (0.016) is shown in (c) and (d) with increased noise. At both contour levels, no density is present that could be assigned to the bound lipids.
Comparison to AcrB in other amphipathic environments
Comparing the refined structure, especially the chain C of the AcrB trimer, in C8-C0-50 to the previously published structures in SMA (6baj)40 and saposin (6sgu)51 using Chimera showed a root mean square deviation (RMSD) of the backbone atoms of 0.7 and 1.5 Å, respectively, reflecting their close similarity. Comparison of the maps (Fig. S3) or overlay of the SMA and CyclAPol structures (Fig. 3d) demonstrates no noticeable difference between structures and only minor variation in loop regions. It is noted that in the reported cryoEM structures of AcrB in SMA or saposin at comparable resolutions, lipids have been identified throughout the transmembrane region. The absence of density observed for lipids in the C8-C0-50 reconstruction (Fig. 4) leads us to investigate the presence of co-purified lipids in the samples by thin-layer chromatography (Fig. S4 and Supplementary methods). This experiment demonstrates the clear presence of lipids in all samples of AcrB solubilised and purified with the different polymers including C8-C0-50. Therefore, the apparent lack of lipids in the EM structure is likely due to their mobility which prevents them from being resolved.
Discussion
Membrane proteins present significant challenges, not least in finding a suitable amphipathic environment that can directly extract the protein from the membrane and stabilise it in an aqueous solution. Although classical APols such as A8-35 are effective in cryoEM, their typical reliance on detergents in the early stages of membrane extraction may be problematic. Using AcrB, we demonstrate that A8-35, as long suspected8, may directly extract proteins from the membrane. However, the yield of AcrB extracted with A8-35 is low, confirming the poor detergency property of A8-35 as previously reported. Solubilisation with A8-35 is also incomplete as large objects similar to small vesicles are observed by negative stain EM (Fig. 2), the size of which perhaps could be fine-tuned by A8-35 concentration (currently 0.5%). Although this makes direct extraction of AcrB with A8-35 unsuitable for single-particle cryoEM, the ability to fragment the membrane into larger rafts may be useful for other techniques, such as AFM or mass spectrometry52, but was beyond the scope of this study. In contrast to A8-35, the two CyclAPols tested are very effective at solubilising AcrB from the membrane at low concentrations (at an estimated total protein/polymer ratio of 1:1 w/w). This is consistent with the previous finding that the CyclAPols are more efficient than A8-35 at extracting proteins from the membrane, regardless of the target protein32. Furthermore, CyclAPol-extracted AcrB (a His-tagged version) can be obtained at a satisfactory level of purity after an affinity purification step. On-going work shows that other affinity tags fused to different bacterial or mammalian membrane proteins including a bacterial ABC transporter (a Strep-tag version) can be also utilised for purification following solubilisation with CyclAPols (Fig. S5).
Importantly, the CyclAPols are compatible with high-resolution cryoEM studies with the resultant 3.2 Å resolution structure of AcrB obtained in CyclAPol C8-C0-50 being in line with the resolutions regularly obtained with classical APols. There is no noticeable change in structure compared to other amphipathic environments, although lipids are not currently visible, and this represents the joint highest resolution AcrB cryoEM structure40,51. We noted a clear improvement in resolution compared to our in-house AcrB-SMA cryoEM reconstructions, the highest resolution of which is ~4.0 Å41 and for which the data acquisition setup and data processing pipelines were comparable.
In this study, the CyclAPol C6-C2-50 appeared less amenable to purification than C8-C0-50, and while two data collections were attempted with this polymer, the best resolution obtained was 4.4 Å (Fig. S2). As previously noted, while less doublets are visible in 2D classification, a higher proportion of the protein appears aggregated or in multimeric chains after purification with this polymer contributing to a highly diffuse transmembrane region (Fig. S2). This highlights how subtle differences in the chemistry between the two CyclAPols can have an effect on the downstream applications, but this effect may be protein-dependent. The overall architecture of AcrB is near indistinguishable between reconstructions in C6-C2-50 and C8-C0-50, although at the lower resolution we may not observe subtle differences.
AcrB is an ideal model protein for such studies as it has been widely characterised with high-resolution cryoEM structures being determined in SMA (3.2 Å),40 saposin (3.2 Å)51 and most recently in liposomes (3.9 Å)53. Studies in saposin51 and liposomes53 involve reconstitution subsequent to detergent purification. Unlike structures determined in both saposin and SMA, AcrB within liposomes does not appear to show closely associated internal lipids. There were also no identifiable lipids in the cryoEM structure of CyclAPol-purified AcrB, despite the clear evidence of co-purified lipids as observed by TLC experiments (Fig. S4). Although lipids have been detected in other cryoEM studies of AcrB40,50, it is possible these are not critical for the function, as AcrZ51 or AcrA54 may mediate interactions between AcrB and lipids, and structures that show no difference simply reflect increased mobility of the lipid within the AcrB system. Furthermore, in contrast to studies in liposomes, in which a great deal of optimisation of cryoEM conditions was required,53 the cryoEM structure obtained here in C8-C0-50 was the result of a single batch of cryoEM grids with no subsequent optimisation.
The CyclAPols represent an important tool in the field of membrane protein structural studies, which may come to represent an important alternative to detergent and SMA. They extract proteins directly from the membrane at low concentrations and provide a clean purification of the target membrane protein. Compared to the AASTY42 polymer, the improved compatibility of CyclAPols with cryoEM shows that changing the chemistry of the polymers can have beneficial effects on sample quality. Moreover, our initial studies have shown that such samples can now be analysed by native MS with the facile release of intact membrane protein complexes for structural analysis with further studies ongoing. We present here the first cryoEM foray into these cycloalkane-modified amphiphile polymer derivatives, anticipating wider applicability to membrane proteins still to be discovered.
Membrane proteins offer great challenges in their study, with a major limitation being in using a solubilising agent that can both solubilise and stabilise the protein of interest and also be applicable to a range of downstream analysis techniques. APols have a strong track record in their applicability to single-particle cryoEM and native MS but have relied on initial detergent extraction which brings with it some limitations. Here we have shown that a modified cycloalkane APol negates the need for initial detergent extraction whilst maintaining the applicability to high-resolution EM structures. This new generation of APols may provide an important addition to the membrane protein toolkit creating more opportunities for membrane protein studies.
Methods
Polymer synthesis
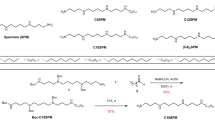
Polymers C6-C2-50 and C8-C0-50 were synthesised and characterised as described fully in Marconnet et al.32 by the grafting of hydrophobic side chains onto polyacrylic acid in the presence of dicyclohexylcarbodiimide. In addition, we used the commercial amphipol A8-35 and DDM from Anatrace. SMA (2:1) supplied unhydrolyzed from Cray Valley which was prepared as previously described in ref. 36. Briefly, the unhydrolyzed polymer is solubilised in 1 M NaOH and heated with reflux for 2 h. Once cooled the polymer is precipitated, centrifuged and washed by multiple rounds of resuspension and centrifugation, first in dH2O, then 0.6 M NaOH pH 8, before being freeze-dried and stored.
Preparation of E. coli membranes
E. coli membranes were prepared according to chapter 3.4 of Postis et al.55. Briefly, the C43(DE3), pRARE2, ΔacrB strain of E. coli was used for overexpression by auto-induction in SB media. Cells were harvested, resuspended for cell disruption, cell debris centrifuged and discarded before centrifugation at 100,000×g for 2 h to collect the membranes. Membranes were resuspended in a minimal volume (~8 mL) of buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 10% (v/v) glycerol) and frozen for storage at −80 °C. The total protein concentration of the resuspended membranes was measured using a BCA assay as per the manufacturer’s recommendations (Thermo Scientific).
AcrB purification
Purification of AcrB in SMA was carried out as described previously44,47 but with 1% (w/v) SMA at the solubilisation step (see below). For purification in SMA, the solubilisation buffer was 50 mM Tris-HCl pH 8.0, 500 mM NaCl, 10% glycerol. Wash and elution buffers additionally contain 20 mM imidazole and 300 mM imidazole, respectively. Purification in amphipols was similar but buffers were modified to reduce the ionic strength. For this, the solubilisation buffer contained 20 mM Tris-HCl pH 8.0, 250 mM NaCl, 5% glycerol. Wash and elution buffers were supplemented with 10 mM imidazole and 300 mM imidazole, respectively. A second purification was also carried out, which provided the sample of AcrB in CyclAPol C6-C2-50 for cryoEM data collections. For this second purification, the protocol was largely similar but an extra resin wash was carried out with buffer containing 50 mM imidazole, and the elution was performed with 500 mM imidazole.
Purification was carried out with membranes homogenised in solubilisation buffer to 1 mg/mL. Typically, 1 g of the membrane (at protein concentration 25 mg/mL), was homogenised in 25 mL. SMA at 1% (w/v) was added directly from powder, while CyclAPols (0.1%) and A8-35 (0.5%) were added from 5% (w/v) stock solutions and samples were incubated for 2 h at room temperature (25 ˚C) before ultracentrifugation at 100,000×g for 1 h at 4 °C to remove insoluble material.
For each gram of solubilised membrane, 1 mL of final cobalt resin was used. HisPur Cobalt Slurry (Thermo) was prepared by 3x washes each with ddH2O, solubilisation buffer and wash buffer (containing 10 mM imidazole). The soluble material was incubated with this equilibrated cobalt resin overnight at 4 °C with rotation. The flow-through was collected, the resin washed with 5 column volumes (5 mL) solubilisation buffer and 5 column volumes wash buffer before elution fractions were collected and analysed by SDS-PAGE. Elution fractions containing purified AcrB were pooled, placed inside SnakeSkin 10 kDa MWCO dialysis tubing (ThermoFisher) and dialysed overnight against 300 mL solubilisation buffer (20 mM Tris-HCl pH 8.0, 250 mM NaCl, 5% glycerol) at 4 °C, with three buffer changes to remove imidazole. Samples were then concentrated using 100 kDa MWCO nitrocellulose concentrator (Merck) and the final concentration was measured using a DS-11 Spectrophotometer (DeNovix). Final samples for A8-35 and CyclAPols were in 20 mM Tris-HCl pH 8.0, 250 mM NaCl, 5% glycerol.
Negative stain electron microscopy
Purified AcrB was diluted to 50 μg/mL in solubilisation buffer. Three microlitres of the sample was applied to a glow-discharged carbon grid, incubated for 30 s and excess removed with blotting paper. The grid was washed with double-distilled water and stained with 1% uranyl acetate. Grids of APol-purified AcrB were imaged at 50k magnification using a Tecnai G2-spirit T12 transmission electron microscope (FEI) fitted with a 120 keV Lab6 electron source and Ultra Scan 4000 CCD camera (Gatan). Grids of SMA-purified AcrB were imaged using a Tecnai F20 transmission electron microscope (FEI) fitted with a 200 keV FEG electron source and a CETA CMOS CCD camera (FEI).
Cryo-electron microscopy
Quantifoil 1.2/1.3 cryo-electron microscopy (cryoEM) grids (QUANTIFOILS) were prepared by glow discharging with a 208-carbon High Vacuum Carbon Coater (Cressington). Purified AcrB at ~1 mg/mL in 20 mM Tris-HCl pH 8.0, 250 mM NaCl, 5% glycerol, after solubilisation with 0.5% A8-35, 0.1% C6-C2-50 and 0.1% C8-C0-50 was applied to grids. CryoEM specimens were prepared with a FEI Vitrobot grid preparation robot at 4 °C and 100% humidity by applying 3 μl of the sample (~1 mg/mL) to glow-discharged grids, blotting for 6 s with a blot force of 6 before freezing in liquid ethane. Grids were stored in liquid nitrogen and imaged subsequently using a Titan Krios G3i cryo transmission electron microscope (FEI) at 300 keV voltage equipped with a Gatan K2 Summit camera at the Astbury Biostructure Laboratory. Grids were screened to assess ice thickness, AcrB concentration, monodispersion and homogeneity.
Electron microscopy data acquisition
Movies were acquired in electron counting mode with a pixel size of 1.07 Å, an exposure rate of 6.6 electrons per pixel per second, and a total exposure time of 10 s divided in 40 frames. Frame alignment and exposure weighting were performed with Motioncor56. Contrast transfer function parameters were estimated from the exposure-weighted averages of movie frames with CTFFIND57.
Image processing
Automated picking of particles was carried out using crYOLO50 with the general model trained on a subset of particles and picking threshold at 0.2. From 1837 micrographs 409,113 particles were picked of which 402,672 were extracted into RELION 51. Two rounds of 2D classification and three rounds of 3D classification were carried out, reducing particle numbers to 100k, prior to further refinement. The map for AcrB structure in SMA40, EMD-7074, in a 256-pixel box and low-pass filtered to 30 Å was used as an initial model. The dataset was also processed in cryoSPARC58, from the raw image stage, obtaining a similar resolution of 3.3 Å at the final stage of refinement. As cryoSPARC’s own algorithms were used for automated picking and model generation this served as an internal control that no bias was imposed. The model was produced by the manual fitting of the AcrB structure obtained in SMA (6baj), with lipids removed, into the map. One round of real-space refinement in Phenix was performed before fitting in Coot. Sidechains were deleted where unambiguous density was not observed. The construct used possesses two additional N-terminal residues and a C-terminus extension including a His-tag. However, these were not seen in the final map, and numbering was matched to the canonical E. coli sequence. Data collection and processing statistics are given in Table 1.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Structural data are available via the Protein Data Bank (PDB 7B5P) and the Electron Microscopy Data Bank (EMDB-12043). Any remaining information can be obtained from the corresponding author upon reasonable request
Change history
05 January 2022
A Correction to this paper has been published: https://doi.org/10.1038/s42003-021-02997-z
References
Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 (2006).
Tate, C. G. Practical considerations of membrane protein instability during purification and crystallisation. Methods Mol. Biol. 601, 187–203 (2010).
Bill, R. M. et al. Overcoming barriers to membrane protein structure determination. Nat. Biotechnol. 29, 335–340 (2011).
Rawson, S., Davies, S., Lippiat, J. D. & Muench, S. P. The changing landscape of membrane protein structural biology through developments in electron microscopy. Mol. Membr. Biol. 33, 12–22 (2016).
Zoonens, M. & Miroux, B. Expression of membrane proteins at the Escherichia coli membrane for structural studies. Methods Mol. Biol. 601, 49–66 (2010).
Denisov, I. G. & Sligar, S. G. Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol. 23, 481–486 (2016).
Carlson, M. L. et al. The peptidisc, a simple method for stabilizing membrane proteins in detergent-free solution. Elife 7, 1–23 (2018).
Popot, J.-L. Membrane Proteins in Aqueous Solutions. From Detergents to Amphipols (Springer, 2018).
Dörr, J. M. et al. The styrene–maleic acid copolymer: a versatile tool in membrane research. Eur. Biophys. J. 45, 3–21 (2016).
Arnold, T. & Linke, D. The use of detergents to purify membrane proteins. Curr. Protoc. Protein Sci. Chapter 4, Unit 4.8.1–4.8.30. (2008).
Cross, T. A., Sharma, M., Yi, M. & Zhou, H. X. Influence of solubilizing environments on membrane protein structures. Trends Biochem. Sci. 36, 117–125 (2011).
Chipot, C. et al. Perturbations of native membrane protein structure in alkyl phosphocholine detergents: a critical assessment of NMR and biophysical studies. Chem. Rev. 118, 3559–3607 (2018).
Hedger, G. & Sansom, M. S. P. Lipid interaction sites on channels, transporters and receptors: recent insights from molecular dynamics simulations. Biochim. Biophys. Acta 1858, 2390–2400 (2016).
Willegems, K. & Efremov, R. G. Influence of lipid mimetics on gating of Ryanodine receptor. Structure 26, 1303–1313.e4 (2018).
Pliotas, C. et al. The role of lipids in mechanosensation. Nat. Struct. Mol. Biol. 22, 991–998 (2015).
Gupta, K. et al. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 541, 421–424 (2017).
Baylon, J. L. et al. Atomic-level description of protein-lipid interactions using an accelerated membrane model. Biochim. Biophys. Acta 1858, 1573–1583 (2016).
Kurauskas, V. et al. How detergent impacts membrane proteins: atomic-level views of mitochondrial carriers in dodecylphosphocholine. J. Phys. Chem. Lett. https://doi.org/10.1021/acs.jpclett.8b00269 (2018).
Dorwart, M. R., Wray, R., Brautigam, C. A., Jiang, Y. & Blount, P. S. aureus MscL is a pentamer in vivo but of variable stoichiometries in vitro: Implications for detergent- solubilized membrane proteins. PLoS Biol. https://doi.org/10.1371/journal.pbio.1000555 (2010).
Gewering, T., Januliene, D., Ries, A. B. & Moeller, A. Know your detergents: a case study on detergent background in negative stain electron microscopy. J. Struct. Biol. 203, 242–246 (2018).
Vinothkumar, K. R. & Henderson, R. Single particle electron cryomicroscopy: trends, issues and future perspective. Q. Rev. Biophys. 49, e13 (2016).
Konijnenberg, A. et al. Global structural changes of an ion channel during its gating are followed by ion mobility mass spectrometry. Proc. Natl Acad. Sci. USA 111, 17170–17175 (2014).
Zoonens, M. & Popot, J. L. Amphipols for each season. J. Membr. Biol. 247, 759–796 (2014).
Tribet, C., Audebert, R. & Popot, J. L. Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc. Natl Acad. Sci. USA 93, 15047–15050. (1996).
Della Pia, E. A., Hansen, R. W., Zoonens, M. & Martinez, K. L. Functionalized amphipols: a versatile toolbox suitable for applications of membrane proteins in synthetic biology. J. Membr. Biol. 247, 815–826 (2014).
Liao, M., Cao, E., Julius, D. & Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013).
Nagy, J. K. et al. Use of amphipathic polymers to deliver a membrane protein to lipid bilayers. FEBS Lett. 501, 115–120 (2001).
Bon, C. Le, Michon, B., Popot, J. L. & Zoonens, M. Amphipathic environments for determining the structure of membrane proteins by single-particle electron cryo-microscopy. Q. Rev. Biophys. 54, e6 (2021).
Owji, A. P. et al. Structural and functional characterization of the bestrophin-2 anion channel. Nat. Struct. Mol. Biol. 27, 382–391 (2020).
Watkinson, T. G. et al. Systematic analysis of the use of amphipathic polymers for studies of outer membrane proteins using mass spectrometry. Int. J. Mass Spectrom. 391, 54–61 (2015).
Le Bon, C., Marconnet, A., Masscheleyn, S., Popot, J. L. & Zoonens, M. Folding and stabilizing membrane proteins in amphipol A8-35. Methods 147, 95–105 (2018).
Marconnet, A. et al. Solubilization and stabilization of membrane proteins by cycloalkane-modified amphiphilic polymers. Biomacromolecules 21, 3459–3467 (2020).
Knowles, T. J. et al. Membrane proteins solubilized intact in lipid containing nanoparticles bounded by styrene maleic acid copolymer. J. Am. Chem. Soc. 131, 7484–7485 (2009).
Jamshad, M. et al. Structural analysis of a nanoparticle containing a lipid bilayer used for detergent-free extraction of membrane proteins. Nano Res. 8, 774–789 (2015).
Gulati, S. et al. Detergent free purification of ABC transporters. Biochem. J. 44, 1–24 (2014).
Lee, S. C. et al. A method for detergent-free isolation of membrane proteins in their local lipid environment. Nat. Protoc. 11, 1149–1162 (2016).
Jamshad, M. et al. Surfactant-free purification of membrane proteins with intact native membrane environment. Biochem. Soc. Trans. 39, 813–818 (2011).
Rajesh, S., Knowles, T. & Overduin, M. Production of membrane proteins without cells or detergents. N. Biotechnol. 28, 250–254 (2011).
Orwick-Rydmark, M. et al. Detergent-free incorporation of a seven-transmembrane receptor protein into nanosized bilayer lipodisq particles for functional and biophysical studies. Nano Lett. 12, 4687–4692 (2012).
Qiu, W. et al. Structure and activity of lipid bilayer within a membrane-protein transporter. Proc. Natl Acad. Sci. USA 115, 12985–12990 (2018).
Johnson, R. M. et al. Cryo-EM structure and molecular dynamics analysis of the fluoroquinolone resistant mutant of the acrb transporter from salmonella. Microorganisms 8, 943. (2020).
Smith, A. A. A. et al. Lipid nanodiscs via ordered copolymers. Chem 6, 2782–2795 (2020).
Hoi, K. K. et al. Detergent-free lipodisq nanoparticles facilitate high-resolution mass spectrometry of folded integral membrane proteins. Nano Lett. 21, 2824–2831 (2021).
Hesketh, S. J. et al. Styrene maleic-acid lipid particles (SMALPs) into detergent or amphipols: an exchange protocol for membrane protein characterisation. Biochim. Biophys. Acta 1862, 183192 (2020).
Popot, J. L. et al. Amphipols from a to Z*. Annu. Rev. Biophys. 40, 379–408 (2011).
Sverzhinsky, A. et al. Amphipol-trapped ExbB–ExbD membrane protein complex from Escherichia coli: a biochemical and structural case study. J. Membr. Biol. 247, 1005–1018 (2014).
Parmar, M. et al. Using a SMALP platform to determine a sub-nm single particle cryo-EM membrane protein structure. Biochim. Biophys. Acta 1860, 378–383 (2018).
Postis, V. et al. The use of SMALPs as a novel membrane protein scaffold for structure study by negative stain electron microscopy. Biochim. Biophys. Acta 1848, 496–501 (2015).
Wagner, T. et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 1–13 (2019).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Du, D. et al. Interactions of a bacterial RND transporter with a transmembrane small protein in a lipid environment. Structure 28, 625–634.e6 (2020).
Chorev, D. S. & Robinson, C. V. “Protein assemblies ejected directly from native membranes yield complexes for mass spectrometry”. Science 362, 829–834 (2018).
Yao, X., Fan, X. & Yan, N. Cryo-EM analysis of a membrane protein embedded in the liposome. Proc. Natl Acad. Sci. USA 117, 18497–18503 (2020).
Shi, X. et al. In situ structure and assembly of the multidrug efflux pump AcrAB-TolC. Nat. Commun. 10, 2635 (2019).
Postis, V. L. G., Rawlings, A. E., Lesiuk, A. & Baldwin, S. A. Ion Channels: Methods and Protocols. Methods in Molecular Biology (Humana Press, 2013).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. CryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Acknowledgements
We would like to acknowledge the Muench and Sobott labs for fruitful discussions, particularly Anton Calabrese and David Klebl. The authors thank Arndt Rohwedder for assistance with TLC, and the Astbury Biostructure Laboratory for their assistance with EM data collection. The FEI Titan Krios microscopes were funded by the University of Leeds (UoL ABSL award) and Wellcome Trust (108466/Z/15/Z). This work and A.J.H. and L.J.M. were funded through a BBSRC grant (BB/R018561/1/). The amphipol development in IBPC Paris was supported by the Centre National de la Recherche Scientifique (CNRS), Université de Paris (Université Paris 7), and the ‘Initiative d’Excellence’ programme from the French State (Grant ‘DYNAMO’, ANR-11-LABX-0011-01).
Author information
Authors and Affiliations
Contributions
Conceptualisation and experimental design: S.P.M., F.S., A.J.H., A.J.F., C.-w.C., T.C., J.L., M.Z. and V.L.G.P.; Polymer design and synthesis: M.Z. and A.M.; Performed experiments: A.J.F. L.J.M. and A.J.H.; Analysis of cryoEM data and model refinement: A.J.H., A.J.F., S.P.M. Analysis and discussion of data and wrote the manuscript: all authors. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Janesh Kumar and Anam Akhtar.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Higgins, A.J., Flynn, A.J., Marconnet, A. et al. Cycloalkane-modified amphiphilic polymers provide direct extraction of membrane proteins for CryoEM analysis. Commun Biol 4, 1337 (2021). https://doi.org/10.1038/s42003-021-02834-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02834-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
