
Abstract
Neurons and sensory cells are particularly vulnerable to oxidative stress due to their high oxygen demand during stimulus perception and transmission. The mechanisms that protect them from stress-induced death and degeneration remain elusive. Here we show that embryonic deletion of the chromodomain helicase DNA-binding protein 7 (CHD7) in auditory neurons or hair cells leads to sensorineural hearing loss due to postnatal degeneration of both cell types. Mechanistically, we demonstrate that CHD7 controls the expression of major stress pathway components. In its absence, hair cells are hypersensitive, dying rapidly after brief exposure to stress inducers, suggesting that sound at the onset of hearing triggers their degeneration. In humans, CHD7 haploinsufficiency causes CHARGE syndrome, a disorder affecting multiple organs including the ear. Our findings suggest that CHD7 mutations cause developmentally silent phenotypes that predispose cells to postnatal degeneration due to a failure of protective mechanisms.
Similar content being viewed by others


Introduction
Sensorineural hearing loss (SNHL) is a common feature of CHARGE syndrome and affects 50–70% of individuals1,2. Mice with heterozygous Chd7 mutations are an excellent model for CHARGE syndrome and, like humans, they exhibit SNHL3,4,5,6. Chd7 plays an important role during neurogenesis both in the brain and the inner ear5,7,8,9,10,11,12. At embryonic day (E) 9.5-E10.5, neuronal progenitors are reduced in the inner ear of Chd7+/− mutants, and Chd7 is necessary for their proliferation5. However, by E11.5, the number of neuronal progenitors is restored and inner ear neurons, as well as the hair cells they innervate, appear normal after birth5,6. Therefore, the cellular function of Chd7 and the mechanisms underlying SNHL have yet to be elucidated.
In the cochlea, inner hair cells are responsible for sound perception, while outer hair cells modulate the sound amplitude13. They are innervated by type I and type II spiral ganglion neurons, respectively, which project to the auditory nuclei in the brain stem14. As the hair cells (and spiral ganglion neurons) lack the capacity to regenerate, oxidative stress caused by loud noise, ageing, or ototoxicity leads to cell death and permanent hearing loss15,16,17,18,19.
Results
In mice, hair cell specification mediated by the transcription factor Atoh1 occurs between E12.5 and E16.520,21. However, their development continues postnatally20 and they reach maturity just before the onset of hearing between postnatal day 10 (P10) and P1422. Chd7 is expressed in hair cells and neurons throughout embryonic and postnatal stages (Fig. 1a, Fig. S1)3,5,6. To investigate its function in hair cells, we deleted Chd7 using the hair cell-specific Atoh1Cre driver (Atoh1Cre/+;Chd7flox). We compared Cre-positive and Cre-negative control with Chd7 heterozygous and homomozygous littermates. In Atoh1Cre/+;Chd7flox mice, Chd7 deletion was initiated in hair cells from E12.5 onwards and we confirmed loss of Chd7 expression in the basal and middle turns of the cochlea at E16.5 (Fig. 1b). Surprisingly, hair cells appeared normal at P8 (n = 10/10; Fig. S2a, b) suggesting that Chd7 function may not be required for their development.

a Immunohistochemistry in wildtype cochlea showing Chd7 expression in hair cells at E16.5 and P7. Expression is weaker at P7 compared to E16.5. Hair cells are stained with F-actin and Calbindin 1. Tectorial membrane is stained with Satb2. b In-situ hybridisation showing loss of Chd7 expression in Atoh1Cre/+;Chd7f/f mutant hair cells within the middle region of the cochlea at E16.5. Atoh1Cre/+ or Chd7floxed siblings were used as controls (indicated at the top of the panel). IHC inner hair cells; OHC outer hair cells; TM tectorial membrane. Scale bars 25 µm.
We, therefore, wished to confirm hair cell integrity and functionality in the early postnatal cochlea. A key feature of hairs cells is their ability to transform mechanical force into electrical energy that in turn elicits action potentials in spiral ganglion neurons. This requires functional mechanotransduction channels located in the hair cell stereocilia23,24. We visualised uptake of the styryl membrane dye FM1-43 through these channels to assess their activity. Cochleae isolated from P3 mice were exposed to FM1-43 for 30 s and imaged using confocal microscopy. Hair cells from both Atoh1Cre/+ (or Chd7f/f) control and Atoh1Cre/+;Chd7f/f homozygous mutant mice showed robust FM1-43 uptake, indicating that mechanotransduction in mutant hair cells was relatively normal (Fig. 2a). To investigate whether mutant inner hair cells established connectivity with spiral ganglia neurons, we assessed the presence and density of ribbon synapses; there was no difference between controls and mutants at P7 (Fig. 2b, c). Together, these results suggest that although Chd7 is deleted at embryonic stages as hair cells are specified, hair cells develop with normal morphology until the first postnatal week with intact mechanotransduction channels and neuronal connections.

a Cochlear explants of control and Chd7 homozygous mutants were treated with FM1-43 for 30 s to assess mechanotransduction. b CtBP2 immunohistochemistry of control and Chd7 homozygous mutants at P7 shows that ribbon synapses at the inner hair cells are unaffected in mutants. c Quantification of CtBP2 puncta (ribbon synapses) per inner hair cell. CTRL = control; HOM = homozygote.
Thereafter we observed rapid degeneration of inner hair cells at P10 (n = 6/8) and P14 (n = 15/16), while outer hair cells degenerated more slowly (Fig. 3a–l, n, Fig. S2a, b). By P21, most inner hair cell nuclei were missing, pyknotic or fragmented, indicating progressive degeneration and cell death (n = 8/8; Fig. 3m, n, Fig. S2b). Chd7 heterozygous mutants showed equally severe phenotypes (Fig. S2c) but at a reduced frequency (n = 5/24). To establish when Chd7 function is critical during hair cell formation, we used inducible Atoh1CreERT2 to temporally delete Chd7. Tamoxifen was administered at E16, with loss of Chd7 protein, particularly in inner hair cells, evident at E18.5 (Fig. S3a, b). Unlike early embryonic Chd7 deletion as soon as hair cells were specified, loss of Chd7 after E16.5 did not cause postnatal hair cell degeneration (n = 6/6; Fig. S3c). These observations suggest that Chd7 loss during early hair cell development predisposes cells to postnatal degeneration which leads to SNHL.

a–l F-actin-stained hair cells in the cochlea of control (a–f) and Chd7 homozygous mutants (g–l) at P14. Dashed boxes in a–c and h, i indicate the zoomed regions shown in (d–f) and (j–l). Scale bars = 20 µm. m Inner hair cells showing pyknotic, fragmented (arrow) and missing (arrow) nuclei in Chd7 mutants at P21. Scale bars = 25 µm. n Average number of hair cells per 200 µm regions in base, middle and apical turns of each cochlea per animal (n = 8 per genotype; each animal is represented by one circle, triangle or square). Separate inner and outer hair cell quantification is provided in Fig. S2. Statistical significance was obtained by performing a nested one-way ANOVA and Dunnett’s multiple comparison test. **P-value = 0.005. o Average number of neurons in the spiral ganglion per section at different postnatal stages in control (CTRL) and Chd7 homozygous mutants (HOM). Student’s t test, P-values: *= < 0.05, **< 0.005, ***= < 0.0005, ****= < 0.00005). p Spiral ganglia neurons stained with NeuN and neurofilament (NF) at P7 in control and mutant animals. Scale bars = 50 µm.
Postmitotic neural progenitors arise in the otic vesicle from ~E9 onwards under the control of NeuroD1 and differentiate into spiral ganglion neurons by E14.525,26,27,28. However, the peripheral auditory circuit is only established in the first 10 days after birth (P0-P10), prior to the onset of hearing14,29,30. To assess Chd7 function in spiral ganglion neurons, we analysed NeuroD1Cre/+;Chd7floxed mutants. In homozygous mutants, ganglion size and neuronal numbers were indistinguishable from controls at P1 (n = 3/3; Fig. 3o, Fig. S4a), but neurons degenerated rapidly to 50% by P7 (n = 3/3; Fig. 3o, p). This phenotype was also observed in Chd7 heterozygous mutants, although neurodegeneration occurred gradually with 50% loss by P21 (n = 3/3; Fig. S4b). Thus, Chd7 controls the survival of a subset of spiral ganglion neurons. Like in hair cells, Chd7 deletion at embryonic stages does not appear to affect neuronal development but leads to delayed neurodegeneration postnatally.
To determine whether Chd7 deletion results in hearing loss, we measured the auditory brainstem response (ABR) of 4- and 8-week-old mutant and control animals. Most Atoh1Cre/+;Chd7flox homozygous mutants exhibited severe-profound hearing loss across all frequencies (n = 6/7; Fig. 4a, Fig. S5), while only 1/7 heterozygous mutants showed a similar ABR profile (Fig. S5). In contrast, NeuroD1Cre/+;Chd7flox mutants exhibited moderate hearing loss (n = 6; Fig. 4b, Fig. S6), presumably due to surviving neurons. Nonetheless, the ABR tests confirm that SNHL correlates with hair cell or neuronal degeneration.

a Auditory brainstem response (ABR) tests of Atoh1Cre/+;Chd7flox mutants and controls at 4 weeks and 8 weeks reveals profound hearing loss in homozygous and mild-moderate hearing loss in heterozygous Chd7 mutants across all frequencies. b ABR tests of NeuroD1Cre/+;Chd7flox mutants and controls at 4 weeks and 8 weeks reveals mild-moderate hearing loss in homozygous mutants. Frequencies where significant threshold elevations were observed are indicated by asterisks (P-values: *< 0.05, **< 0.005, ***< 0.0005, ****< 0.000005). See Figs. S5 and S6 for ABR profiles of each mouse. Error bars represent the standard error of mean (see Figs. S5 and S6). CTRL = control; HET = heterozygote; HOM = homozygote.
Chd7 controls transcription through regulation of chromatin architecture9,11, but how it exerts its function in auditory hair cells and neurons is poorly understood. We, therefore, examined the earliest transcriptional changes resulting from Chd7 deletion by comparing gene expression of FAC-sorted hair cells or spiral ganglia neurons from mutant and control animals (Fig. 5a, Supplementary Data 1, 4). Using the non-inducible (Atoh1Cre/+) and inducible (Atoh1CreERT2/+) mediated deletion of Chd7 enabled us to pinpoint the earliest developmental window in which Chd7 function is required (i.e., between E12.5 and E16.5). By E16.5, most hair cells have formed20,21; by this time point Chd7 expression was lost in mutant cochleae. Therefore, we analysed changes in hair cell gene expression at E16.5. We were unable to determine the precise time point for Chd7 function in neurons due to the lack of an inducible NeuroD1CreERT2/+ transgenic line. We, therefore, chose P4 for transcriptional profiling to capture the time point when spiral ganglion neurons actively undergo cell death in mutants (Fig. 3o) and when they normally establish synaptic connections with hair cells14,29,30.

a Schematic showing the experimental approach used for RNA sequencing. DP = double positive. n = 6 cochleae or ganglia were pooled for RNA-seq in three independent experiments per genotype. b Comparison of the number of differentially expressed genes between hair cells and neurons in homozygotes. c qPCR expression analysis of genes in controls and homozygous FAC-sorted hair cells at E16.5. Error bars represent the standard error. P-values: *= < 0.05, **< 0.005, ***< 0.0005. ns = not significant. d Plot of Odds ratio by −log10 of the P-value for human diseases identified by disease ontology. Odds ratio is used to determine the relative odds of the occurrence and magnitude of disease, given exposure to the risk factor (e.g., genes). Complete disease ontology is provided in Supplementary Data 5 and 6. e Gene ontology for differentially regulated genes. Complete gene ontology is provided in Supplementary Data 7 and 8.
Differential gene expression analysis revealed significant changes (FDR ≤ 0.05, fold change >2) in 2437 transcripts in hair cells and 1653 transcripts in neurons (Fig. 5b, Fig. S7a–d, Supplementary Data 1–4). We selected genes with high fold-change in expression (see Supplementary Data 2) as well as genes unaffected by Chd7 deletion (e.g., Atoh1) and transcripts that are normally not expressed (e.g., Satb2) for validation by qRT-PCR (Fig. 5c) and protein by immunohistochemistry (Fig. S8). These results confirmed our RNAseq data. Analysis of Disease Ontology terms for all differentially expressed genes showed enrichment of Chd7-associated syndromes as well as hearing loss. Surprisingly, there was also a strong association with neurodegenerative diseases including dementia (Fig. 5d, Supplementary Data 2, 4, 5, 6) pointing towards a common mechanism underlying these conditions.
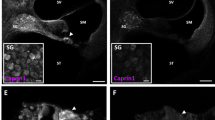
Gene Ontology terms for RNA processing and stress pathways were strongly associated with all differentially expressed genes (Fig. 5e, Supplementary Data 3, 4), while RNA-binding proteins were among the most prominent transcripts deregulated by Chd7 deletion (Fig. 6a, b). Interestingly, previous reports showed that Chd7 binds to many of their promoters in neuronal progenitors (Fig. S7e, Supplementary Data 9; ref. 7) suggesting that they may be direct Chd7 targets. RNA-binding proteins are critical regulators of cellular stress, controlling the assembly and disassembly of stress granules31,32,33. As transient membrane-less compartments, they assemble in the cytoplasm under oxidative stress conditions to allow cells to survive, while their prolonged persistence triggers apoptosis31,32,33,34,35. The metabolic demands of sound detection and amplification elicits oxidative stress in neurons and hair cells that causes cell death unless tightly regulated16,36. We, therefore, tested the hypothesis that Chd7 mutant hair cells are hypersensitive to oxidative stress by exploiting a cochlear explant system in which oxidative stress can be induced by treatment with aminoglycosides37,38,39. When P6 control explants from Atoh1Cre/+;mTmG mice were exposed to gentamicin for 5 h (100 µM), hair cells were intact along the entire length of the cochlea, as were untreated Atoh1Cre/+;Chd7f/f homozygous mutant hair cells (n = 5/5 each; Fig. 7a, b). In contrast, gentamicin treatment of Atoh1Cre/+;Chd7f/f mutant explants resulted in a reduction of hair cells by more than 50% across all regions of the cochlea (n = 6/6; Fig. 7a, b). These findings show that Chd7 mutant hair cells are hypersensitive to oxidative stress, causing degeneration in response to stress inducers. Thus, in vivo sound exposure at the onset of hearing may trigger cell death in Chd7-deficient hair cells. Our data suggest that SNHL in CHARGE syndrome may partly be due to misregulation of RNA-binding proteins as key regulators of stress granules thereby altering the response of neurons and hair cells to normal sound.

a Heatmap of representative functional categories of differentially expressed genes in hair cells. b Heatmap of representative functional categories of differentially expressed genes in neurons. Highlighted blocks of genes: green = upregulated; red = downregulated.

a Cochlear explants of control and Chd7 homozygous mutants were treated with gentamicin to induce oxidative stress. Rapid hair cell death is observed in mutants within 5 h whereas control hair cells and untreated mutant hair cells survive. Green = Cre recombined cells expressing membrane GFP, magenta = all hair cells stained with Myo7a, blue = DAPI stained nuclei. b Quantification of Myo7a+ and GFP+ hair cells per 200 µm region in untreated and treated explants in both controls (CTRL) and mutants (HOM). Two-tailed unpaired t test shows the significant difference between mutant and control. P-value = 0.0025. Scale bars = 25 µm.
Discussion
In humans, CHD7 haploinsufficiency is the major cause of CHARGE syndrome, a complex developmental disorder associated with growth retardation and defects of the eye, heart, genitals and the ear1,2. 50–70% of CHARGE patients present with sensorineural hearing loss, however, the cellular and molecular mechanisms involved are poorly understood. Here we demonstrate that Chd7 plays an essential role in the survival of neurons and hair cells in the murine inner ear. Deleting Chd7 at early developmental stages, when hair cells or neurons are being specified and start to differentiate, does not result in overt developmental phenotypes but rather has longlasting effects on their ability to survive postnatally. What are the underlying molecular mechanisms? Other members of the Chd family are important components in DNA damage response and repair pathways; relocating dynamically to sites of DNA damage, they play critical roles in cell survival40,41,42,43. The fact that Chd7 function is critical during early embryonic development long before hair cell and neuronal degeneration argues against a direct role for Chd7 in these programmes. It is more likely that Chd7 is required to maintain an epigenomic state that allows a balanced response when exposed to oxidative stress. In support of this idea, we show that loss of Chd7 predisposes embryonic cells to cell death. In the absence of Chd7 function, hair cells are hypersensitive to oxidative stress and key components of these stress response pathways are misregulated. In neuronal progenitor cells, their promoters are occupied by Chd7 suggesting that embryonic loss of Chd7 leads to prolonged epigenomic changes, ultimately preventing the execution of efficient survival programmes necessary to counter oxidative stress. Our findings, therefore, provide a possible explanation and molecular mechanism for SNHL in CHARGE syndrome patients and may pave the way for new therapeutic interventions.
A recent study corroborates our finding that Atoh1Cre/+;Chd7f/f mutant hair cells develop with seemingly normal morphology at early postnatal stages44. We show that from the second postnatal week when hair cells begin to dectect sound and encounter oxidative stress14,18,19,29,30,35,39, they rapidly degenerate. While a critical role for Chd7 in promoting neural progenitor expansion has been demonstrated using Neurog1Cre/+;Chd7f/f mutants, the deletion of Chd7 using ShhCre/+ in maturing spiral ganglion neurons has no effect on their ongoing development, at least until P144. Whether abnormalities in spiral ganglion neurons arise thereafter is unknown. Our results show that despite normal morphology at P1 in NeuroD1Cre/+;Chd7f/f mutants, rapid neurodegeneration is observed between P2-P7. This is a crucial and metabolically demanding period for neurons as they establish synaptic connections with hair cells and refine their activity in preparation for the onset of hearing (i.e., P10+)14,29,30.
Single-cell transcriptomic data reveal that type I spiral ganglion neurons can be divided into molecularly distinct subtypes45,46,47. Identifying whether specific subtypes undergo neurodegeneration following Chd7 deletion and gaining insights into the disruption of tonotopy may guide improvements in cochlear implants for CHARGE syndrome patients. Interestingly, ~50% of NeuroD1Cre/+;Chd7f/f mutant spiral ganglion neurons escape degeneration. Although neurons project to the cochlear epithelium and navigate towards hair cells, our transcriptomic data indicate that subtle axonal phenotypes may contribute to hearing loss since a number of genes encoding proteins involved in axon guidance, axon elongation and fasciculation are altered in the mutants.
In summary, Chd7 emerges as a key coordinator of cellular stress proteins. Its embryonic deletion leads to an imbalance in stress pathways that does not affect the normal development of neurons or hair cells. Axons navigate towards hair cells and ribbon synapses are correctly established. However, as cells mature and encounter environmental stress, they begin to degenerate. Our findings suggest that some neurodegenerative diseases arise from neurodevelopmental abnormalities that go undetected, and that SNHL may be an early indicator for these conditions.
Methods
Animals
The Atoh1Cre (B6.Cg-Tg(Atoh1-cre)1Bfri)48, Chd7flox (B6.tm1c(EUCOMM)Wtsi)8 and NeuroD1Cre (B6.Cg-Tg(NeuroD1-cre)RZ24Gsat49 mice were maintained on a C57BL/6J genetic background. The mTmG (tm4(ACTB-tdTomato,-EGFP)Luo)50 mice were maintained on a 129S6/SvEv background. The Atoh1CreERT2 (Tg(Atoh1-cre/Esr1*)14Fsh, JAX #007684) used in Fig. S4 was maintained on an FVB/NJ background. The Chd7floxed mice were crossed with the relevant Cre/reporter lines and backcrossed to C57BL/6J for three generations. All mice were maintained in either C57BL/6J or mixed genetic background. For tamoxifen-induced Cre recombination, a single dose of 20 mg/ml tamoxifen (Sigma, T5648) dissolved in corn oil (Sigma, C8267) was administered to pregnant Atoh1CreERT2;Chd7flox;mTmG dams (80 mg/kg of body weight) by intraperitoneal injection. To minimise abortion, 10 mg/ml of progesterone (Sigma: P0130) was simultaneously administered at half tamoxifen dose (40 mg/kg of body weight). One injection gave a consistent recombination efficiency of 95–99%. Upon Cre-mediated recombination, targeted cells expressed membrane GFP. All animal work was performed in accordance with UK Home Office regulations. Experiments were performed on male and female littermates maintained in the same environment to avoid bias. Animals were randomly allocated to experimental groups.
Immunohistochemistry
Dissected inner ear tissue was fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) and processed for whole-mount immunostaining or frozen sectioning. For whole-mount immunostaining, following permeabilisation with 0.2% Triton X-100/PBS (3 × 10 m) and blocking with 0.2% Triton X-100/5% serum/PBS (1 h), the cochleae were incubated overnight at 4 °C in primary antibodies and then washed in 0.2% Triton X-100 (3 × 10 m). Fluorescent secondary antibodies were applied for 1 h at room temperature. After staining with DAPI, the cochleae were washed extensively prior to mounting onto slides in 50% glycerol/PBS. For immunostaining on cryoprotected sections, following washes in PBS (2 × 10 m), permeabilisation in 0.1% TritonX-100/PBS (1 × 10 m) and blocking in 0.1% TritonX-100/5% serum/PBS (30 m), sections were incubated overnight at 4 °C with primary antibodies. After several washes in 0.1% TritonX-100/PBS, the sections were incubated for 1 h at room temperature with fluorescent secondary antibodies, subsequently washed in PBS, stained with DAPI and mounted onto slides with 50% glycerol/PBS. Primary antibodies used were: rabbit Myo7a (1:1000, Proteus, 25-6790); rabbit NeuN (1:1000, Abcam, ab177487); mouse NF-M (1:200, ThermoFisher Scientific, 13-0700); rabbit Sptbn1 (1:500, Bethyl Laboratories, A300-936A); rabbit Lmx1a (1:100, Abcam, ab139726); rabbit Epha3 (1:100, St John’s Laboratory, STJ110712); mouse Calbindin (1:50, Abcam, ab82812); mouse Parvalbumin (1:100, Sigma, P3088); mouse Satb2 (1:100, Abcam, ab51502); rabbit Chd7 (1:100, ThermoFisher Scientific, PA5-72964); mouse CtBP2 (1:100, BD Biosciences, 612044). Secondary antibodies were: goat anti-rabbit Alexa Fluor 635 (1:500, Invitrogen, A31576); goat anti-mouse Alexa Fluor 488 (1:1000, Invitrogen, A11001). F-actin were stained with Phalloidin 488 (1:1000, Invitrogen, A12379) or 546 (1:500, Invitrogen, A22283).
In-situ hybridisation
E16.5 inner ears were fixed in 4% PFA overnight at 4 °C followed by cochlea dissection in DEPC-treated PBS. In-situ hybridisation was performed using dig-labelled mouse Chd7 riboprobe.
Auditory brainstem response (ABR)
ABR measurements were performed as described in ref. 51. An audiometric profile for each mouse at 4 and 8 weeks old was obtained across a range of sound frequencies (3, 6, 12, 18, 24, 30, 36 and 42 kHz). The mice were on a mixed genetic background (C57BL/6J x 129S6/SvEv). Statistical significance was obtained using Kruskal–Wallis non-parametric ANOVA and Bonferroni-corrected significance in GraphPad Prism 9.0.0.121.
Isolation of hair cells and neurons by FAC-sorting
For RNA-sequencing, samples were collected for three biological replicates on independent occasions. E16.5 cochlear duct from Atoh1Cre;Chd7flox mice or P4 spiral ganglia neurons from NeuroD1Cre;Chd7flox mice were isolated from inner ears in cold L-15 medium (Thermofisher, 21083027). Tissues were cut into 3–6 pieces depending on stage and collected into low-binding tubes with L-15 on ice. Per experiment, a total of 6 cochleae or ganglia from three siblings were pooled into one tube. Excess L-15 was removed and 100 µl of 20 U/ml Papain (27 mg/ml, Sigma, P3125) and 1 U/ul RNase-free DNAse (Promega, M6101) in L-15 medium was added to each tube. Cells were dissociated at 37 °C in a heated shaker, triturating using a filtered low-binding tip (Alpha Laboratories, LP200NFRS) every 5 m for a total of 40 m for hair cells and 1 h for neurons. The dissociation reaction was stopped by adding 1:1 volume of prewarmed sample buffer (1% fetal bovine serum in L-15). Cells were strained using a 40 µm nylon sterile cell strainer (Falcon, 352340) into a 50 ml low-binding tube (VWR, 5250403) and transferred to a 5 ml FACS tube (Falcon, 352235). DAPI (1 mg/ml) was added (1:1000) immediately prior to FAC-sorting using the BD FACSAria sorters into 1.5 ml low-binding tubes with 100 µl of sample buffer. FAC-sorted cells were centrifuged at 4 °C for 4 m at 8000 relative centrifugal force (Eppendorf centrifuge 54415R), frozen in liquid nitrogen and stored at −80 °C or immediately processed for RNA extraction and first strand cDNA synthesis.
RNA purification, library preparation and RNA Sequencing
FACS-sorted cells were processed using the NEB Monarch kit (T2010S/L) for polyA+ RNA isolation and NEBNext Single Cell/Low Input RNA Library Prep Kit for Illumina (E6420S/L) and NEBNext Multiplex Oligos for Illumina (Index Primers Set 1, E7335S/L) was used for library preparation (as per kit instructions). RNA and cDNA quality were analysed using Agilent Total RNA 6000 Pico or High Sensitivity DNA Assay on a Bioanalyser (Agilent, 2100). Additional library quality control was performed by the Oxford Genomic Centre at the Wellcome Centre for Human Genetics (funded by the Wellcome Trust, grant 203141/Z/16/Z) and sequenced using Illumina HiSeq 4000 75 bp paired-end reads. Following quality control, paired reads were aligned to mouse MM10 genome assembly. Alignment was performed using HiSAT2 version 2.1.0 with the default parameters in Galaxy version 2.1.052,53. To facilitate quantitative gene expression analysis, aligned reads for each sample were counted using featureCounts version1.6.454.
Differential gene expression analysis
Differential gene expression analysis was performed using DESeq2 version 2.11.40.6, applying parametric fit55. Prior to differential gene expression analysis, a number of filters were applied. We considered the RPKM values of genes that are not normally expressed in E16.5 hair cells (i.e., Satb2) or P4 spiral ganglia neurons (i.e., Atoh1) and removed all genes with an RPKM value equivalent to or less than Satb2 or Atoh1. This resulted in 6910 transcripts for genes expressed in hair cells and 11,293 genes expressed in neurons. We performed a pairwise comparison between controls and homozygotes and controls and heterozygotes. Considering an adjusted p-value (FDR) of ≤0.05 and linear fold change of >2 in either direction, we found a total of 2437 genes in hair cells (1014 upregulated, 1423 downregulated) and 1653 genes in neurons (609 upregulated, 1044 downregulated) that were differentially expressed in Chd7 homozygous mutants compared to controls (Fig. 3b, Supplementary Data 1–4, see Fig. S7a–d for heterozygotes).
Gene Ontology and Disease Ontology analysis were performed both separately and together on up- or downregulated genes using the R interface (https://cran.r-project.org/web/packages/enrichR/vignettes/enrichR.html) to the Enrichr (https://maayanlab.cloud/Enrichr/) databases (see Supplementary Data 5–8 for specific databases for each analysis). Heatmaps and bubble plots were generated with the R packages pheatmap and GOplot. Volcano plots were generated in GraphPad Prism 9.0.0.121.
Quantitative (q) RT-PCR
cDNA from RNA extracted from FAC-sorted hair cells or neurons were subjected to qPCR analysis with the AriaMx Real-Time PCR System (Agilent Technologies) using SYBR green and gene specific primers. Reactions were repeated in triplicates. Relative expression levels were calculated using 2−ΔΔCT method using Ppp6r3 as an endogenous housekeeping gene. Differences between experimental groups were compared using an unpaired two-tailed Student’s t-test and P-value ≤0.05 was considered statistically significant.
Cochlear explants
Cochlear explants from postnatal day 6 Atoh1Cre/+;mTmG and Atoh1Cre/+;Chd7f/f;mTmG mice were cultured in MatTek dishes coated with CellTak (BD Biosciences). Culture medium comprised of L-15 medium (Thermofisher, 21083027), 5% FBS, 0.2% N2, and 0.001% ciprofloxacin. Explants were maintained at 37 °C under 5% CO2 for 1 h prior to gentamicin exposure. Explants were incubated with or without 100 µM gentamicin for 5 h. At 5 h, explants were rinsed in PBS, fixed in 4% PFA (20 m at room temperature) and rinsed again in PBS (3 × 10 m) prior to immunohistochemistry. Experiments were performed on explants of basal and middle turns of the cochlea.
FM1-43 uptake by hair cells
Cochleae from postnatal day 3 Atoh1Cre/+ (or Chd7f/f) and Atoh1Cre/+;Chd7f/f mice were dissected in L-15 medium, placed on MatTek dishes and incubated in FM1-43 (Invitrogen) diluted to 3 µM in L-15 for 30 s. Following repeated washes (6×) with L-15, the explants were imaged live under confocal microscopy.
Microscopy and Imaging
Confocal z stack images were obtained using a TCS SP5 confocal (Leica) microscope, projected using Fiji and further processed using Photoshop (Adobe). Figures were assembled in Photoshop.
Statistics and reproducibility
Statistical significance for hair cell and neuronal quantification was obtained by performing one-way ANOVA and Dunnett’s multiple comparison test or paired t-test. Differences between experimental groups in Fig. 5 were compared using two-tailed unpaired t-tests. All statistical tests were conducted using Microsoft Excel and GraphPad Prism 9.0.0.121. Further detail on statistical analyses including sample size and replicates are provided in the relevant method sections and figure legends. Sample size (n = 7) is based on power calculations, with an effect size of 1.75, a power of 0.8 and significance level of 0.05.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Sequencing data that support the findings of this study have been deposited in Gene Expression Omnibus with the accession code GSE163798. Source data underlying graphs and charts are provided in Supplementary Data 10.
Change history
04 January 2022
In the original version of this article, the given and family names of M. Albert Basson were incorrectly structured. The name was displayed correctly in all versions at the time of publication. The original article has been corrected.
References
Vissers, L. E. et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957 (2004).
Zentner, G. E., Layman, W. S., Martin, D. M. & Scacheri, P. C. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am. J. Med. Genet. A. 152A, 674–686 (2010).
Bosman, E. A. et al. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet 14, 3463–3476 (2005).
Randall, V. et L. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J. Clin. Invest 119, 3301–3310 (2009).
Hurd, E. A. et al. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development 137, 3139–3150 (2010).
Hurd, E. A. et al. Mature middle and inner ears express Chd7 and exhibit distinctive pathologies in a mouse model of CHARGE syndrome. Hear Res 282, 184–195 (2011).
Engelen et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat. Genet. 43, 607–611 (2011).
Jones, K. M. et al. CHD7 Maintains neural stem cell quiescence and prevents premature stem cell depletion in the adult hippocampus. Stem Cells 33, 196–210 (2015).
Feng, W. et al. Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat. Commun. 8, 14758 (2017).
Donovan, A. P. A. et al. Cerebellar vermis and midbrain hypoplasia upon conditional deletion of Chd7 from the embryonic mid-hindbrain region. Front Neuroanat. 11, 86 (2017).
Whittaker, D. E. et al. Distinct cerebellar foliation anomalies in a CHD7 haploinsufficient mouse model of CHARGE syndrome. Am. J. Med Genet C. Semin. Med. Genet. 175C, 465–477 (2017).
Whittaker, D. E. et al. The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression. J. Clin. Invest 127, 874–887 (2017).
Dallos, P. et al. Prestin-based outer hair cell motility is necessary for mammalian cochlear amplification. Neuron 58, 333–339 (2008).
Koundakjian, E. J., Appler, J. L. & Goodrich, L. V. Auditory neurons make stereotyped wiring decisions before maturation of their targets. J. Neurosci. 27, 14078–14088 (2007).
Feghali, J. G., Liu, W. & Van De Water, T. R. L-n-acetyl-cysteine protection against cisplatin-induced auditory neuronal and hair cell toxicity. Laryngoscope 111, 1147–1155 (2001).
Henderson, D., Bielefeld, E. C., Harris, K. C. & Hu, B. H. The role of oxidative stress in noise-induced hearing loss. Ear Hear 27, 1–19 (2006).
Jiang, H., Talaska, A. E., Schacht, J. & Sha, S. H. Oxidative imbalance in the aging inner ear. Neurobiol. Aging 28, 1605–1612 (2007).
Cheng, L. et al. Moderate noise induced cognition impairment of mice and its underlying mechanisms. Physiol. Behav. 104, 981–988 (2011).
Fujimoto, C. & Yamasoba, T. Oxidative stresses and mitochondrial dysfunction in age- related hearing loss. Oxid. Med Cell Longev. 2014, 582849 (2014).
Chen, P. et al. The role of Math1 in inner ear development: Uncoupling the establishment of the sensory primordium from hair cell fate determination. Development 129, 2495–2505 (2002).
Ahmed, M. et al. Eya1-Six1 interaction is sufficient to induce hair cell fate in the cochlea by activating Atoh1 expression in cooperation with Sox2. Dev. Cell 22, 377–390 (2012).
Mikaelian, D. & Ruben, R. J. Development of hearing in the normal Cba-J mouse: correlation of physiological observations with behavioral responses and with cochlear anatomy. Acta Otolaryngol. 59, 451–461 (1965).
Beurg, M., Evans, M. G., Hackney, C. M. & Fettiplace, R. A large-conductance calcium-selective mechanotransducer channel in mammalian cochlear hair cells. J. Neurosci. 26, 10992–11000 (2006).
Beurg, M., Nam, J. H., Crawford, A. & Fettiplace, R. The actions of calcium on hair bundle mechanics in mammalian cochlear hair cells. Biophys. J. 94, 2639–2653 (2008).
Kim, W. Y. et al. NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development 128, 417–426 (2001).
Evsen, L. et al. Progression of neurogenesis in the inner ear requires inhibition of Sox2 transcription by Neurogenin1 and Neurod1. J. Neurosci. 33, 3879–3890 (2013).
Fritzsch, B., Pirvola, U. & Ylikoski, J. Making and breaking the innervation of the ear: neurotrophic support during ear development and its clinical implications. Cell Tissue Res. 295, 369–382 (1999).
Sanchez-Calderon, H., Milo, M., Leon, Y. & Varela-Nieto, I. A network of growth and transcription factors controls neuronal differentation and survival in the developing ear. Int. J. Dev. Biol. 51, 557–570 (2007).
Huang L. C. et al. Synaptic profiles during neurite extension, refinement and retraction in the developing cochlea. Neural Dev. B, 7:38 (2012).
Michanski, S. et al. Mapping developmental maturation of inner hair cell ribbon synapses in the apical mouse cochlea. Proc. Natl Acad. Sci. 116, 6415–6424 (2019).
Gilks, N. et al. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15, 5383–5398 (2004).
Leeuw, F. D. et al. The cold-inducible RNA-binding protein migrates from the nucleus to cytoplasmic stress granules by a methylation-dependent mechanism and acts as a translational repressor. Aging Cell 19, e13136 (2020).
Gopal, P. P. et al. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl Acad. Sci. USA 114, 2466–2475 (2017).
Hansen, J. M., Jacob, B. R. & Piorczynski, T. D. Oxidative stress during development: chemical-induced teratogenesis. Curr. Opin. Toxicol. 7, 110–115 (2018).
Neal, M. & Richardson, J. R. Time to get personal: a framework for personalized targeting of oxidative stress in neurotoxicity and neurodegenerative disease. Curr. Opin. Toxicol. 7, 127–132 (2018).
Ohlemiller, K. K., Wright, J. S. & Dugan, L. L. Early elevation of cochlear reactive oxygen species following noise exposure. Audio. Neurootol. 4, 229–236 (1999).
Hirose, K., Hokenbery, D. M. & Rubel, E. W. Reactive oxygen species in chick hair cells after gentamicin exposure in vitro. Hear Res 104, 1–14 (1997).
Towers, E. R. et al. Caprin-1 is a target of the deafness gene Pou4f3 and is recruited to stress granules in cochlear hair cells in response to ototoxic damage. J. Cell Sci. 124, 1145–1155 (2011).
Goncalves et al. Drug-induced stress granule formation protects sensory hair cells in mouse cochlear explants during ototoxicity. Sci. Rep. 9, 12501 (2019).
Moore, S. et al. The CHD6 chromatin remodeler is an oxidative DNA damage response factor. Nat. Commun. 10, 241 (2019).
Luijsterburg, M. S. et al. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol. Cell 61, 547–562 (2016).
Larsen, D. H. et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell. Biol. 190, 731–740 (2010).
Kari, V. et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 17, 1609–1623 (2016).
Balendran, V. et al. Chromatin remodeler CHD7 is critical for cochlear morphogenesis and neurosensory patterning. Dev. Biol. 477, 11–21 (2021).
Shrestha, B. R. et al. Sensory neuron diversity in the inner ear is shaped by activity. Cell 174, 1229–1246 (2018).
Petitpré, C. et al. Neuronal heterogeneity and stereotyped connectivity in the auditory afferent system. Nat. Commun. 9, 3691 (2018).
Grandi, F. C., Tomasi, L. D. & Mustapha, M. Single-cell RNA analysis of type I spiral ganglion neurons reveals a Lmx1a population in the cochlea. Front. Mol. Neurosci. 13, 83 (2020).
Matei, V. et al. Smaller inner ear sensory epithelia in Neurog1 null mice are related to earlier hair cell terminal mitosis. Dev. Dyn. 234, 633–650 (2005).
Gong, S. et al. Targeting CRE recombinase to specific neuron populations with bacterial artificial chromosome constructs. J. Neurosci. 27, 9817–9823 (2007).
Muzumdar, M. D. et al. A global double‐fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Ingham, N. J., Pearson, S. & Steel, K. P. Using the auditory brainstem response (ABR) to determine sensitivity of hearing in mutant mice. Curr. Protoc. Mouse Biol. 1, 279–287 (2011).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46, W537–W544 (2018).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Liao, Y., Gordon, K. S. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
We thank Karen Steel, Claudio Stern and Jemeen Sreedharan for critical reading of the manuscript, Owen Harrison and Kimberley Riegman for excellent technical assistance, Zoe Mann and members of the Streit group for discussions. We thank Mary Beth Hatten for the NeuroD1Cre line. This work was supported by the MRC MR/R004625/1 and by Action on Hearing Loss (S39).
Author information
Authors and Affiliations
Contributions
M.A. conceptualised and designed the study together with M.A.B. and A.S. M.A. performed most experiments and data analysis; R.M. analysed the neuronal Chd7 phenotype, while E.J. performed ABR tests. R.P. assisted in sequencing alignment and bioinformatics. M.A., A.S. and M.A.B. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Biology thanks Angelika Doetzlhofer and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editor: Karli Montague-Cardoso.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmed, M., Moon, R., Prajapati, R.S. et al. The chromatin remodelling factor Chd7 protects auditory neurons and sensory hair cells from stress-induced degeneration. Commun Biol 4, 1260 (2021). https://doi.org/10.1038/s42003-021-02788-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02788-6
This article is cited by
-
Whole-exome sequencing reveals genetic variants that may play a role in neurocytomas
Journal of Neuro-Oncology (2024)
-
Advance and Application of Single-cell Transcriptomics in Auditory Research
Neuroscience Bulletin (2023)
-
The spectrum of cochlear malformations in CHARGE syndrome and insights into the role of the CHD7 gene during embryogenesis of the inner ear
Neuroradiology (2023)
-
CHD7 regulates otic lineage specification and hair cell differentiation in human inner ear organoids
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
