
Abstract
Genome-wide association studies have identified SLC16A13 as a novel susceptibility gene for type 2 diabetes. The SLC16A13 gene encodes SLC16A13/MCT13, a member of the solute carrier 16 family of monocarboxylate transporters. Despite its potential importance to diabetes development, the physiological function of SLC16A13 is unknown. Here, we validate Slc16a13 as a lactate transporter expressed at the plasma membrane and report on the effect of Slc16a13 deletion in a mouse model. We show that loss of Slc16a13 increases mitochondrial respiration in the liver, leading to reduced hepatic lipid accumulation and increased hepatic insulin sensitivity in high-fat diet fed Slc16a13 knockout mice. We propose a mechanism for improved hepatic insulin sensitivity in the context of Slc16a13 deficiency in which reduced intrahepatocellular lactate availability drives increased AMPK activation and increased mitochondrial respiration, while reducing hepatic lipid content. Slc16a13 deficiency thereby attenuates hepatic diacylglycerol-PKCε mediated insulin resistance in obese mice. Together, these data suggest that SLC16A13 is a potential target for the treatment of type 2 diabetes and non-alcoholic fatty liver disease.
Similar content being viewed by others


Introduction
Obesity is the major risk factor for type 2 diabetes (T2D) and is associated with ectopic fat accumulation in the liver (non-alcoholic fatty liver disease, NAFLD) and insulin resistance. T2D is an expanding international epidemic. Even with modern therapies, diabetes-associated mortality is 2–3 times higher as compared with non-affected individuals1. Therefore, more efficient therapies to target the root cause of NAFLD are an urgent clinical need, and research on novel therapeutic targets is of high interest. More than 140 gene loci have been linked to T2D, including members of the solute carrier (SLC) superfamily2. In 2014, a novel locus was identified in people of Mexican origin that associates with T2D at the genome-wide significance and spans the two adjacent genes SLC16A11 and SLC16A13, encoding for solute carriers SLC16A11/MCT11 and SLC16A13/MCT133. The SLC16 family of monocarboxylate transporters (MCT) mediates fluxes of monocarboxylates, e.g., l-lactate, pyruvate, and ketone bodies, across the plasma membrane and is crucial for cellular energy metabolism4,5. While SLC16A11 is known to be a pyruvate transporter6, SLC16A13 is one of the orphan members of the SLC16 family. The strongest association signal in Mexicans with T2D was ultimately localized to the SLC16A11 gene, establishing it as a gene of great interest to the broader scientific community. Subsequent genome-wide association studies (GWAS) recently validated the SLC16A11 risk haplotype7,8,9,10,11,12 and most research focused on providing evidence for a causal relationship between the development of diabetes and changes in SLC16A11 function. Yet, in mouse models, this relationship is currently a subject of controversy. While AAV-mediated Slc16a11 knockdown in high-fat diet (HFD)-fed wild-type mice leads to improvement in glucose tolerance and hepatic insulin signaling and lowered hepatic lipid accumulation13, AAV-mediated reconstitution of mutated Slc16a11 in the liver of knockout mice worsens the organismal response to high-fat diet14. Especially disagreements whether T2D-associated SLC16A11 variants reflect loss- or gain-of-function mutations arose15,16. Therefore, it seems reasonable to also investigate SLC16A13 in the context of T2D susceptibility; besides, the function and role of SLC16A13 in metabolic disease are completely unknown. Of note, SLC16A13 was linked to T2D in another GWAS, describing the polymorphism rs312457 associated with T2D in people of Japanese origin17. Upregulation of murine Slc16a13 in the small intestine after treatment with an agonist of PPARα18, a key regulator of hepatic lipid metabolism, supports the idea that SLC16A13 may also be involved in energy homeostasis. Since the human phenotype associated with the SLC16A13 polymorphisms reflects higher susceptibility to T2D, we hypothesized that changes in SLC16A13 abundance will affect hepatic lipid deposition and insulin sensitivity. Our objective was to determine the physiological function of the SLC16A13 transporter and to elucidate how transporter deficiency affects the development of metabolic dysfunction. Therefore, we studied SLC16A13 expression in human and mouse tissue and generated gain- and loss-of-function models, including Slc16a13 knockout mice that were metabolically characterized in the context of diet-induced obesity.
Results
Hepatic SLC16A13 is upregulated under conditions of metabolic dysfunction in humans and mice
SLC16A13/Slc16a13 is expressed in human and mouse liver and only marginally expressed in other metabolically active organs such as adipose tissue, brain, and skeletal muscle (Fig. 1a and Supplementary Fig. 1a). This expression pattern is similar to published human data showing the highest abundance of SLC16A13 expression in liver tissue3. Of note, in mice but not in humans, Slc16a13 expression is highest in the kidney. However, in our study, we focused on the role of SLC16A13 in the liver for three reasons: First, the overall aim of the study is to investigate the potential role of SLC16A13 in human metabolic disease; second, human SLC16A13 expression is the highest in the liver, and third, the liver is crucial in glucose and lipid metabolism. Because the human SLC16A13 polymorphism is associated with T2D, we speculated that SLC16A13 expression and/or protein function might also be dysregulated in the context of metabolic disease independent of the known polymorphism. To address this point, we analyzed Slc16a13 gene expression under conditions of obesity. In diet-induced obese mice, Slc16a13 expression is differentially regulated in the liver (Fig. 1b). Hepatic Slc16a13 expression was up to 1.8-fold higher in obese animals compared to mice fed a normal-chow diet (NCD), an effect that was observed both under fed and fasted conditions. The observed increase by high-fat diet strongly suggests the possibility that the SLC16A13 transporter is induced by an excess of the nutritional substrate during periods of high caloric supply. Strikingly, we observed a fasting-feeding difference in Slc16a13 gene expression in HFD-fed mice, while there was no such nutritional dependence in NCD-fed mice, demonstrating a diet-dependent difference in the molecular regulation of this gene. Diet-induced obesity may lead to dysregulation of Slc16a13 expression, making it impossible to regulate it normally during fasting. In contrast, Slc16a13 expression in other tissues than the liver is not or only marginally upregulated in the tested dietary conditions (Supplementary Fig. 1b), suggesting a key role of the liver in Slc16a13-mediated metabolic dysregulation. To further investigate the potential role of the transporter in human metabolic disease, we analyzed SLC16A13 gene expression in human liver samples with different stages of NAFLD and insulin resistance. Liver specimens were obtained from 45 human patients after partial hepatectomy; regions of the liver without pathological findings were biopsied and were processed for gene expression analysis. Physical and clinical parameters have been recorded for all patients and tissue NAFLD-activity score was determined by a sample-masked pathologist (Supplementary Table 1). Hepatic SLC16A13 gene expression significantly correlated with waist circumference (Fig. 1c), BMI (Fig. 1d), HOMA-IR (Fig. 1e), and histologically assessed liver steatosis (Fig. 1f) as well as with quantitatively measured liver TAG content (Fig. 1g). Interestingly, the highest positive correlation (r = 0.731) was observed for liver steatosis (Fig. 1f). The association with liver steatosis remained significant after adjustment for main confounders (Supplementary Table 2). These human data provide evidence for an association between SLC16A13 gene expression and the development of adiposity, fatty liver, and insulin resistance independent of the described human polymorphism. Understanding the link between increased SLC16A13 expression and these metabolic phenotypes will help to uncover its potential contribution to T2D development.

a SLC16A13 mRNA expression in one human sampler relative to mRNA expression in the liver (n = 5). Slc16a13 mRNA expression in mice fed normal-chow diet (n = 7–8) relative to mRNA expression in the liver. b Relative hepatic Slc16a13 mRNA expression in C57BL/6J mice fed with NCD or HFD for 15 weeks. Animals are fed (n = 7–10 per group) or fasted for 16 h (n = 6–9 per group). a, b Bars represent means ± SEM. *p < 0.05, **p < 0.01 determined using two-tailed unpaired Student’s t-test. c–g Correlation of relative hepatic SLC16A13 mRNA expression and adiposity-related parameters in humans. Linear regression curve is shown. c Waist circumference, d BMI (Body Mass Index), e HOMA-IR (Homeostatic Model Assessment for Insulin Resistance), f Logarithm liver steatosis, g Hepatic TAG (triacylglycerol). r = Spearman-Rho correlation coefficient.
Slc16a13 transports l-lactate across the plasma membrane
As an orphan transporter, the precise function and role of SLC16A13 remain to be determined. Knowledge of substrate specificity and localization would assist in the development of hypotheses regarding its role in metabolic control. To characterize cellular localization and substrate specificity of the Slc16a13 transporter, we stably transfected HEK293 cells with a vector containing Slc16a13 cDNA. Human SLC16A13 was previously described to be located at the Golgi apparatus but has not been detected at the plasma membrane3. Here, we could confirm that mouse Slc16a13 localizes to the Golgi apparatus (Supplementary Fig. 2), but importantly, based on immunofluorescence co-staining with the plasma membrane marker wheat germ agglutinin, we observed that mouse Slc16a13 also localizes to the plasma membrane (Fig. 2a and Supplementary Fig. 2). To further characterize the biology of the Slc16a13 transporter, substrate uptake experiments were performed with the HEK-Slc16a13 cell line. l-lactate is a prominent monocarboxylate transported by other SLC16 family members. At l-lactate concentrations ranging from 1 to 100 µM, Slc16a13-dependent l-lactate transport was increased up to 2-fold in HEK-Slc16a13 compared to the HEK-control cell line (Fig. 2b). The net uptake of l-lactate, defined by the difference in uptake between HEK-Slc16a13 and HEK-control cells, was used to calculate the Km (0.47 ± 0.35 µM) and Vmax values (2.02 ± 0.26 pmol × mg protein−1 × min−1) (Fig. 2c). These results suggest that Slc16a13 is a high-affinity and low-capacity transporter for l-lactate.

a Immunofluorescence co-staining of the plasma membrane marker wheat germ agglutinin and FLAG-tagged Slc16a13 in HEK-Slc16a13-FLAG cells. One representative staining is shown. b l-lactate uptake into HEK-control and HEK-Slc16a13 cells (n = 3 for each concentration). Bars represent means ± SEM. **p < 0.01, ***p < 0.001 determined using one-way ANOVA followed by Dunnett’s multiple comparison test. c Net l-lactate uptake as the difference between the uptake into HEK-control and HEK-Slc16a13 cells. Nonlinear regression curve and means ± SEM are displayed.
Generation of Slc16a13 knockout mice
Both human and mouse expression data (Fig. 1b–g) do not indicate whether SLC16A13 deregulation is causal or just a consequence of metabolic alterations in the context of obesity. Since SLC16A13 transports l-lactate across the plasma membrane, we hypothesize that the transporter causally contributes to the development of NAFLD and insulin resistance through changes in hepatic monocarboxylate transport, affecting cellular energy metabolism, hepatic lipid content, and insulin sensitivity. To test this hypothesis in an in vivo model, we generated constitutive Slc16a13 knockout mice using CRISPR/Cas9 technology (Fig. 3a). The homozygous knockout of Slc16a13 generates viable offspring in Mendelian ratios. To exclude that CRISPR/Cas9-mediated Slc16a13 gene editing targets other genes, off-target analysis was performed in the G1 generation and mRNA expression of Slc16a11 and Bcl6b, located adjacently to Slc16a13 on mouse chromosome 11, was analyzed in the G3 generation (Supplementary Fig. 3). Efficient deletion of Slc16a13 was confirmed by gene expression analysis of multiple tissues, with the liver having <1% of remaining Slc16a13 expression compared to wild-type mice (Fig. 3b), and by western blot analysis of wild-type and knockout liver lysates that manifests Slc16a13 protein in the plasma membrane of wild-type, but not of knockout livers (Fig. 3c and Supplementary Fig. 12a). Histological comparison of liver, kidney, white adipose tissue, brain, and skeletal muscle did not reveal any morphological abnormalities due to Slc16a13 deletion in knockout mice (Fig. 3d).

a Deletion of 4.2 kb including 1.8 kb of the proximal promotor region, exon 1, exon 2, and exon 3 of the mouse Slc16a13 gene as indicated by the red arrow. To distinguish between the wild-type (WT) and Slc16a13 knockout (KO) allele on the DNA level, a forward primer (P1) binding upstream of junction 1 is used in combination with the two reverse primer P2 binding within and P3 binding downstream the deleted region as indicated by the gray arrows. mRNA expression levels in WT and KO tissue are analyzed by qRT-PCR primer binding at the exon-exon junctions of exon 1/2 and exon 2/3 (not displayed in the figure). b Slc16a13 mRNA expression in different organs of Slc16a13 KO mice relative to WT (n = 4–5 each group). Relative expression calculated as fold change to the liver. Bars represent means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 determined using two-tailed unpaired Student’s t-test. c Western blot analysis of cytosolic and plasma membrane fraction extracted from liver lysates of Slc16a13 KO and WT mice. d Histological comparison of Slc16a13 KO mice and WT controls at a mean age of 20 weeks. Representative H&E stainings of the liver, kidney (cortex), white adipose tissue, brain (cortex), and skeletal muscle (quadriceps) at ×10 magnification.
Slc16a13 knockout mice show mild metabolic changes
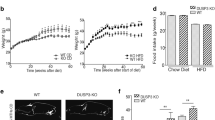
Slc16a13 knockout and wild-type control mice were fed either normal-chow (NCD) or high-fat diet (HFD) for 15 weeks. Slc16a13 knockout mice showed a body weight equal to wild-type mice when fed NCD (Fig. 4a). Interestingly, the deletion of Slc16a13 led to a reduction in fat mass up to 20% compared to wild-type mice (Fig. 4b). However, energy expenditure, locomotor activity, food intake, and respiratory exchange ratio (RER) were not different between the genotypes at 9 weeks of NCD (Fig. 4c–f, respectively). Furthermore, deletion of Slc16a13 did not affect glucose metabolism as indicated by similar glucose tolerance in knockout and wild-type mice at 7 weeks of NCD (Fig. 4g). Plasma insulin levels were unaltered after glucose administration (Fig. 4h), excluding a compensatory hyperinsulinemia to maintain blood glucose levels. In the context of diet-induced obesity, the knockout of Slc16a13 did not significantly affect body weight, body composition, or energy expenditure (Fig. 5a–c), suggesting obesity might mask the Slc16a13-dependent effect on body composition seen in NCD-fed mice. Locomotor activity, food intake, and respiratory exchange ratio (RER) were also unaltered at 9 weeks of HFD (Fig. 5d–f). In contrast, already after 7 weeks of HFD-feeding, loss of Slc16a13 function improved glucose tolerance, demonstrated by significantly decreased blood glucose levels in Slc16a13 knockout mice 60 min after oral glucose administration and significantly lower area under the curve (AUC) (Fig. 5g, h), while insulin levels were unchanged between the genotypes (Fig. 5i). These data suggest a modulatory role of SLC16A13 in glucose metabolism in the context of diet-induced obesity.

a Body weight of WT and Slc16a13 KO mice over a time course of 18 weeks. b–f Body composition, energy expenditure, locomotor activity, food intake, and respiratory exchange ratio (RER) of WT, and Slc16a13 KO mice at an age of 14 weeks. g, h Blood glucose levels and plasma insulin levels of WT and Slc16a13 KO mice after oral glucose administration at an age of 12 weeks. Slc16a13 WT (n = 9), Slc16a13 KO (n = 8–11). a, g, h Means ± SEM are displayed. b–f Bars represent means ± SEM. *p < 0.05 determined using two-tailed unpaired Student’s t-test.

a Body weight of WT and Slc16a13 KO mice over a time course of 18 weeks. b–f Body composition, energy expenditure, locomotor activity, food intake, and respiratory exchange ratio (RER) of WT, and Slc16a13 KO mice at an age of 14 weeks. g–i Blood glucose levels (g), related area under the curve (AUC) (h) and plasma insulin levels (i) of WT and Slc16a13 KO mice after oral glucose administration at an age of 12 weeks. Slc16a13 WT (n = 9–14), Slc16a13 KO (n = 8–11). a, g, i Means ± SEM are displayed. b–f, h Bars represent means ± SEM. **p < 0.01 determined using two-way ANOVA followed by Sidak’s multiple comparison test (g) and *p < 0.05 determined using two-tailed unpaired Student’s t-test (h).
Slc16a13 knockout attenuates insulin resistance in HFD-fed mice
Since an oral glucose tolerance test is only a rough marker of insulin sensitivity, we investigated whole body and tissue-specific insulin action by hyperinsulinemic–euglycemic (HE) clamp after 15 weeks of HFD-feeding. During HE clamp, blood glucose and insulin levels did not differ significantly between Slc16a13 knockout and wild-type mice (Fig. 6a, b). Consistent with the lower blood glucose levels seen in the oral glucose tolerance test (Fig. 5g, h), the glucose-infusion (GINF) rate was higher in Slc16a13 knockout mice (Fig. 6c) and significantly increased by 20% in the clamp steady-state (Fig. 6d). To address the tissue-specific contributions to the improved insulin sensitivity, we determined endogenous hepatic glucose production and glucose uptake into peripheral organs. Insulin-mediated suppression of endogenous glucose production, a reflection of hepatic insulin sensitivity, was markedly better in Slc16a13 knockout mice than wild-type mice (Fig. 6e, f), demonstrating attenuation of HFD-induced hepatic insulin resistance. Accordingly, insulin downstream signaling was improved in Slc16a13 knockout mice as addressed by Akt phosphorylation in liver tissue (Supplementary Fig. 4a), whereas liver glycogen storage was unaltered (Supplementary Fig. 4b). Insulin-stimulated glucose clearance was not different between Slc16a13 knockout and wild-type mice, demonstrating no effect on peripheral insulin sensitivity (Fig. 6g). Consistent with this, 2-deoxyglucose uptake into skeletal muscle and adipose tissue was not affected by genotype (Fig. 6h). Therefore, attenuation of insulin resistance in obese Slc16a13 knockout mice could be attributed to improved hepatic insulin sensitivity.

a Plasma glucose concentration during HE clamp. b Basal and clamp plasma insulin levels. c, d Glucose-infusion rate during HE clamp and in the clamped state (120–140 min) in WT and Slc16a13 KO mice. All three steady-state data points per mouse are shown (d). e, f Basal and clamp hepatic endogenous glucose production and suppression of basal endogenous glucose production in WT and Slc16a13 KO mice. g Plasma glucose clearance in WT and Slc16a13 KO mice. h 2-deoxyglucose uptake in quadriceps muscle and epigonadal white adipose tissue. Slc16a13 WT (n = 8), Slc16a13 KO (n = 8). a, c Means ± SEM are displayed. b, d–h Bars represent means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 determined using two-tailed unpaired Student’s t-test, except for e, f *p < 0.05 determined using one-tailed unpaired Student’s t-test.
HFD-fed Slc16a13 knockout mice are protected from hepatic lipid accumulation
Ectopic lipid accumulation in the liver causes fatty liver disease and insulin resistance19,20. In accordance with improved insulin sensitivity, H&E and Oil-Red-O staining of liver sections revealed less lipid droplets in Slc16a13 knockout mice compared to controls after 15 weeks of HFD-feeding (Fig. 7a). Hepatic lipid accumulation includes many species but mainly triglycerides, which seem to be metabolically inert. In contrast, diacylglycerols (DAGs) and ceramides have been shown to be bioactive mediators of insulin resistance in mice and humans21,22. Interestingly, the hepatic content in triglycerides, as well as diacylglycerols, was significantly reduced in Slc16a13 knockout mice (Fig. 7b, c), whereas that of ceramides was unaltered (Fig. 7d). DAGs are known to mediate insulin resistance via activation of atypical protein kinases, such as PKCε in the liver. Once activated, cytosolic PKC converts into an active membrane-associated form that phosphorylates the insulin receptor on threonine 1160 (in mouse threonine 1150), which, in turn, inhibits the receptor intrinsic tyrosine kinase activity21,23. Compared to wild-type mice, the amount of membrane-associated PKCε was reduced more than 3-fold in the livers of Slc16a13 knockout mice (Fig. 7e and Supplementary Fig. 12b). Together, our data suggest that Slc16a13 deletion attenuates the ectopic deposition of lipids in the liver, leading to a decrease in hepatic DAG content, lower PKCε activation, and, in turn, reduces diet-induced hepatic insulin resistance. In this model, our data also dissociate hepatic ceramide content from changes in hepatic insulin sensitivity.

a Representative H&E and Oil-Red-O stainings of WT and Slc16a13 KO mice. b Hepatic triglycerides in WT (n = 20) and Slc16a13 KO mice (n = 14). Two cohorts of mice have been analyzed. c, d Hepatic diacylglycerol and ceramide level in WT and Slc16a13 KO mice (n = 11 each group). e PKCε protein expression determined by western blot shown for WT (n = 6) and Slc16a13 KO mice (n = 5). PKCε translocation assessed as the ratio of membrane to cytosol PKCε in WT and Slc16a13 KO mice (n = 11 each group). Bars represent means ± SEM. **p < 0.05, **p < 0.01, ***p < 0.001 determined using two-tailed unpaired Student’s t-test.
Reduced hepatic lipid accumulation is caused by increased mitochondrial respiration in Slc16a13 knockout mice
Hepatic lipid content is regulated by four mechanisms: β-oxidation, de novo lipogenesis, lipid export as VLDL particles, and uptake of circulating fatty acids20,24. Loss of SLC16A13 function might counteract one of these mechanisms in the context of diet-induced obesity. To identify the underlying mechanism for reduced fat content in the livers of Slc16a13 knockout mice, we measured oxygen consumption of Slc16a13 knockout and wild-type hepatocytes isolated from NCD-fed females at a mean age of 20 weeks. During Seahorse XF Cell Mito Stress Test using the Seahorse Analyzer, cells were treated with the ATP synthase (complex V) inhibitor oligomycin, the mitochondrial uncoupler FCCP and rotenone/antimycin A, inhibitors of complexes I and III of the mitochondrial respiratory chain, respectively. Deletion of Slc16a13 was associated with higher mitochondrial respiration, indicated by a higher oxygen consumption rate (OCR) and a significant 25% increase in maximal respiration (Fig. 8a). To investigate this observation in more detail, we performed high-resolution respirometry in the Oroboros O2k oxygraph using the substrate-uncoupler-inhibitor titration (SUIT) protocol on liver tissue isolated from mice fed HFD for 15 weeks. The application of the substrates malate, octanoylcarnitine, ADP, glutamate, and succinate, the uncoupler FCCP, and the inhibitor antimycin A allows the analysis of β-oxidation-linked mitochondrial respiration. Mitochondrial function measured as O2 flux per mass was increased in livers of Slc16a13 knockout mice and consistent with the Seahorse data showing a 60% greater maximum capacity of the electron transport chain (ETS) in knockout livers (Fig. 8b). Furthermore, respiratory control ratios calculated from respirometry data indicate that mitochondria in Slc16a13 deficient liver become more effective at generating ATP from fatty acids (Supplementary Fig. 5). The fatty acid OXPHOS coupling control factor is significantly increased with loss of Slc16a13 and indicates that this is primarily due to higher OXPHOS capacity and not caused by reduction of the respiratory LEAK, suggesting improved control and/or capacity of the ADP phosphorylation system regarding the respiratory flux after fatty acid utilization (Supplementary Fig. 5a). In line with this, the respiratory control ratio is increased and the LEAK control ratio is decreased in livers of Slc16a13 knockout mice (Supplementary Fig. 5b, c, respectively). These ratios suggest a more effective ATP synthesis from fatty acids, what may be the reason for increased fatty acid oxidation. To investigate whether other mechanisms might contribute to the reduced hepatic lipid accumulation, VLDL export, de novo lipogenesis, and FFA uptake were analyzed in these mice. Plasma triglyceride levels addressing hepatic VLDL production, assessed by injection of the lipoprotein lipase (LPL)-inhibiting detergent Poloxamer-407 in fasted mice at 13 weeks of HFD, were unaltered between Slc16a13 knockout and wild-type mice (Fig. 8c). Hepatic de novo lipogenesis, assessed by application of deuterated water, and plasma non-esterified fatty acid concentrations did not differ between the genotypes at 15 weeks of HFD (Fig. 8d, e, respectively), and we observed no difference at the mRNA level of hepatic fatty acid transporters (Fig. 8f). Together, our data suggest that increased mitochondrial β-oxidation is the main mechanism protecting against fatty liver development in Slc16a13 knockout mice.

a Mitochondrial respiration of hepatocytes isolated from Slc16a13 WT (n = 27) and Slc16a13 KO (n = 25) mice at a mean age of 20 weeks, both groups females fed NCD. Time course and calculated parameters of oxygen consumption rate (OCR) during mitochondrial stress test performed in the Seahorse Analyzer XF96. b Mitochondrial respiration of liver tissue isolated from 20 weeks old HFD-fed Slc16a13 WT (n = 5) and Slc16a13 KO (n = 7) mice. Time course and calculated parameters of oxygen flux during high-resolution respirometry performed in the Oroboros Oxygraph-2k. LEAK = proton leak, ion leak, and slip compensatory state; FAO = fatty acid oxidation; OXPHOS = oxidative phosphorylation; CIOXPHOS = complex I-related oxidative phosphorylation capacity; CI + IIOXPHOS = complex I and II-related oxidative phosphorylation capacity; ETS = electron transfer system; ROX = residual oxygen consumption. c Plasma triglyceride levels after intraperitoneal injection of the LPL-inhibitor Poloxamer-407 at an age of 18 weeks. d Hepatic de novo lipogenesis in WT and Slc16a13 KO mice at 20 weeks of age. e Plasma level of free-fatty acids (FFA) in a fasted state. f Relative hepatic mRNA expression of fatty acid transporters. Slc16a13 WT (n = 5–11), Slc16a13 KO (n = 5–11). a–c Means ± SEM are displayed. a, b, d–f Bars represent means ± SEM. **p < 0.01, ***p < 0.001 determined using two-way ANOVA followed by Sidak’s multiple comparison test.
Deletion of Slc16a13 increases mitochondrial function by AMPK activation
To investigate mitochondrial function at the molecular level, we performed western blot analysis on liver lysates and found the protein expression of respiratory chain complexes to be significantly higher in Slc16a13 knockout mice compared to wild-type littermates after 15 weeks of HFD-feeding (Fig. 9a and Supplementary Fig. 12c). This is consistent with transcriptomic data showing significantly upregulated oxidative phosphorylation in knockout compared to wild-type liver as determined by gene set enrichment analysis (Fig. 9b). In contrast, neither OXPHOS protein levels nor gene set enrichment analysis indicated deregulated oxidative phosphorylation in skeletal muscle between the two genotypes (Supplementary Fig. 6a, b, respectively). Thus, we propose Slc16a13 deletion in the liver is a key causal factor for the increase in oxidative phosphorylation and finally, the improvement in metabolic phenotype observed in HFD-fed Slc16a13 knockout mice.

a Hepatic protein expression of respiratory chain complexes I–V determined by western blot. b Gene set enrichment analysis of liver transcriptomic data from WT and Slc16a13 KO mice. c AMPK phosphorylation as a ratio of phospho-AMPK/total AMPK determined by western blot. d ACC phosphorylation as ratio of phospho-ACC/total ACC determined by western blot. Slc16a13 WT (n = 6–7), Slc16a13 KO (n = 6–7). e Relative lactic acid levels determined by untargeted metabolomics in the livers of WT (n = 5) and Slc16a13 KO mice (n = 6). a, c–e Bars represent means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 determined using two-tailed unpaired Student’s t-test, Welch correction for unequal variance was used in e.
Given that mitochondrial respiration is increased in Slc16a13 knockout mice, AMPK, the main sensor of cellular energy status and regulator of mitochondrial homeostasis, could be causative. AMPK is activated by phosphorylation at low energy status, defined by high cellular AMP and low ATP level, and promotes mitochondrial biogenesis and expression of respiratory proteins25. Here, protein expression analysis of liver extracts showed a significant increase in the ratio of phospho-AMPK/total AMPK in Slc16a13 knockout mice (Fig. 9c and Supplementary Fig. 12d). AMPK phosphorylates and thereby inactivates acetyl-CoA carboxylase (ACC), an enzyme that exists in two isoforms, ACC-1 and ACC-2, both highly expressed in the liver26,27. AMPK phosphorylation of ACC-1 at serine 212 and ACC-2 at serine 79 reduces fatty acid synthesis and increases fatty acid oxidation through activation of carnitine palmitoyltransferase 1 (CPT1)28,29. Importantly, it was shown that both, ACC-1 and ACC-2 regulate fatty acid oxidation, whereas ACC-1 mainly regulates lipogenesis30. To further establish the link between AMPK activation and mitochondrial function, we found the ratio of phospho-ACC/total ACC protein was significantly higher in Slc16a13 knockout liver extracts (Fig. 9d and Supplementary Fig. 12e), in agreement with cellular activation of AMPK. To confirm these results, mouse primary hepatocytes isolated from Slc16a13 knockout and wild-type mice were treated with the saturated free-fatty acid palmitate to induce insulin resistance mimicking HFD-feeding. Also in these hepatocytes, phosphorylation of AMPK and ACC was significantly increased in cells deficient for Slc16a13 (Supplementary Fig. 7a). In contrast, BSA control-stimulated hepatocytes did not show this difference in protein phosphorylation (Supplementary Fig. 7b), suggesting that an insulin-resistant state induced by palmitate in vitro, or by HFD-feeding in vivo, is mandatory to provoke these changes in the context of Slc16a13 deficiency. Since we showed that Slc16a13 is a plasma membrane l-lactate transporter (Fig. 2a–c), we postulated the energy deficit in the liver of Slc16a13 knockout mice stems from the reduction in intracellular lactate level31,32. Metabolome analysis of the liver was consistent with this hypothesis, showing a significant reduction of l-lactate level by 25% in Slc16a13 knockout compared to wild-type liver (Fig. 9e). In contrast, monocarboxylates transported by other SLC16 family members did not differ between wild-type and knockout livers (Supplementary Fig. 8a). To finally test the hypothesis that reduced lactate transport in the liver is the primary metabolic impact of Slc16a13 deficiency, intrahepatocellular lactate utilization was analyzed in Slc16a13 knockout and wild-type primary hepatocytes (Fig. 10). In palmitate-treated hepatocytes, lactate, and pyruvate-driven glucose production were significantly reduced if Slc16a13 is missing. The reason for reduced gluconeogenesis might be reduced Slc16a13-mediated uptake of lactate into hepatocytes and consequently reduced availability of this gluconeogenic substrate. Furthermore, glucose production was significantly decreased in palmitate-treated Slc16a13 knockout hepatocytes, but not in BSA control-treated hepatocytes, suggesting that Slc16a13 deficiency only has a metabolic impact under conditions of excess dietary nutrients and linked insulin resistance, as seen for AMPK and ACC phosphorylation (Supplementary Fig. 7). Most importantly, this reduced intrahepatocellular lactate availability will not exclusively affect glucose metabolism, but impact the overall energy status of the cell. According to our hypothesis, intracellular energy deficit may drive AMPK activation and further downstream signaling leading to improved insulin sensitivity in diet-induced obese Slc16a13 knockout mice. Taken together, these results identify a mechanism for SLC16A13 function in energy homeostasis: loss of SLC16A13 reduces l-lactate uptake in the liver, thereby limiting intracellular substrate availability and reducing intracellular energy levels that lead to activation of AMPK, which promotes mitochondrial respiration. Increased β-oxidation attenuates lipid (DAG) accumulation and the development of hepatic insulin resistance in the context of obesity.

Glucose production of primary hepatocytes isolated from Slc16a13 WT (n = 8) and Slc16a13 KO (n = 7) mice at a mean age of 20 weeks. Hepatocytes were stimulated with 500 µM BSA-coupled palmitate (6:1 molar ratio) or BSA control and 100 nM glucagon or vehicle control as indicated. Cell culture supernatant was collected after 8 h of incubation in glucose-free media containing 20 mM lactate and 2 mM pyruvate. Bars represent means ± SEM. *p < 0.05 determined using two-tailed unpaired Student’s t-test.
Discussion
SLC16A13 has been linked to T2D in humans by two independent GWAS3,17. However, the biology and function of the SLC16A13 transporter, and whether alterations in SLC16A13 are a cause or consequence of T2D, is unknown. Here, we describe a causal relationship between SLC16A13 function and the development of insulin resistance and fatty liver, reflecting the phenotype associated with the SLC16A13 polymorphism in a cohort of Japanese people. Our hypothesized link between intracellular l-lactate availability and hepatic insulin resistance is supported by the characterization of the transporter localization and substrate specificity in vitro, and by the physiological phenotype of our knockout mouse model in vivo.
Importantly, cellular fractionation of mouse liver lysates clearly demonstrated that endogenous Slc16a13 protein localizes to the plasma membrane (Fig. 3c). In addition, Slc16a13 was identified as a plasma membrane transporter for l-lactate using Slc16a13-expressing HEK cells (Fig. 2 and Supplementary Fig. 2). Although l-lactate transport into HEK-Slc16a13 cells was only increased 2-fold compared to the HEK-control cells (Fig. 2b), recently published data on the SLC16A11 transporter also showed a comparably mild change in SLC16A11-dependent pyruvate transport that still may have an effect on T2D risk6. Most importantly, high levels of endogenous transport in HEK cells due to high SLC16A1/MCT1 activity may impede the detection of a stronger Slc16a13-dependent effect. While our investigation only addressed mouse Slc16a13, it is important to highlight that human and mouse protein share 87% protein identity and the human transporter may also localize to the plasma membrane and not only intracellular membranes as presented before by Williams et al.3. The description of l-lactate as a primary Slc16a13 substrate is supported by an unbiased metabolomics assessment of the liver that showed lactate was reduced in the livers of Slc16a13 knockout mice, while other monocarboxylates were not (Fig. 9e and Supplementary Fig. 8a). Although lactate levels are reduced by only 25%, which may be partially explained by MCT1-mediated compensation of lactate transport, these data strongly argue for lactate being a major substrate for Slc16a13. However, we did not investigate transport kinetics using additional substrates and, therefore, cannot rule out the possibility that substrates other than l-lactate are physiologically relevant or contribute to the SLC16A13-associated diabetic phenotype. Further studies will clarify if SLC16A13 also transports other substrates.
SLC16A1 encodes MCT1, which transports l-lactate, pyruvate, and ketone bodies across the plasma membrane4, and it is broadly expressed in contrast to SLC16A13. MCT1, along with two other prominent SLC16 family members MCT2 and MCT4, catalyzes the proton-linked influx or efflux of lactic acid in various tissues. Lactic acid is a product of glycolytic cells at low oxygen levels and substrate for gluconeogenesis and lipogenesis in the liver, kidney, and adipose tissue as well as for oxidative phosphorylation in the heart, skeletal muscle, and brain5,33. Increased lactate production in adipose tissue due to hypoxia34 and decreased oxidative capacity in insulin-resistant muscle35,36 contribute to increased circulating lactate levels associated with obesity and T2D37,38,39. Plasma lactate levels are unaltered in Slc16a13 knockout mice (Supplementary Fig. 8b, c), suggesting that loss of Slc16a13 can be at least partially compensated by other monocarboxylate transporters (e.g., MCT1, MCT2, and MCT4) that facilitate plasma lactate uptake into organs, or that the uptake of liver lactate is insufficient to affect plasma levels. Consistent with unaltered plasma lactate concentrations, muscle insulin sensitivity is unchanged in Slc16a13 knockout mice (Fig. 6h). An increased expression of hepatic monocarboxylate transporter encoding Slc16 genes was observed in the liver and kidney, what may have served compensatory purposes (Supplementary Fig. 9), indirectly supporting the finding that SLC16A13 transports l-lactate (Fig. 2b). Multiple Slc16 genes are dysregulated in the kidney of Slc16a13 knockout mice, probably due to the higher expression of Slc16a13 in the kidney compared to the liver (Fig. 1a). Just two Slc16 genes are significantly upregulated in Slc16a13 knockout liver under conditions of diet-induced obesity: Slc16a1 and Slc16a11. Both genes encode pyruvate transporter and lactate transport is well characterized for SLC16A1/MCT1. Therefore, it is likely that all three transporter share the same substrates: l-lactate and pyruvate. However, it is unlikely that this compensatory upregulation affects the Slc16a13 knockout phenotype to a major extend. Slc16a1 haploinsufficient mice are resistant to diet-induced obesity and associated metabolic perturbations40, vice versa Slc16a1 overexpression would rather result in a detrimental metabolic phenotype, supported by the finding of exercise-induced hyperinsulinemia in human patients with promotor-activating mutations in SLC16A141,42 and in a transgenic mouse line with beta cell-specific Slc16a1 overexpression43. Although reduced SLC16A11 activity is proposed to induce metabolic changes associated with a higher risk of T2D6, contrary data exist13,14, and the significance of Slc16a11 overexpression remains unclear. Furthermore, both, Slc16a1 and Slc16a11 show only slight increases in the liver, and although Slc16a11 is stronger dysregulated in the kidney, overexpression of SLC16 transporter in the kidney is to our knowledge not linked to metabolic phenotypes of improved insulin sensitivity.
Assuming that SLC16A13 transports l-lactate in the liver, the key question is why loss of SLC16A13 function might be (patho)physiologically relevant, if MCT1 and SLC16A11 show the same substrate specificity and at least MCT1-mediated l-lactate transport is able to compensate SLC16A13 loss. One reason might be the distinct capacity and affinity of substrate transport. Here, we describe Slc16a13 as a low-capacity, but high-affinity transporter for l-lactate with a Km value of approximately 0.47 µM in HEK293 cells (Fig. 2c). Compared to MCT2 and MCT4, MCT1 is described as intermediate affinity transporter with Km values between 3.5 and 4.5 mM4,33,44,45 for the human protein, reflecting lower affinity for l-lactate compared to mouse Slc16a13. Human plasma concentrations of l-lactic acid are ~1.0–1.5 mM46, whereas plasma concentrations in our C57BL/6N wild-type mice are higher (Supplementary Fig. 8c). Therefore, SLC16A13 may be a lactate transporter that is quickly saturated, but whose high affinity enables to react to minor changes in plasma lactate concentrations in the early stages of metabolic disease. Compared to MCT1, we speculate that SLC16A13 function is more relevant in liver disease and it may be the transporter mediating hepatic l-lactate influx during specific pathophysiological states. This idea is supported by our gene expression analyses: whereas hepatic Slc16a13 expression is upregulated in the context of diet-induced obesity (Fig. 1b), Slc16a1 expression was not found to be induced by HFD- compared to NCD-feeding (Supplementary Fig. 10a). In obese mice, the significant increase in Slc16a1 expression during fasting might reflect a compensatory upregulation in periods of low substrate supply, even though its hepatic expression is not induced by obesity per se. Furthermore, also Slc16a11 expression differs under distinct dietary conditions and food deprivation (Supplementary Fig. 10b), pointing towards distinct roles of these SLC16 transporters in diet-induced obesity. However, this hypothesis needs further investigation and to date, the distinct roles of both transporters remain unclear. Importantly, transport kinetics of the human SLC16A13 protein would allow the direct comparison of the Km values of both transporters.
Slc16a13 deletion is significantly associated with lower fat mass in NCD-fed (Fig. 4b), but not in HFD-fed mice (Fig. 5b, p = 0.13). Potentially, obesity-related increase in fat mass and decrease in energy expenditure mask the effect seen during normal-chow feeding. Although Slc16a13 has no effect on body weight and energy expenditure, and only a mild effect on fat mass in lean mice fed normal chow, loss of Slc16a13 function results in improved glucose metabolism in obese mice fed HFD as determined by oral glucose tolerance test (Fig. 5g, h) and revealed in more detail using HE clamp (Fig. 6). The significantly higher insulin sensitivity of Slc16a13 knockout mice can exclusively be explained by increased hepatic insulin sensitivity (Fig. 6e, f) since we did not observe any effects on insulin-stimulated peripheral glucose uptake (Fig. 6h). DAG accumulation due to reduced mitochondrial β-oxidation predisposes to the development of insulin resistance and T2D21,47,48,49. Conversely, we observed increased mitochondrial respiration that reduces hepatic lipid content and protects from diet-induced ectopic lipid accumulation in our Slc16a13 knockout mouse model (Figs. 7 and 8). These results are consistent with previous studies that have demonstrated that elevation of liver-targeted mitochondrial uncoupling can reduce HFD-induced hepatic steatosis and hepatic insulin resistance50,51. In our model, hepatic Slc16a13 seems to be causative in this protection, as this transporter is highly expressed in the liver, but only marginally expressed in other metabolically active tissues, such as skeletal muscle (Fig. 1a). Furthermore, adipose and muscle insulin sensitivity is unaltered between Slc16a13 knockout and wild-type mice, as determined by 2-deoxyglucose uptake during HE clamp (Fig. 6h). Most importantly, the proposed liver-specific effect is supported by the difference between the genotypes in mitochondrial respiration of primary hepatocytes and liver tissue (Fig. 8a, b), liver transcriptomics (Fig. 9b), and protein expression analyses (Fig. 9a). Opposite to these changes in liver metabolism, we could not detect altered mitochondrial function in skeletal muscle of knockout mice, indicated by transcriptomic data and respiratory protein expression (Supplementary Fig. 6). This may also explain the trend seen in whole-body energy expenditure whereby high oxygen consumption in the liver, which accounts for up to 50% of whole-body energy expenditure in mice52, but not skeletal muscle, may have a non-significant impact on total energy expenditure (Fig. 5c), which is consistent with prior observations using liver-specific mitochondrial uncoupling agents50,51. Although we assert altered hepatic β-oxidation to be causative for metabolic changes seen in the Slc16a13 knockout mice, the respiratory exchange ratio addressed by indirect calorimetry is unchanged between the genotypes (Fig. 5f). However, as assessed for whole-body energy expenditure, also fatty acid oxidation may not be upregulated on a whole-body level if our proposed mechanism is liver-specific. Finally, the overall mild phenotype of Slc16a13 knockout mice may also be at least partially explained by the observed upregulation of various Slc16 genes (Supplementary Fig. 9), maintaining hepatic lactate level above a critical threshold that may affect hepatic lipid and glucose metabolism more dramatically. Therefore, also biomarkers of liver and kidney function may not be altered in the Slc16a13 knockout mouse model (Supplementary Fig. 11).
Our whole-body knockout mouse model cannot distinguish between Slc16a13 action in different organs and its organ-specific phenotypical consequences. In this study, we did not focus on the role of Slc16a13 in organs other than the liver. However, in obese mice, our data do not point to any impact of Slc16a13 deficiency on adipose tissue and skeletal muscle. In contrast to the liver of obese mice, adipose tissue and skeletal muscle did not show differences in lipid accumulation, as addressed by adipose tissue weights (Supplementary Table 3) and absolute triglyceride quantification in quadriceps muscle (Supplementary Fig. 6c). Moreover, insulin sensitivity is unaltered in adipose tissue and skeletal muscle (Fig. 6h), and finally, oxidative phosphorylation is altered in the liver, but not in skeletal muscle (Supplementary Fig. 6a, b), which accounts for ~10% of whole-body energy expenditure in mice52. Importantly, Slc16a13 is only marginally expressed in adipose tissue and skeletal muscle (Fig. 1a and Supplementary Fig. 1a), and not differentially expressed comparing lean and obese mice (Supplementary Fig. 1c, d), speculating that the impact of Slc16a13 deficiency on these tissues may be low compared to tissues of high Slc16a13 abundance and/or dysregulation in obese mice, as seen in the liver. Although our data clearly show an effect in the liver, we cannot completely rule out the possibility that deletion of Slc16a13 in the kidney contributes to the effects on metabolism. Contrary to mice, human SLC16A13 shows its highest expression in the liver, and kidney SLC16A13 may not be crucial in human metabolic disease. Additionally, we and others3 only investigated mRNA expression. Further investigations on SLC16A13 protein levels could help uncover the physiological function of the transporter in different tissues more clearly.
Our results are similar in some aspects to published data on Slc16a1 knockout mice32,40. Although homozygous Slc16a1 knockout mice are not viable, heterozygous knockout mice are protected from diet-induced obesity, insulin resistance, and hepatic steatosis40. Reduced weight gain was associated with decreased hepatic fat accumulation and proposed to be mediated by lower liver lactate metabolism and subsequent AMPK activation32. There are several advantages of SLC16A13/MCT13 over MCT1 in terms of a pharmacological target for the treatment of metabolic disease: First, homozygous deletion in mice leads to viable offspring, which is not the case in Slc16a1 knockout mice. Second, Slc16a1 is ubiquitously expressed and thus, MCT1 inhibition could have broader physiological side effects. Finally, and most importantly, GWAS identified SLC16A13 as novel loci for T2D in one large cohort17.
Regarding the improved metabolic phenotype associated with loss of Slc16a13 function in our knockout mouse model, we propose the described human SLC16A13 polymorphism linked to T2D to reflect a gain-of-function mutation. Interestingly, this may be opposite to previously reported SLC16A11 polymorphisms where loss-of-function mutations leading to T2D are postulated6,16. The SLC16A11 T2D risk variants have been shown to impact the expression of SLC16A11 but not of SLC16A136. Our work proposes SLC16A13 gain-of-function genetic variants to increase T2D risk and raises the question about a potential causative role for SLC16A13 other than SLC16A11, highlighting the need of additional studies to better understand the distinct mechanism through which these SLC16 transporters affect the development of T2D. Finally, our study demonstrates that targeting SLC16A13 represents a promising therapeutic strategy for the treatment of fatty liver and insulin resistance at the same time.
Methods
Animals
Slc16a13 heterozygous mice on C57BL/6N genetic background were generated by Taconic Biosciences using CRISPR/Cas9 technology. Mice were bred to homozygosity in the animal facility at the Experimental Centre of the Medical Faculty, TU Dresden. Animals were housed under standard conditions with a room temperature of 22 °C, humidity of 50–60%, 12 h light-dark cycle, and free access to water and food. Slc16a13 knockout and littermate wild-type males were fed with normal-chow diet (NCD, 15% calories from fat, sniff, Soest) or high-fat diet (HFD, 60% calories from fat, sniff, Soest) at an age of 5 weeks to 20 weeks. In vivo experiments were performed after an overnight fast at the indicated ages and if not stated otherwise, mice were killed after an overnight fast at 20 weeks of age. If not stated otherwise, parameters were investigated at 15 weeks of HFD-feeding and one data set represents one experiment on a single cohort of animals, whereat multiple cohorts have been analyzed. All procedures were approved by the Landesdirektion Sachsen (TVV 59/2015).
Metabolic monitoring
Fat and lean mass were determined by 1H nuclear magnetic resonance using the minispec LF50 body composition analyzer (Bruker). The TSE labmaster caging system (TSE systems) was used to analyze O2 consumption (VO2) and CO2 production (VCO2) by indirect calorimetry, energy expenditure as calculated by (VO2 × 15.818) + (VCO2 × 5.176), locomotor activity as well as food intake.
Oral glucose tolerance test
40% glucose solution was administered by gavage (2 mg glucose per gram body weight) and blood glucose was determined prior and 15, 30, 60, and 120 min after application. Blood was taken from the tail by massage technique.
Hyperinsulinemic euglycemic clamp
Insulin sensitivity of the animals was investigated using the hyperinsulinemic–euglycemic clamp as described previously53. Intravenous lines were placed into the right vena jugularis externa. One week after surgery, mice were continuously infused with 0.05 μCi/min [3-3H]glucose (Perkin Elmer) over 2 h to assess basal glucose turnover. Hyperinsulinemic euglycemic clamps were conducted over 140 min with a primed insulin bolus of 21 mU/kg over 3 min, followed by continuous infusion of 3 mU/(kg-min) insulin and a variable infusion of 20% dextrose to maintain euglycemia (120 mg/dl). 0.1 μCi/min [3-3H]glucose was continuously infused to determine whole-body glucose uptake and insulin suppression of endogenous glucose production after the basal period. 10 μCi bolus of 2-deoxy-d-[1-14C]glucose (Perkin Elmer) was injected after 85 min for the determination of the insulin-stimulated tissue glucose uptake into individual insulin-sensitive organs, i.e., skeletal muscle and adipose tissue.
De novo lipogenesis
Hepatic de novo lipogenesis was assessed as reported previously54. Mice were injected i.p. with 23.4 µl deuterated saline (0.9% saline dissolved in 99% 2H2O-enriched water, Sigma-Aldrich) per gram body weight. At this time, drinking water was replaced with 6% 2H2O, 1% sucrose. After three nights, mice were fasted for 6 h, killed and liver tissue and whole blood were collected. Triglycerides were extracted from the frozen liver by a chloroform-methanol extraction. Samples in chloroform were purified by silica gel thin-layer chromatography. Triglyceride-fatty acids were analyzed by GC–MS as fatty acid methyl esters following derivatization with methanolic boron trifluoride. The plasma 2H2O pool was assessed by exchange of hydrogens from plasma to acetone in the presence of sodium hydroxide; acetone deuterium enrichment was analyzed by GC–MS.
VLDL export
Hepatic VLDL-triglyceride secretion was analyzed as described before55. Briefly, blood was taken from the tail prior to and 1, 2, 3, and 4 h after i.p. injection of 0.001 g Poloxamer-407 (Sigma-Aldrich, dissolved in 0.9% saline) per gram body weight. Plasma triglycerides were determined by an enzymatic method.
Insulin stimulation
Mice were injected i.p. with 7.5 mU insulin per gram body weight or 0.9% saline as control. Liver tissue was sampled 20 min after injection.
Generation of HEK-Slc16a13 cells
The cDNA encoding mouse Slc16a13 was cloned using a PCR-based approach. The primer pair 5′-TGT-TCC-TTT-CGG-AAT-GGT-GCG-3′ and 5′-CGG-TGG-AGT-TTA-GTT-CTC-TCC-C-3′ for HEK-Slc16a13 or 5′-TCA-CTT-GTC-GTC-ATC-GTC-TTT-GTA-GTC-GTT-CTC-TCC-CAG-CCC-CTC-C-3′ for HEK-Slc16a13-FLAG was used to amplify Slc16a13 from mouse liver cDNA. Amplified fragments were cloned into pcDNA3.1(+) expression plasmid. HEK293 cells were stably transfected with mouse Slc16a13 expression plasmid (HEK-Slc16a13), Slc16a13-FLAG expression plasmid (HEK-Slc16a13-FLAG) or the empty vector (HEK-control) using Effectene transfection reagent (Qiagen) and selected using geneticin (800 µg/ml). Overexpression of Slc16a13 in the HEK-Slc16a13 and HEK-Slc16a13-FLAG cell line compared to HEK-control cells was determined by qRT-PCR.
Immunofluorescence staining
HEK-Slc16a13-FLAG or HEK-control cells were seeded onto poly-D-lysine coated cover glasses, fixed in 4% PFA, blocked with 10% goat normal serum in PBS, and incubated with the following antibodies and dyes: FLAG (ThermoFisher Scientific, PA1-984B, 1:500), Golgin-97 (ThermoFisher Scientific, A-21270, 1:100), Calnexin (Novus Biologicals, NB300-518, 1:100), goat anti-rabbit Alexa Fluor 488 secondary antibody (ThermoFisher Scientific, A-11008, 1:500), goat anti-mouse Alexa Fluor 594 (ThermoFisher Scientific, R37121, 1:500), Alexa Fluor 594 wheat germ agglutinin (ThermoFisher Scientific, W11262, 1:100), MitoTracker CMXRos (ThermoFisher Scientific, M7512, 100 nM) and Hoechst 33342 (Invitrogen, 1:2000). Microscopy has been performed at the Leica TCS SP5 confocal laser scanning microscope (Leica Microsystems).
l-lactate uptake
Uptake experiments were performed as previously described56. In brief, HEK-Slc16a13 and HEK-control cells were seeded onto poly-d-lysine coated 12-well plates. After three days, cells are washed twice with pre-warmed uptake buffer (120 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 5 mM glucose, 1.8 mM CaCl2, 25 mM HEPES, pH 7.3) and incubated in uptake buffer for 2 min at 37 °C with the indicated concentrations of unlabeled l-lactic acid spiked with l-[14 C(U)]-Lactic Acid (Perkin Elmer). Cells were washed three times with ice-cold uptake buffer before lysing the cells with 0.2% SDS. 14C l‑lactic acid in the whole-cell lysate was detected by liquid scintillation counting. Uptake experiments were performed three times in triplicates. All transport rates were normalized to protein content. The transport of l-lactate in HEK-control cells was set to 100% to calculate l-lactate uptake of HEK-Slc16a13 cells (%). Net l-lactate uptake was calculated as the difference in transport between HEK-Slc16a13 cells and HEK-control cells. Km value was assessed using nonlinear regression curve fit (GraphPad Prism 7).
Isolation of mouse primary hepatocytes
Isolation of hepatocytes was performed as previously described57. Hepatocytes were isolated from NCD-fed female mice at a mean age of 20 weeks. Mice were anesthetized by i.p. injection of ketamine-xylazine. Livers were perfused via the vena cava with pre-warmed perfusion buffer (Earle’s Balanced Salt Solution w/o CaCl2/MgCl2) supplemented with 0.5 mM EGTA, followed by perfusion with pre-warmed digestion buffer (5000 U collagenase in phenol red-free Hank’s Salt Solution). The liver was resected and mashed in Dulbecco’s modified Eagle’s medium (DMEM) w/o pyruvate containing 25 mM glucose, 10% fetal calf serum (FCS), and 1% penicillin/streptomycin. The cell suspension was filtered through a 100 µM mesh and hepatocytes were purified by density gradient centrifugation using Percoll solution (GE Healthcare).
Cell culture
Primary hepatocytes and HEK293 cells were cultured in DMEM containing 25 mM glucose, 10% FCS, and 1% penicillin/streptomycin. For the culture of HEK cell lines, geneticin (800 µg/ml) was added to the medium. Cells were cultured in an incubator at 37 °C and 5% CO2. Cell culture media, supplements, and buffers are purchased from Gibco unless otherwise stated.
Human liver samples
Liver biopsies from patients undergoing partial hepatectomy were taken from a segment without pathological findings as reported previously57,58,59. The tissue was snap-frozen immediately and processed for mRNA expression analysis. Medical history, physical parameters like age, BMI, fat mass, and waist circumference as well as clinical parameters including fasting blood glucose and insulin levels to calculate the HOMA-IR have been recorded for all patients. Histopathology was carried out by evaluating the NAFLD-activity score (NAS), which uses the criteria steatosis, inflammation, hepatocyte ballooning, and fibrosis. Subjects were then characterized according to steatosis severity: steatosis score = 0 (0–<5% steatosis); steatosis score = 1 (5–33% steatosis); and steatosis score ≥ 2 (>33% steatosis)60. Excessive ethanol consumption was considered to be >20 g/day for women and >40 g/day for men. Metabolic syndrome and type 2 diabetes were diagnosed according to accepted criteria. Clinical chemistry was measured in fasting blood samples in certified laboratories. Whole-body insulin resistance was estimated by the homeostasis model assessment of insulin resistance61. Characteristics of the 45 included patients from the cross-sectional INSIGHT study are shown in Supplementary Table 1. Informed consent was obtained from each patient included in the study, and the study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in approval by the ethics committee of the Charité-Universitätsmedizin Berlin (ethics approval number: EA2/135/08, German Clinical Trials Register: DRKS00005450).
qRT-PCR
Total RNA was extracted from frozen liver tissue homogenates using Trizol reagent (Sigma-Aldrich). cDNA was synthesized using the RevertAid First Strand cDNA Synthesis Kit and the qRT-PCR reaction was performed using SYBR Green (both ThermoFisher Scientific) and the CFX384 Touch Real-Time PCR Detection System (Bio-Rad). Procedures were performed according to the manufacturer’s instructions. Ct-values were normalized to the housekeeper HPRT. Primer sequences are shown in Supplementary Table 4.
H&E- and Oil-Red-O staining
Liver tissue was fixed in 4% PFA, embedded in paraffin, and cut into 3 µM sections. Liver sections were deparaffinized, rehydrated, and stained with hematoxylin–eosin (H&E) for histological evaluation of liver morphology. For visualization of liver lipids, the liver tissue was fixed, dehydrated in 20% sucrose solution, and embedded in TissueTek (Sakura Finetek) for cryostat sectioning. 15 µM-thick liver sections were stained with freshly prepared Oil-Red-O solution (Sigma-Aldrich, 3 mg/ml). Microscopy has been performed at the Axio Observer Z.1 widefield microscope using the Zen 2.3 software (Carl Zeiss microscopy).
Triglyceride extraction
Liver and muscle tissue were homogenized in 0.9% saline. Triglycerides were isolated using a 2:1 chloroform-methanol solution. Evaporated lipid phase was dissolved in isopropanol and quantified using an enzymatic assay.
Diacylglycerol and ceramide extraction
Hepatic diacylglycerols and ceramides were analyzed using liquid chromatography and tandem mass spectrometry as described before62.
PKCɛ translocation assay
Membrane-associated and cytosolic fractions were prepared as previously described23. Following western blotting, the ratio of membrane PKCε intensity (normalized to Na-K ATPase) to cytosolic PKCε intensity (normalized to GAPDH) was calculated.
Palmitate stimulation of mouse primary hepatocytes
Hepatocytes were seeded in collagen-coated 6-well plates (700,000 cells/well). After incubation for 5 h, DMEM was changed and cells were incubated overnight. The next day, the cell culture medium was replaced by DMEM without FCS and supplemented with 5% lipid-depleted serum and 0.5% fatty acid-free BSA or 500 µM fatty acid-free BSA-coupled palmitate (~6:1 molar ratio). After incubation for 18 h, cells were harvested for western blot analysis of AMPK and ACC phosphorylation.
Glucose production of mouse primary hepatocytes
In vitro gluconeogenesis was analyzed as previously described63 with minor changes. In short, hepatocytes were seeded in collagen-coated 12-well plates (300,000 cells/well) in DMEM high-glucose medium (25 mM glucose). After incubation for 5 h, cells were washed with PBS and incubated for 16 h in low-glucose DMEM (5.55 mM glucose) supplemented with 0.5% fatty acid-free BSA or 500 µM fatty acid-free BSA-coupled palmitate (~6:1 molar ratio). The next morning, cells were washed twice with PBS and cell culture media was replaced with 0.5 ml glucose production medium (DMEM base medium supplemented with 20 mM sodium lactate, 2 mM sodium pyruvate, and 10 mM HEPES, pH 7.4) in the presence of 500 µM BSA-coupled palmitate or 0.5% BSA control, and 100 nM glucagon or vehicle control. After incubation for 8 h, the cell culture medium was collected for analysis of glucose, and cells were harvested for protein normalization.
Oxygen consumption of mouse primary hepatocytes
Mouse primary hepatocytes were seeded in collagen-coated XF96 cell culture microplates (10,000 cells/well) and incubated overnight. On the day of the experiment, the cell culture medium was replaced by Seahorse assay medium (DMEM base medium supplemented with 31.6 mM NaCl, 1 mM sodium pyruvate, 2 mM l-glutamine, 10 mM glucose, pH 7.4). The Seahorse XF Cell Mito Stress Test was performed on the Seahorse XF96 Extracellular Flux Analyzer (Agilent Technologies) according to the manufacturer’s instructions using 3 µM oligomycin, 0.5 µM FCCP, 5 µM rotenone, and 5 µM antimycin A (Seahorse Bioscience). The oxygen consumption rate was normalized to protein content. The following states have been analyzed: basal respiration = OCR at baseline conditions, ATP production = OCR calculated as oligomycin-inhibited OCR subtracted from basal respiration, maximal respiration = OCR after FCCP-mediated uncoupling, spare respiratory capacity = OCR calculated as basal OCR subtracted from maximal OCR, and non-mitochondrial respiration = rotenone/ antimycin A-inhibited OCR.
Oxygen flux of mouse liver tissue
2 mg fresh liver was used for high-resolution respirometry with the Oroboros O2k oxygraph (Oroboros Instruments). SUIT protocol was performed according to the manufacturer’s instructions using 2 mM malate, 1 mM octanoylcarnitine, 2.5 mM ADP, 10 mM glutamate, 5 mM succinate, 0.25 µM steps of FCCP titration, and 5 µM antimycin A. Oxygen flux was normalized to tissue mass. The respiration states were defined according to the following substrate injections: LEAK = proton leak, ion leak and slip compensatory state (O2 flux after malate-octanoylcarnitine injection), FAO = fatty acid oxidation capacity (O2 flux after ADP injection), CIOXPHOS = complex I-related oxidative phosphorylation capacity (O2 flux after glutamate injection), CI + IIOXPHOS = complex I and III-related oxidative phosphorylation capacity (O2 flux after succinate injection), ETS = electron transfer system capacity (maximum O2 flux after FCCP titration), and ROX = residual oxygen consumption (minimum O2 flux after antimycin A injection).
Biochemical assays
Blood glucose levels were determined using the Accu-Chek Aviva Plus test system (Roche Diagnostics). Plasma insulin levels were analyzed using the Ultra Sensitive Mouse Insulin ELISA (Crystal Chem). Liver and plasma triglyceride levels were measured using the LabAssay Triglyceride kit (FUJIFILM Wako Pure Chemical Corp.). Plasma-free fatty acids were quantified using the NEFA-HR(2) assay (FUJIFILM Wako Pure Chemical Corp.). Plasma l-lactic acid concentrations were determined using the coloriometric l-lactate assay kit (Abcam). Plasma ALT, AST, creatinine, urea, and albumin as well as glucose in cell culture supernatant were measured using the cobas 8000 modular analyzer (Roche Diagnostics). All measurements were performed according to the manufacturer’s instructions.
Western blot
Tissue was lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease and phosphatase inhibitors (Life Technologies). 10–50 µg protein lysate was separated on 10% SDS-PAGE gels. For the Slc16a13 western blot, the plasma membrane was extracted using the Abcam kit ab65400 according to the manufacturer’s instructions, and 90 µg membrane protein was loaded on the SDS-PAGE gel. Protein gels were transferred to methanol-activated PVDF membranes using the wet electrophoretic transfer system (Bio-Rad). The membrane was blocked with 5% BSA or milk powder in TBS/0.1% Tween, primary antibodies were incubated at 4 °C overnight and secondary antibodies for one hour at room temperature. Peroxidase-conjugated secondary antibodies and Western Bright Chemiluminescence Substrate Sirius (Biozym) were used for protein visualization using the Fusion FX (Vilber Lourmat) and Fusion FX7 Advance imaging software. Densitometric analysis was carried out using ImageJ software (NIH). The following antibodies have been used: SLC16A13 (ThermoFisher Scientific, PA5-39249, 1:500), Vinculin (Abcam, ab129002, 1:10,000), GAPDH (Abcam, ab8245, 1:20,000), Na-K ATPase (Abcam, ab7671; 1:1000), PKCε (BD Biosciences, 610086, 1:1000), Acetyl Coenzyme A Carboxylase (Abcam, ab45174, 1:2000), Acetyl Coenzyme A Carboxylase (phospho S79) (Abcam, ab68191, 1:5000), total OXPHOS Rodent WB Antibody Cocktail (Abcam, ab110413, 1:1000), AMPK alpha 1 + AMPK alpha 2 antibody (Abcam, ab207442, 1:1000), AMPK alpha 1 (phospho T183) + AMPK alpha 2 (phospho T172) (Abcam, ab133448, 1:1000), Akt (pan) (Cell Signaling, 4691, 1:1000), Phospho-Akt (Ser473) (Cell Signaling, 4060, 1:1000), goat anti-rabbit IgG, HRP conjugated (Millipore, 401353, 1:10,000), goat anti-mouse IgG, HRP conjugated (Millipore, 401253, 1:10,000).
Sequencing
For next-generation sequencing, RNA quality and concentration were measured using a High Sensitivity Total RNA kit on a fragment analyzer (Agilent Technologies, Santa Clara, CA). RNA quality was high with RNA Integrity (RIN) values greater than 8.5 for all samples. For the library preparation, 50 ng of total RNA was used with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (E7760L) using the NEBNext Poly(A) mRNA Magnetic Isolation Module from (NEB #E7490) and the dual index plate (NEB # E6440L) from New England BioLabs Inc. We obtained 350 bp fragments including adapters in average size. For multiplexing, 11 individual libraries were normalized and pooled. The pooled libraries were clustered using the Illumina cBot Instrument (TruSeq SR Cluster Kit v3—cBot—HS (cat# GD-401-3001; Illumina Inc.)). Single-end reads were sequenced at a length of 85 bp with 8 bases dual index reads on an Illumina HiSeq4000 instrument (TruSeq SBS Kit HS- v3, 50-cycle, cat# FC-401-3002; Illumina Inc.). All samples reached a sequencing depth of 30–35 million reads per sample.
The limma package64 in the R programming environment was used to convert raw counts to log2-counts per million, normalized to account for the systematic mean-variance relationships often present in microarray and RNA-Seq data. A Wilcoxon Rank Sum-based gene set enrichment analysis was performed using the algorithm provided in the gage package65, which was designed for use on microarray and RNA-Seq data. All p-values were corrected for False Discovery Rate.
Metabolomics
Metabolomics analysis on mouse liver extracts and serum was performed by the U.C. Davis West Coast Metabolomics Center as previously described66. In brief, liver tissue (4 mg) was homogenized in extraction solution (acetonitrile:isopropanol:water, 3:3:2), vortexed for 45 s, and incubated for 5 min at 4 °C. Following centrifugation at 14,000×g for 2 min, aliquots were dried via evaporation overnight. The dried aliquot was resuspended in 50% acetonitrile (degassed) and centrifuged at 14,000×g for 2 min. The supernatant was evaporated again to dryness. Internal standards (C8-C30 fatty acid methyl esters) were added and the sample was derivatized by methoxyamine hydrochloride in pyridine and subsequently by N-methyl-N-trimethylsilyl trifluoro-acetamide for trimethylsilylation of acidic protons. Data were acquired using the method as described67, which is briefly summarized in Mitchell et al.68. Data were presented as a ratio of the metabolite to the total metabolites returned. Metabolite profiles were analyzed using MetaboAnalyst versions 3.0 and 4.069,70, utilizing univariate and multivariate built-in analytical methods from modules of this web-based platform, as specified. Differential expression analysis was performed with the limma package64 in the R programming environment and an unpaired two-tailed t-test with Welch’s correction of individual metabolites was performed by considering the genotype.
Statistics and reproducibility
Data analysis was performed using GraphPad Prism 7 software. A two-tailed unpaired Student’s t-test was used to compare the mean of two groups. One-tailed unpaired Student’s t-test was used for hypothesis testing of higher or lower than the control mean. One-way ANOVA followed by Dunnett’s multiple comparison test was used to compare means of more than three groups to the control group. Two-way ANOVA followed by Sidak’s multiple comparison test was used to compare two groups with repeated measurements. Statistical significance is stated as follows: *p < 0.05, **p < 0.01, ***p < 0.001. Different cohorts of Slc16a13 knockout and wild-type littermates were included in this study in order to conform animal welfare and secure the reproducibility of the metabolic phenotype.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Source data are available in Supplementary Data 1. RNA-Seq data have been deposited to the Gene Expression Omnibus (GEO) and can be accessed via the accession number GSE152937. The metabolomics data set has been deposited to the Metabolomics Workbench (study ID ST001643, project https://doi.org/10.21228/M8V702). Other data that support the findings of the study are available from the corresponding author on reasonable request.
Change history
14 July 2021
A Correction to this paper has been published: https://doi.org/10.1038/s42003-021-02425-2
References
Engelmann, J. et al. Determinants of mortality in patients with type 2 diabetes: a review. Rev. Endocr. Metab. Disord. 17, 129–137 (2016).
Morris, A. P. Progress in defining the genetic contribution to type 2 diabetes susceptibility. Curr. Opin. Genet. Dev. 50, 41–51 (2018).
Williams, A. L. et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506, 97–101 (2014).
Halestrap, A. P. & Price, N. T. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem. J. 343, 281–299 (1999).
Halestrap, A. P. & Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflug. Arch. 447, 619–628 (2004).
Rusu, V. et al. Type 2 diabetes variants disrupt function of SLC16A11 through two distinct mechanisms. Cell 170, 199–212.e120 (2017).
Hidalgo, B. A. et al. Associations between SLC16A11 variants and diabetes in the Hispanic Community Health Study/Study of Latinos (HCHS/SOL). Sci. Rep. 9, 843 (2019).
Almeda-Valdes, P. et al. The SLC16A11 risk haplotype is associated with decreased insulin action, higher transaminases and large-size adipocytes. Eur. J. Endocrinol. 180, 99–107 (2019).
Miranda-Lora, A. L. et al. Associations of common variants in the SLC16A11, TCF7L2, and ABCA1 genes with pediatric-onset type 2 diabetes and related glycemic traits in families: a case-control and case-parent trio study. Pediatr. Diabetes 18, 824–831 (2017).
Traurig, M. et al. Analysis of SLC16A11 variants in 12,811 American Indians: genotype-obesity interaction for type 2 diabetes and an association with RNASEK expression. Diabetes 65, 510–519 (2016).
Lara-Riegos, J. C. et al. Diabetes susceptibility in Mayas: evidence for the involvement of polymorphisms in HHEX, HNF4alpha, KCNJ11, PPARgamma, CDKN2A/2B, SLC30A8, CDC123/CAMK1D, TCF7L2, ABCA1 and SLC16A11 genes. Gene 565, 68–75 (2015).
Huerta-Chagoya, A. et al. Genetic determinants for gestational diabetes mellitus and related metabolic traits in Mexican women. PLoS ONE 10, e0126408 (2015).
Zhang, T., Qi, Z., Wang, H. & Ding, S. Adeno-associated virus-mediated knockdown of SLC16A11 improves glucose tolerance and hepatic insulin signaling in high fat diet-fed mice. Exp. Clin. Endocrinol. Diabetes https://doi.org/10.1055/a-0840-3330 (2019).
Zhao, Y. et al. Gain-of-function mutations of SLC16A11 contribute to the pathogenesis of type 2 diabetes. Cell Rep. 26, 884–892.e884 (2019).
Zhao, Y., Feng, Z. & Ding, Q. Type 2 diabetes variants in the SLC16A11 coding region are not loss-of-function mutations. Cell Rep. 29, 781–784 (2019).
Hoch, E., Florez, J. C., Lander, E. S. & Jacobs, S. B. R. Gain-of-function claims for type-2-diabetes-associated coding variants in SLC16A11 are not supported by the experimental data. Cell Rep. 29, 778–780 (2019).
Hara, K. et al. Genome-wide association study identifies three novel loci for type 2 diabetes. Hum. Mol. Genet. 23, 239–246 (2014).
Hirai, T., Fukui, Y. & Motojima, K. PPARalpha agonists positively and negatively regulate the expression of several nutrient/drug transporters in mouse small intestine. Biol. Pharm. Bull. 30, 2185–2190 (2007).
Petersen, M. C. & Shulman, G. I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 98, 2133–2223 (2018).
Shulman, G. I. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N. Engl. J. Med. 371, 1131–1141 (2014).
Birkenfeld, A. L. & Shulman, G. I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 59, 713–723 (2014).
Turpin, S. M. et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 20, 678–686 (2014).
Petersen, M. C. et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Invest. 126, 4361–4371 (2016).
Ipsen, D. H., Lykkesfeldt, J. & Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. life Sci. 75, 3313–3327 (2018).
Bergeron, R. et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 281, E1340–E1346 (2001).
Barber, M. C., Price, N. T. & Travers, M. T. Structure and regulation of acetyl-CoA carboxylase genes of metazoa. Biochim. Biophys. Acta 1733, 1–28 (2005).
Harwood, H. J. Jr. Acetyl-CoA carboxylase inhibition for the treatment of metabolic syndrome. Curr. Opin. Investig. Drugs 5, 283–289 (2004).
Wakil, S. J., Stoops, J. K. & Joshi, V. C. Fatty acid synthesis and its regulation. Annu. Rev. Biochem. 52, 537–579 (1983).
McGarry, J. D., Leatherman, G. F. & Foster, D. W. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem. 253, 4128–4136 (1978).
Savage, D. B. et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J. Clin. Invest. 116, 817–824 (2006).
Sun, S., Li, H., Chen, J. & Qian, Q. Lactic acid: no longer an inert and end-product of glycolysis. Physiology 32, 453–463 (2017).
Carneiro, L. et al. AMPK activation caused by reduced liver lactate metabolism protects against hepatic steatosis in MCT1 haploinsufficient mice. Mol. Metab. 6, 1625–1633 (2017).
Halestrap, A. P. The SLC16 gene family - structure, role and regulation in health and disease. Mol. Asp. Med. 34, 337–349 (2013).
Hosogai, N. et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56, 901–911 (2007).
Petersen, K. F., Dufour, S., Befroy, D., Garcia, R. & Shulman, G. I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 350, 664–671 (2004).
Stump, C. S., Short, K. R., Bigelow, M. L., Schimke, J. M. & Nair, K. S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl Acad. Sci. USA 100, 7996–8001 (2003).
Avogaro, A. et al. Intracellular lactate- and pyruvate-interconversion rates are increased in muscle tissue of non-insulin-dependent diabetic individuals. J. Clin. Invest. 98, 108–115 (1996).
Chen, Y. D., Varasteh, B. B. & Reaven, G. M. Plasma lactate concentration in obesity and type 2 diabetes. Diabete Metab. 19, 348–354 (1993).
Crawford, S. O. et al. Association of blood lactate with type 2 diabetes: the Atherosclerosis Risk in Communities Carotid MRI Study. Int. J. Epidemiol. 39, 1647–1655 (2010).
Lengacher, S. et al. Resistance to diet-induced obesity and associated metabolic perturbations in haploinsufficient monocarboxylate transporter 1 mice. PLoS ONE 8, e82505 (2013).
Otonkoski, T. et al. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am. J. Hum. Genet. 81, 467–474 (2007).
Otonkoski, T. et al. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes 52, 199–204 (2003).
Pullen, T. J. et al. Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic beta-cells leads to relative hyperinsulinism during exercise. Diabetes 61, 1719–1725 (2012).
Broer, S. et al. Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem. J. 333, 167–174 (1998). Pt 1.
Perez-Escuredo, J. et al. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 1863, 2481–2497 (2016).
Psychogios, N. et al. The human serum metabolome. PLoS ONE 6, e16957 (2011).
Samuel, V. T., Petersen, K. F. & Shulman, G. I. Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375, 2267–2277 (2010).
Goedeke, L., Perry, R. J. & Shulman, G. I. Emerging pharmacological targets for the treatment of nonalcoholic fatty liver disease, insulin resistance, and type 2 diabetes. Annu Rev. Pharm. Toxicol. 59, 65–87 (2019).
Camporez, J. P. et al. Mechanism by which arylamine N-acetyltransferase 1 ablation causes insulin resistance in mice. Proc. Natl Acad. Sci. USA 114, E11285–E11292 (2017).
Perry, R. J. et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 18, 740–748 (2013).
Perry, R. J., Zhang, D., Zhang, X. M., Boyer, J. L. & Shulman, G. I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256 (2015).
Kummitha, C. M., Kalhan, S. C., Saidel, G. M. & Lai, N. Relating tissue/organ energy expenditure to metabolic fluxes in mouse and human: experimental data integrated with mathematical modeling. Physiol. Rep. 2, https://doi.org/10.14814/phy2.12159 (2014).
Birkenfeld, A. L. et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 14, 184–195 (2011).
Vatner, D. F. et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc. Natl Acad. Sci. USA 112, 1143–1148 (2015).
Lee, H. Y. et al. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology 54, 1650–1660 (2011).
Seithel, A. et al. The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab. Dispos. 35, 779–786 (2007).
Heidenreich, S. et al. Retinol saturase coordinates liver metabolism by regulating ChREBP activity. Nat. Commun. 8, 384 (2017).
von Loeffelholz, C. et al. The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology 66, 616–630 (2017).
Neuschafer-Rube, F. et al. The mammalian INDY homolog is induced by CREB in a rat model of type 2 diabetes. Diabetes 63, 1048–1057 (2014).
Kleiner, D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005).
Matthews, D. R. et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419 (1985).
Yu, C. et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 277, 50230–50236 (2002).
Perry, R. J. et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 579, 279–283 (2020).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Luo, W., Friedman, M. S., Shedden, K., Hankenson, K. D. & Woolf, P. J. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161 (2009).
Mitchell, S. J. et al. Nicotinamide improves aspects of healthspan, but not lifespan, in mice. Cell Metab. 27, 667–676.e664 (2018).
Fiehn, O. et al. Quality control for plant metabolomics: reporting MSI-compliant studies. Plant J. 53, 691–704 (2008).
Mitchell, S. J. et al. Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab. 23, 1093–1112 (2016).
Chong, J. et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486–w494 (2018).
Xia, J. & Wishart, D. S. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinformatics 55, 14.10.11–14.10.91 (2016).
Acknowledgements
We thank Doreen Ussath, Julia Paditz, Stephan Friebe, and David Arturo Juárez López for excellent technical assistance. We thank Mario Kahn for the analysis of DAGs and ceramides, Werner Rust, and Dr. Tobias Hildebrandt for conducting the next-generation sequencing and Taconic Biosciences for the generation of Slc16a13 knockout mice. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—BI 1292/10-1 and the Deutsche Diabetes Gesellschaft (DDG, German Diabetes Association) as well as an unrestricted grant from Boehringer Ingelheim Pharma GmbH & Co. KG. This work was also supported, in part, by the Intramural Research Program of the National Institute on Aging, NIH.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
The study was designed by A.L.B. and T.S. Experiments have been performed by T.S., J.K., C.v.L., D.F.V., D.Z., R.J.P, M.B., E.S., C.H., A.K., and N.N.E.-A. and supervised by B.S.H., R.d.C., D.P., G.I.S., and A.L.B. Human material from the INSIGHT study was provided by C.v.L. Transcriptomics and metabolomics data sets have been analyzed by E.S., M.B., J.C., and J. F.O’S. The manuscript was written by T.S. and further input was given by all authors.
Corresponding author
Ethics declarations
Competing interests
Eric Simon and Bradford S. Hamilton are employees of Boehringer Ingelheim Pharma GmbH & Co. KG. The remaining authors declare no competing interests.
Additional information
Peer review information Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schumann, T., König, J., von Loeffelholz, C. et al. Deletion of the diabetes candidate gene Slc16a13 in mice attenuates diet-induced ectopic lipid accumulation and insulin resistance. Commun Biol 4, 826 (2021). https://doi.org/10.1038/s42003-021-02279-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02279-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
