
Abstract
The presence of FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) is one of the most frequent mutations in acute myeloid leukemia (AML) and is associated with an unfavorable prognosis. FLT3 inhibitors, such as midostaurin, are used clinically but fail to entirely eradicate FLT3-ITD + AML. This study introduces a new perspective and highlights the impact of RAC1-dependent actin cytoskeleton remodeling on resistance to midostaurin in AML. RAC1 hyperactivation leads resistance via hyperphosphorylation of the positive regulator of actin polymerization N-WASP and antiapoptotic BCL-2. RAC1/N-WASP, through ARP2/3 complex activation, increases the number of actin filaments, cell stiffness and adhesion forces to mesenchymal stromal cells (MSCs) being identified as a biomarker of resistance. Midostaurin resistance can be overcome by a combination of midostaruin, the BCL-2 inhibitor venetoclax and the RAC1 inhibitor Eht1864 in midostaurin-resistant AML cell lines and primary samples, providing the first evidence of a potential new treatment approach to eradicate FLT3-ITD + AML.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) is a genetically heterogeneous disease characterized by the clonal expansion of immature myeloid progenitor cells in the bone marrow (BM). Mutations of the FMS-like tyrosine kinase 3 (FLT3) gene occur in ~30% of AML cases, with internal tandem duplications (ITDs) being the most common type of mutation. These mutations lead to constitutive autophosphorylation of the FLT3 receptor; consequently, growth factor signaling pathways are activated. Thus, therapies that inhibit FLT3 kinase activity are highly warranted to disrupt disease progression. Drug discovery efforts have resulted in first- and second-generation FLT3 inhibitors, which can be classified as type I and type II based on their mechanism of interaction with the FLT3 receptor1. Type I inhibitors, including sunitinib (first-generation), midostaurin (first-generation), lestaurtinib (first-generation), crenolanib (second-generation), and gilteritinib (second-generation) interact with the ATP-binding site when the receptor is in the active conformation, while type II inhibitors, such as sorafenib (first-generation), ponatinib (first-generation), and quizartinib (second-generation), interact with a hydrophobic region directly adjacent to the ATP-binding domain that is accessible only when the receptor is inactive and prevent receptor activation. Unlike type II inhibitors, type I inhibitors reduce FLT3 phosphorylation in FLT3-ITD- and FLT3-TKD-mutated AML cells, while type II inhibitors can target FLT3-ITD but lack efficacy against TKD mutations, which exhibit secondary mechanism resistance to type II inhibitors1,2,3. Gilteritinib and midostaurin have been approved by the US Federal Drug Administration (FDA) to treat individuals with FLT3-mutant AML. Nevertheless, FLT3-ITD AML is associated with unfavorable prognosis, and patients develop drug resistance, with the underlying mechanisms remaining largely unexplained4,5.
FLT3 receptor glycosylation and its autophosphorylation have been associated with resistance to FLT3 inhibitors in FLT3-ITD AML6,7,8,9. The exogenous expression of FLT3-ITD or FLT3-ITD mutations leads to Ras-related C3 botulinum toxin substrate 1 (RAC1) activation10. RAC1 belongs to the RHO GTPase family and is involved in many cellular functions, such as actin polymerization, cytoskeletal organization, migration, and apoptosis11. The polymerization of actin monomers (known as G-actin) is mainly carried out in four steps: (1) RAC1-GTP and phosphatidylinositol 4,5-bisphosphate (regulated positively by RAC1) bind to WAVE2 and N-WASP; (2) WAVE2 and N-WASP undergo conformational changes to an activated state; (3) the activated forms of N-WASP and WAVE2 recruit the ARP2/3 complex; and (4) the multiprotein complex comprising RAC1 + P-WAVE2 or P-N-WASP + ARP2/3 initializes actin polymerization to form actin filaments (known as F-actin)11,12,13. Moreover, profilin-1 (PFN1), which binds to N-WASP or WAVE proteins, can accelerate actin polymerization14. Deregulation of this process through increasing the number of actin filaments and cell stiffness enhances the invasiveness and therapy resistance in solid tumor15,16. Moreover, RAC1 inhibits apoptosis by binding to and stabilizing antiapoptotic BCL-2 family proteins (BCL-2 and MCL1) in colon cancer, cervical cancer, and B-cell lymphoma17,18,19.
We hypothesized that FLT3 receptor phosphorylation and glycosylation, which have been associated with FLT3 inhibitor resistance, may induce RAC1 hyperactivation, which consequently deregulates of actin dynamics and the antiapoptotic BCL-2 family and may confer midostaurin resistance in FLT3-mutant AML.
Results
FLT3-dependent RAC1 hyperactivation in FLT3-ITD + midostaurin-resistant cells
To study the role of RAC1-regulated cellular processes in response to FLT3 inhibitors, we developed midostaurin-resistant AML cell lines from the FLT3-ITD homozygous and heterozygous AML cell lines MV4-11 and MOLM-13, respectively. Parental cells are designated MID-Sens, and midostaurin-resistant cells are designated MID-Res. The IC50 of midostaurin in MV4-11 cells increased from 15.09 to 55.24 nM in MID-Sens and MID-Res cells; that of MOLM-13 cells increased from 29.41 to 87.83 nM (Fig. 1a). Afterwards, we characterized FLT3 receptor glycosylation and phosphorylation by western blotting, flow cytometry and direct stochastic optical reconstruction microscopy (dSTORM). In MV4-11 and MOLM-13 MID-Res cells, FLT3 receptor phosphorylation at tyrosine 969 (residue) was upregulated compared to that in their respective MID-Sens cell lines (Figs. 1b and S5). Additionally, FLT3 receptor glycosylation was upregulated in MV4-11 MID-Res cells versus MV4-11 MID-Sens cells (Figs. 1b and S5). To determine the impact of midostaurin on FLT3 receptor activity, MV4-11 and MOLM-13 MID-Sens and MID-Res cells were treated with 50 nM midostaurin for 24 h. A single cycle of midostaurin treatment increased FLT3 receptor glycosylation in MID-Sens and MID-Res cells. However, the reduction in tyrosine 969 phosphorylation was more pronounced in MID-Sens than in MID-Res cells (Figs. 1b and S5), with MID-Res cells showing no reduction in FLT3 phosphorylation. The FLT3 glycosylation level was correlated with FLT3 surface expression. As expected, FLT3 surface expression as measured by flow cytometry was increased in MV4-11 and MOLM-13 MID-Res cells compared with the respective MID-Sens cells (Fig. 1c). Moreover, dSTORM revealed the existence of FLT3 clusters in the plasma membrane of MID-Res cells, which was not observed in MID-Sens cells (Fig. 1d).

a Midostaurin (Mid) IC50/cell viability and FLT3 glycosylation/phosphorylation analysis in MV4-11/MOLM-13 Mid sensitive (MID-Sens) and resistant (MID-Res) cells. b FLT3 phosphorylation/glycosylation was analyzed in MV4-11/MOLM-13 MID-Sens ± MID and MID Res ± MID by Western blot. FLT3 blot, G means glycosylated band (160 kDa) and NG, non glycosylated band (130 kDa). c FLT3 surface expression was analyzed in MV4-11/MOLM-13 MID-Sens ± MID and MID Res ± MID by flow cytometry. d FLT3 receptor was analyzed in MV4-11 MID-Res versus MID-Sens by super-resolution microscopy dSTORM. e RAC1 activation was analyzed in MV4-11/MOLM-13 MID-Res ± Mid 50 nM compared to MID-Sens cells ± Mid 50 nM (MID-Sens + Mid 50 nM versus MID-Sens − Mid (MV) p = 0.059, t = 3.4, df = 2.3 (MO) p = 0.07, t = 3.1, df = 2.3; MID-Res − Mid versus MID-Sens − Mid (MV) p < 0.01, t = 6.1, df = 3.8 (MO) p < 0.01, t = 5.7, df = 3.9; MID-Res + Mid versus MID-Sens (MV) p < 0.01, t = 10.03, df = 2.7 (MO) p < 0.01, t = 6.5, df = 3.4). f RAC1 activation was analyzed in FLT3 KD MV4-11/MOLM-13 MID-Res compared to scramble MV4-11 MID-Res cells ((MV) p < 0.01, t = 8.56, df = 2.9, (MO) p < 0.01, t = 5.1, df = 3.5). Relative light units (RLU). The western blot results are normalized by loading control (GAPDH) and are expressed as fold change relative to the control. Data are shown as means ± SDs (error bars) from three independent experiments.
Next, we quantified RAC1 activation (RAC1-GTP) in our two model cell lines: RAC1-GTP levels were increased in MV4-11 and MOLM-13 MID-Res cells compared to their respective MID-Sens cells (Fig. 1e). A single dose of midostaurin significantly decreased RAC1 activation in MV4-11 and MOLM-13 MID-Sens cells (Fig. 1e), but RAC1 activation was not reduced by 50 nM midostaurin in MID-Res cells (Fig. 1e). To show that FLT3 was responsible for RAC1 hyperactivation in MV4-11 and MOLM-13 MID-Res cells, we performed siRNA knockdown (KD) of FLT3 (Figs. 1f). FLT3 KD significantly reduced RAC1 activation in MV4-11 and MOLM-13 MID-Res cells (Fig. 1f). Moreover, FLT3 KD reversed midostaurin resistance in MV4-11 and MOLM-13 MID-Res cells (Fig. S1A).
The RAC1 inhibitor Eht1864 overcomes midostaurin resistance by inducing cell cycle arrest in G1 and apoptosis in MID-Res cell lines and primary samples
To define the functional role of RAC1 activation in resistance to midostaurin, we treated cells with the specific RAC1 inhibitor Eht1864. Eht1864 binds to RAC1 and keeps it in an inert and inactive RAC1 state, which prevents GEF-mediated nucleotide exchange as well as RAC1 binding to downstream effectors20. In our experiments, Eht1864 in combination with midostaurin synergistically reduced cell viability in MV4-11 and MOLM-13 MID-Res cells (Fig. 2a). This combination induced cell cycle arrest in G1 phase after 24 h of treatment (Fig. 2b). Subsequently, apoptosis activation and cell death induction in MID-Res cell lines were observed by caspase-3 cleavage protein and annexin V/PI assays upon treatment with this combination for 24 and 48 h, respectively (Figs. 2c).

a The cell viability and synergy analysis of Mid + Eht1864 (Eht) combination was done in MV4-11/MOLM-13 MID-Res treated with these compounds during 48 h. b The cell cycle analysis was carried out in MV4-11 MID-Res treated with Mid and Eht alone/combination during 24 h. c The apoptosis was studied by caspase-3 cleavage monoclonal antibody and cell death % was defined by Annexin V + /PI− + Annexin V + PI+ in MV4-11/MOLM-13 MID-Res cells treated with Mid and Eht alone/combination during 24 and 48 h, respectively. d Cell viability and synergy analysis of Eht + Mid combination was done in Refractory (REF) REF001/REF002 samples treated with these compounds during 48 h. e The apoptosis analysis in REF001/REF002 and in PBMCs obtained from healthy donors treated with Mid and Eht alone/combination during 24 h. Data are shown as means ± SDs (error bars) from three independent experiments.
Once we confirmed that RAC1 inhibition overcomes midostaurin resistance, we explored the effect of this combination in cells obtained from patients with refractory FLT3-ITD+ and FLT3-TKD + AML. A 24-h treatment with Eht1864 in combination with midostaurin synergistically reduced the viability of relapsed cells by inducing cell death (Fig. 2d, e). In addition, this combination did not induce toxicity in peripheral blood mononuclear cells (PBMCs) obtained from healthy donors (Fig. 2e).
Midostaurin-dependent actin polymerization machinery is overexpressed in midostaurin-resistant cells
To understand the underlying mechanisms of RAC1-induced midostaurin resistance, we focused on the genes that regulate actin polymerization. We found that the protein levels of total and phosphorylated N-WASP, WAVE2, and PFN1 were enhanced in MV4-11 and MOLM-13 MID-Res cells compared to their respective MID-Sens cells (Fig. 3a). Additionally, the expression of WASP protein (48% homology to N-WASP), which possesses lower actin polymerization activity than N-WASP, was upregulated in MV4-11 and MOLM-13 MID-Res versus their respective MID-Sens cells (Figs. 3a and S6). To determine the impact of midostaurin on the regulation of the actin polymerization machinery, MV4-11 and MOLM-13 MID-Sens and MID-Res cells were treated for 24 h with 50 nM midostaurin. A single treatment cycle of midostaurin reduced WAVE2 phosphorylation but not N-WASP phosphorylation in MV4-11 and MOLM-13 MID-Res cells (Figs. 3a and S6). Treatment with FLT3 KD and the RAC1 inhibitor Eht1864 reversed midostaurin resistance, reduced N-WASP phosphorylation in MV4-11 and MOLM-13 MID-Res cells (Figs. 3b). To determine whether the midostaurin response was regulated via actin polymerization, WAVE2, N-WASP, WASP, the ARP2/3 complex, and PFN1 were specifically targeted by siRNAs. The inhibition of N-WASP, the ARP2/3 complex, PFN1 and WASP reduced the proliferation of MV4-11 and MOLM-13 MID-Res cells over four days (Figs. 3c and S2A). However, WAVE2 KD did not induce any change in the proliferation of MV4-11 and MOLM-13 MID-Res cells (Fig. 3c). The IC50 of midostaurin was significantly reduced in MV4-11 and MOLM-13 MID-Res cells with N-WASP/ARP2/PFN1 KD compared to cells treated with scramble (Fig. 3c). No changes in the IC50 value of midostaurin were observed in MV4-11 and MOLM-13 MID-Res cells with WAVE2 and WASP KD (Figs. 3c, d and S2A). The decrease in cell viability observed upon N-WASP/ARP2/3/PFN1 inhibition was due to cell death induction, and this decrease was enhanced by midostaurin treatment (Fig. S2B). Additionally, the actin cytoskeleton disruptor Cytochalasin D and ARP2/3 small-molecule inhibitor CM-636 reversed midostaurin resistance in MID-Res cells via cell death induction (Fig. S2C).

a The protein expression analysis of actin polymerization positive (red) and negative (green) regulators by western blot in MV4-1/MOLM-13 MID-Res compared to parental cells, in MV4-11/MOLM-13 MID-Res ± 50 nM Mid compared to MID-Sens ± 50 nM Mid. b In FLT3 KD MV4-11/MOLM-13 MID-Res compared to scramble MV4-11 MID-Res and in MV4-11 MID-Res treated with Mid and Eht alone/combination. GAPDH was used as housekeeping. c Proliferation and Mid IC50 (after 4 days) in MV4-11/MOLM-13 MID-Res N-WASP KD ((MV) p < 0.0001, t = 25.7, df = 3.8, (MO) (p < 0.001, t = 10.5, df = 3.9)), WAVE2 KD ((MV) p = 0.32, t = 1.1, df = 2.93, (MO) p = 0.4, t = 1, df = 2.6), ARP2/3 complex KD ((MV) p < 0.001, t = 39.6, df = 2.06, (MO) p < 0.001, t = 11.8, df = 3.6), and PFN1 KD ((MV) p < 0.001, t = 25.1, df = 3.9, (MO) p < 0.001, t = 9.8, df = 3.5) compared to scramble MID-Res cells. d Actin polymerization positive regulators protein expression analysis targeting them by siRNAs in MV4-11/MOLM-13 MID-Res. e Correlation analysis between the mean of actin polymerization positive regulators (N-WASP, WAVE2, ARP2, PFN1) and FLT3 pathway genes expression in 639 AML patients by using microarray expression data (E-MTAB-3444). The western blot results are normalized by loading control (GAPDH) and are expressed as fold change relative to the control. Data are shown as means ± SDs (error bars) from three independent experiments.
To determine whether this regulation occurs in AML patients, correlation analysis between the expression of actin polymerization positive regulators (N-WASP, WAVE2, ARP2/3 complex, PFN1) and FLT3 signaling genes was performed by using publicly available microarray expression data (E-MTAB-3444). A positive correlation between the expression of these four inducers of actin polymerization and FLT3 signaling genes was observed in patients with FLT3-ITD (r = 0.67, n = 178) and FLT3 WT (r = 0.57, n = 461) de novo AML (Fig. 3e). Correlating the expression of each activator of actin polymerization individually with FLT3 pathway gene expression showed a moderate positive correlation for WAVE2/PFN1/ARP2 in FLT3-ITD and FLT3 WT de novo AML patients (Fig. S3A–C), but no correlation was observed between N-WASP and FLT3 pathway gene expression (Fig. S3D).
RAC1 hyperactivation increases cell stiffness and enhances adhesion forces to mesenchymal stromal cells in midostaurin-resistant FLT3-ITD+ AML cells
ARP2/3 activation is essential in the induction of actin polymerization21. The grade of actin polymerization positively correlates with actin filament growth, which is an important regulator of cell mechanical properties such as cell stiffness22,23,24. To show that the ARP2/3 complex was overactivated in MV4-11 and MOLM-13 MID-Res cells, we analyzed the actin filament load and stiffness in MV4-11 and MOLM-13 MID-Sens and MID-Res cells. We first visualized the load of actin filaments by high-resolution structured illumination microscopy (SIM) and observed an increased number of actin filaments in MV4-11/MOLM-13 MID-Res cells compared to their respective MID-Sens cells (Fig. 4a). Then, MV4-11 and MOLM-13 MID-Res cells were treated with the combination of Eht1864 and midostaurin for 24 h, and we observed a drastic reduction in the number of actin filaments in MV4-11 and MOLM-13 MID-Res cells (Fig. 4a). To assess changes in cell stiffness, we applied FluidFM®, a new microfluidic technology adapted to traditional atomic force microscopy (AFM)25, which confirmed significant increases in cell stiffness in MV4-11 and MOLM-13 MID-Res cells compared to MID-Sens cells (Fig. 4b). Likewise, the combination of Eht1864 and midostaurin for 24 h reduced cell stiffness in MV4-11 and MOLM-13 MID-Res cells (Fig. 4b). Moreover, as previously described by others, RAC1 activation increases the adhesion forces of cancer cells to mesenchymal stromal cells (MSCs), which negatively impacts the therapeutic response26. Accordingly, we independently co-cultured MID-Sens and MID-Res cells with the MSC line HS-5 for 24 h. The adhesion forces of MV4-11 MID-Res cells to HS-5 cells as well as those of MID-Sens cells to HS-5 cells were assessed by FluidFM®, and MV4-11 MID-Res cells were attached more strongly to HS-5 cells than were the MID-Sens cells (Fig. 4c and Supplementary Movies 1–2). Strikingly, the combination of Eht1864 and midostaurin (incubated for 24 h) was able to detach MV4-11 MID-Res cells from the MSCs (Supplementary Movies 3–5).

a Actin filaments visualization in MV4-11/MOLM-13 MID-Res versus MID-Sens cells and MV4-11/MOLM-13 MID-Res treated with the combination 50 nM Mid + 7.5 µM Eht versus MID-Res treated with 50 nM Mid + DMSO during 24 h. b Cell stiffness analysis in MV4-11/MOLM-13 MID-Res compared to MID-Sens cells ((MV) p < 0.001, F = 14.8, df = 1, (MO) p < 0.05, F = 6.4, df = 1) and in MV4-11/MOLM-13 MID-Res treated with the combination 50 nM Mid + 7.5 µM Eht versus MID-Res treated with 50 nM Mid + DMSO (vehicle control) during 24 h ((MV) p < 0.05, F = 5.3, df = 1, (MO) p < 0.01, F = 9.3, df = 1)*. c Cell adhesion forces quantification in MV4-11 MID-Sens/HS-5 cells and MV4-11 MID-Res/HS-5 (p < 0.05, F = 4.3, df = 1)*. Data are shown as means ± SDs (error bars) from three independent experiments. *Due to technical limitations, EHT1864 monotherapy was not performed in stiffness/cell adhesion assays.
The combination of Eht1864 and venetoclax reverses midostaurin resistance by blocking the antiapoptotic axis BCL-2/MCL1
As RAC1 is a positive regulator of BCL-2/MCL1, we studied the role of the BCL-2 family in the response to midostaurin in FLT3-mutant AML. BCL-2 and MCL1 expression was strongly upregulated in MV4-11 MID-Res cells compared to their parental cells (Figs. 5a); no changes were observed in MOLM-13 cells. To determine the role of midostaurin in BCL-2/MCL1 regulation, MV4-11 and MOLM-13 MID-Sens and MID-Res cells were treated with 50 nM midostaurin for 24 h. MCL1 expression was decreased by midostaurin in MV4-11 and MOLM-13 MID-Sens and MID-Res cells (Figs. 5a); however, BCL-2 expression was increased by midostaurin in MID-Sens cells (Figs. 5a). In addition, MV4-11 and MOLM-13 MID-Res cells were more sensitive to the BCL-2 inhibitor venetoclax than were the respective MID-Sens cell lines (Fig. 5b). Notably, the combination of midostaurin and Eht1864 decreased MCL1 expression but not BCL-2 expression in MV4-11 and MOLM-13 MID-Res cells (Figs. 5c). BCL-2 in the absence of MCL1 sequesters the proapoptotic proteins BIM/BAX/BAK and partially inhibits apoptosis27. Thus, BCL-2 expression could induce secondary resistance to the combination of midostaurin and Eht1864 in AML. Thus, we hypothesized that venetoclax would exhibit synergistic effects of with the midostaurin/Eht1864 combination therapy. Notably, venetoclax in combination with either midostaurin or Eht1864 synergistically reduced the viability of MV4-11 and MOLM-13 MID-Res cells by significantly abrogating MCL1/BCL-2 expression and inducing cell death (Figs. 5d, e and S4A, B). The results were confirmed in primary samples from two patients with refractory FLT3-ITD AML; cell death was induced in the majority of cells (>65%) (Fig. 5f). Of note, this triple combination was not toxic in PBMCs obtained from three healthy donors (Fig. 5f).

a BCL-2 and MCL1 protein expression analysis in MV4-11/MOLM-13 MID-Res compared to MID-Sens cells, in MV4-11/MOLM-13 MID-Res ± 50 nM Mid compared to MID-Sens ± 50 nM Mid. b Venetoclax (Ve) IC50 analysis in MV4-11/MOLM-13 MID-Sens and MID-Res cells treated with Ve during 48 h. c BCL-2 and MCL1 protein expression analysis in MV4-11/MOLM-13 MID-Res treated with Mid and Eht alone/combination. d Cell viability and synergy analysis in MV4-11/MOLM-13 MID-Res treated with Mid, Ve, and Eht alone/combination during 48 h. e BCL-2 and MCL1 protein expression and cell death analysis (Annexin V + /PI- + Annexin V + PI+) in DMSO (control), Mid (p = 0.76, t = 0.34, df = 2.3), Ve (p = 0.054, t = 2.7, df = 3.6), Eht (p < 0.05, 3.1, df = 3.7) and Mid + Eht (p < 0.01, t = 10.29, df = 3) Mid + Ve (p < 0.01, t = 8, df = 3.8) Ve + Eht (p < 0.001, t = 14, df = 3.9) and Eht + Ven + Mid (p < 0.001, t = 16.1, df = 3.2) during 24 h. f Cell death analysis by flow cytometry in REF001/REF002 AML samples and in PBMCs obtained from Healthy Donors HD4/HD5/HD6 treated with Mid, Eht, and Ve alone/combination during 24 h. The western blot results are normalized by loading control (GAPDH) and are expressed as fold change relative to the control. Data are shown as means ± SDs (error bars) from three independent experiments.
Discussion
Activation of the PI3K and JAK/STAT pathways is a known resistance mechanism against FLT3 inhibition in FLT3-ITD AML28,29,30,31. RAC1 is a positive regulator of both pathways and interacts with FLT310,32. This cross talk mediates drug resistance against classical chemotherapy via regulation of DNA repair pathways10,19. However, the role of RAC1 in the efficacy of FLT3 inhibitors in the treatment of FLT3-ITD leukemia has not yet been determined. Our study provides the first evidence that RAC1-dependent actin cytoskeleton remodeling and inhibition of apoptosis may underlie midostaurin resistance in FLT3-ITD AML.
FLT3-ITD mutations, which increase FLT3 autophosphorylation, lead to the activation of RAC1 in AML10,33. Clinical responses to FLT3 inhibitors have been correlated with the inhibition of FLT3 phosphorylation7. Moreover, FLT3 receptor glycosylation that is induced by FLT3 inhibitors, has been associated with FLT3 inhibitor resistance9. Accordingly, we observed increases in FLT3 phosphorylation/glycosylation and RAC1 hyperactivation in MV4-11 and MOLM-13 cells resistant to midostaurin, which was reversed by specifically targeting the FLT3 receptor. We used Eht1864, a specific RAC1 inhibitor, to investigate the effects of its hyperactivated state on midostaurin resistance. This compound is 20-fold more potent than other RAC1 inhibitors (Nsc23766) used in other AML studies10. Eht1864 reversed midostaurin resistance by inducing cell cycle arrest in G1 and activating apoptosis in established FLT3-ITD AML cell lines and in primary cells obtained from refractory AML patients with FLT3-ITD and FLT3-TKD D835 mutations. Weisberg et al. demonstrated that midostaurin reduces the viability of FLT3-mutated Ba/F3 leukemia cell lines and mouse models by inducing G1 arrest and apoptosis6. Therefore, this study corroborates our result that RAC1 inhibition overcomes the antitumor activity of midostaurin in FLT3-mutant clones resistant to midostaurin. Of note, Eht1864 and midostaurin either alone or in combination did not show any toxicity in PBMCs obtained from healthy donors. These results are in accordance with other studies, where no cell death was observed in healthy cells treated with these compounds 17,34.
RAC1 is a master regulator of actin cytoskeleton rearrangement35. RAC1 is activated by binding to N-WASP and WAVE2, which induces the conformational activation of these two proteins. Then, N-WASP and WAVE2 can bind to the ARP2/3 complex and activate it, resulting in actin polymerization36,37. The phosphorylation of N-WASP and WAVE2 is positively correlated with binding to the ARP2/3 complex and actin polymerization activation36,37. Any dysregulation in actin polymerization regulators favors cancer cell proliferation, migration, metastases and drug resistance in solid tumor15,38. However, their role in treating hematological malignancies has not yet been described. Here, we provide evidence that in FLT3-ITD AML, RAC1-dependent actin cytoskeleton remodeling plays a substantial role in the acquisition of resistance to midostaurin in vitro. N-WASP and WAVE2 phosphorylation is upregulated in midostaurin-resistant cells. N-WASP-specific KD but not WAVE2 KD reversed midostaurin resistance. Moreover, ARP2/3 complex inhibition by the small-molecule inhibitor CK-636 and siRNAs restored midostaurin activity. These results show the importance of N-WASP phosphorylation and ARP2/3 complex activation in midostaurin resistance in FLT3-mutant AML. Regarding WAVE2, because midostaurin is able to reduce its phosphorylation, we think that WAVE2 is not critical for midostaurin resistance in AML. In addition, proteins that stimulate actin polymerization, such as PFN1, have been associated with resistance to the proteasome inhibitor bortezomib in multiple myeloma.39. Additionally, we demonstrated that PFN1 expression is correlated with midostaurin resistance in FLT3-mutant AML cells.
Overactivation of actin polymerization leads to increased production of actin filaments40. As expected, MV4-11 and MOLM-13 midostaurin-resistant cells showed an increase in the number of actin filaments and cell stiffness compared to their respective midostaurin-sensitive parental cells. In line with our results, recent studies have described that tumor stiffening promotes resistance to the tyrosine kinase inhibitor sorafenib in triple-negative breast cancer cells and that cell rigidity is higher in cisplatin-/paclitaxel-resistant ovarian cancer cells versus parental ovarian cancer cells41,42,43. Moreover, massive actin filament accumulation has been associated with resistance to natural killer cell-based immunotherapy and chemotherapy in ovarian and breast cancers, respectively43,44. These results suggest that the cytoskeletal architecture and physical properties of cancer cells could play a critical role in drug resistance and could be novel parameters to evaluate therapy response.
It has been hypothesized that remodeling of the actin cytoskeleton is a strategy by which tumor cells evade apoptosis45. In fact, RAC1 blocks apoptosis by stabilizing the antiapoptotic proteins BCL-2 and MCL1 and upregulating their expression in cancer17,18. In line with that, MV4-11 MID-Res cells showed higher BCL-2 and MCL1 expression than did the parental cells. A single dose of midostaurin decreased MCL1 expression in sensitive and resistant cell lines. However, BCL-2 expression was upregulated under midostaurin treatment. As RAC1 activation is reduced under Midotaurin treatment in MV4-11 and MOLM-13 parental cell lines, RAC1 might regulate MCL-1 stability but not BCL2. Further investigation is needed to elucidate the role of RAC1 in the stability of BCL2/MCL-1 in MID-Sens/MID-Res cells. Due to BCL2 overexpression, higher sensitivity to the BCL-2 inhibitor venetoclax was observed in resistant cells than in sensitive cells. Recently, Ma et al. described that FLT3 inhibitors such as midostaurin and gilteritinib sensitize FLT3-ITD de novo AML cells to venetoclax by reducing MCL1 expression34. To induce full apoptosis in cancer cells, inhibition of MCL1 and BCL-2 is needed46. The combination of midostaurin and Eht1864 degrades MCL1 but increases BCL-2 expression. BCL-2 sequesters proapoptotic genes such as BIM and partially inhibits apoptosis47. Therefore, we believe that BCL-2 expression may contribute to secondary resistance to Eht1864 and midostaurin combination therapy. Consequently, we decided to add venetoclax, which was recently approved by the FDA in combination with hypomethylating agents or low-dose cytarabine for elderly individuals (75 years or older) with newly diagnosed AML or those who have comorbidities precluding intensive induction chemotherapy, to investigate whether BCL-2 inhibition enhanced the antitumor activity of the Eht1864/midostaurin combination therapy in FLT3-mutant AML. Strikingly, this triple combination comprising Eht1864, venetoclax and midostaurin synergistically reduced the viability of FLT3-ITD midostaurin-resistant cells and cells obtained from patients with FLT3-ITD refractory AML by reducing MCL1 and BCL-2 expression. Notably, no apoptosis was observed in PBMCs obtained from healthy donors treated with the triple combination therapy.
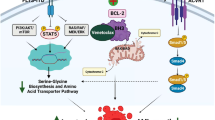
In summary, we propose a new midostaurin resistance mechanism in FLT3-mutated AML cells, in which actin cytoskeleton remodeling by FLT3-dependent RAC1 activation, N-WASP phosphorylation and ARP2/3 complex activation favors AML cell survival and proliferation. Moreover, RAC1 hyperactivation inhibits apoptosis by affecting MCL1/BCL2 expression, changes the mechanical properties of these cells and increases the adhesion forces of FLT3-ITD AML cells to MSCs (Fig. 6a). Based on these findings, under the condition that this combination is safe and feasible, we see clinical potential in combining midostaurin, venetoclax and Eht1864 as a novel therapeutic strategy to treat FLT3-mutant AML patients (Fig. 6b).

a Overactivation of actin polymerization machinery and antiapoptotic BCL-2 family in FLT3-ITD Midostaurin-resistant AML cells. b The triple combination Eht1864/Venetolax/Midostaurin by targeting Actin polymerization and BCL-2 reversed Midostaurin resistance.
Material and methods
Human samples and cell lines
The human AML cell line MV4-11 (homozygous FLT3-ITD) and MOLM-13 (heterozygous FLT3-ITD) were used for in vitro studies. Midostaurin-resistant cell lines were developed as follows: (1) 1 × 106/ml MV4-11 and MOLM-13 cells were plated in 24-well plates in 2 ml cell culture medium and treated with 10 nM Midostaurin, (2) every 7 days, leukemia cells were adjusted to 1 × 106/ml in fresh medium and 2 ml of this cell suspension plated per well in 24-well plates, and (3) MV4-11 and MOLM-13 cells were initially exposed to 10 nM Midostaurin and concentration was increased serially (increased by 10 nM weekly) up to 50 nM. MV4-11 and MOLM-13 parental cell lines were purchased from ATCC and DSMZ, respectively. They were maintained in culture in RPMI-1640 medium supplemented with 10% fetal bovine serum with 1% penicillin-streptomicin and 1% L-Glutamine at 37 °C in a humid atmosphere containing 5% CO2 at a concentration of 2 × 105 to 1 × 106. HS-5 MSC line was kindly provided by Patricia Maiso laboratory and were maintained in DMEM medium supplemented with 10% fetal bovine serum with 1% penicillin-streptomicin 1% L-Glutamine at 37 °C in a humid atmosphere containing 5% CO2 at a concentration of 5 × 105 to 1 × 106. The cell lines were tested for mycoplasma contamination monthly. BM mononuclear cells were obtained from refractory FLT3-mutated AML patients after an informed consent was signed by the patients or the patient’s guardians, in accordance with the Declaration of Helsinki. Clinical characteristics of relapsed FLT3-mutated AML patients used in this study are explained in Table S1. To test the toxicity of the compounds that were used in this work, we used PBMCs obtained from three healthy donors.
dSTORM analysis of FLT3 receptor expression
Localization data were analyzed by custom-made code written in Mathematica 11.1 (Wolfram Research Inc.). From each dSTORM image an appropriate region of interest was identified and localizations within this region of interest were selected. A DBSCAN clustering algorithm (Elki Data Mining) was applied to group localizations according to their local density48. Parameters were chosen by scanning the parameter space for weakest dependency of the resulting clustering set (neighborhood radius or epsilon = 20; minPoints = 3).
Dye conjugation to antibodies
After buffer exchange using 0.5 ml 7 kDa Spin Desalting Columns (Thermo Fisher, 89882, Erlangen, Germany) in 100 mM NaHCO3 50 µg of the purified monoclonal anti-FLT3 (BD Biosciences 558995, Heidelberg, Germany) were incubated in a five molar excess with Alexa Fluor 647 NHS (Thermo Fisher Scientific, A20101MP, Erlangen, Germany) at RT for 3 h in the dark. To exchange the buffer to 0.02 NaN3 and to remove unreacted dyes the antibodies were purified with 0.5 ml 7 kDa Spin Desalting Columns. Lastly, the degree of labeling of the conjugated antibody was measured by a UV–vis spectrophotometer (Jasco V-650). The conjugated antibodies were stored at 4 °C.
Live cell immunostaining
For super-resolved imaging 1.5 × 105 MV4-11 cells per well were seeded in poly-D-lysine coated chambers (Lab-Tek II, Nunc, Thermo Fisher Scientific, Erlangen, Germany) and the cells adhered at 37 °C at 5% CO2 for 1 h. For live cell staining the cells were incubated for 30 min on ice in an antibody concentration of 2.5 µg/ml in PBS. Following by washing of the cells and fixation for 15 min in 2% formaldehyde and 0.2% glutaraldehyde. After three more washing steps the cells were stored at 4 °C until in PBS imaging.
dSTORM imaging
The dSTORM samples were imaged on a homebuilt widefield setup with an inverted microscope (Olympus IX-71) using an oil immersion objective (Olympus APON 60xO TIRF, NA 1.49). The dyes were excited with a laser of the wavelength 639 nm (Genesis MX639-1000, Coherent) The excitation light was filtered from the fluorescence light by a beam splitter (ZT405/514/635rpc, Chroma) and an emission filter (Brightline HC 679/41, Semrock). Imaging was carried out with an EMCCD camera (iXon Ultra 897, Andor) for 15,000 frames at a rate 50 Hz at ~7 kW/cm in 100 mM ß-mercaptoethylamin pH 7.4. The super-resolved dSTORM images were reconstructed with the open source software rapidSTORM 3.349.
RAC1 activation assay
RAC1 activation was measured by RAC1 G-LISATM kit (Cytoskeleton BK126, Denver, CO, USA). This kit provides RAC1-GTP-binding protein linked to the wells of a 96 wells plate. The bound active RAC1 is detected with a RAC1 specific antibody and luminescence. The RAC1 activation was analyzed in MV4-11/MOLM-13 MID-Res ±50 nM Midostaurin compared to their parental cells ±50 Nm Midostaurin and FLT3 KD MV4-11/MOLM-13 MID-Res versus scramble MV4-11/MOLM-13 MID-Res. Following the kit instructions, these cells were incubated in serum free medium during 24 h to inactivate RAC1 In the case of FLT3 KD and scramble MV4-11/MOLM-13 MID-Res cells serum starvation was done 48 h after siRNAs transfection, to be sure that FLT3 was knocked down. Then, 5.106 MID-Sens/MID-Res cells ±50 nM Midostaurin during 30 min (At this time were observed the highest differences). In FLT3 KD experiment, 5.106 FLT3 KD/scramble MID-Res cells after serum starvation, were placed in supplemented RPMI-1640 medium at 37 °C in a humid atmosphere during 30 min. Then, the cells were harvested, lysates and protein was quantified by Precision RedTM Advanced Protein Assay Reagent (Cytoskeleon GL50). The final concentration used of equalized lysates was 1 mg/ml. Anti-RAC1 primary and the secondary HRP labeled antibody were diluted 1:5000. This antibody dilution allowed that readings were in the linear range of the luminometer. RAC1 activation Relative Light Units (RLU) was calculated by resting the lysis Buffer RLU mean (used as reference blank) to MID-Sens/MID-Res ±Midostaurin RLU mean MID-Res scramble/MID-Res FLT3 KD RLU mean.
Cell death and cell cycle analysis
The cell death and cell cycle assay were carried out as described previously 50.
Cell viability assay and cell treatments
Cell viability was analyzed by CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI, USA) following manufacturer’s instruction. All experiments were repeated three times. ABT-199 (Venetoclax), EHT 1864 2HCl (Eht1864), PKC412 (Midostaurin) and CK-636 compounds were purchased from selleckhem (Munich, Germany). Cytochalasin D was purchased from Sigma-Aldrich (Munich, Germany). To understand the mechanism by which the combination of Midostaurin + Eht1864, Midostaurin + Venetoclax, and Eht1864 + Midostaurin + Venetoclax overcome Midostaurin resistance, MV4-11 and MOLM-13 MID-Res cells were treated at the half maximal inhibitory concentration (IC50) and maintained in culture for one day. Then, the cells were washed in PBS and used for different assays. The IC50 value was determined using GraphPad Prism software, log (inhibitor) versus response variable slope from dose-response inhibition (version 5, La Jolla, CA, USA). The synergistic between these compounds in Midostaurin-resistant cells was evaluated by Compusyn software.
Correlation analysis between the expression of actin polymerization inducers and FLT3 signaling in AML patients
Meta information and gene expression data of 639 AML patients were obtained from ArrayExpress (E-MTAB3444)51. Raw data were RMA normalized using the {affy} package and multiple probes per gene were aggregated by mean. FLT3 target gene expression was correlated with the expression of N-WASP, WASF2, PFN1, ARP2 or the mean expression of these four genes (“inducers of actin polymerization”) dependent on the FLT3 mutation status of the patient. FLT3 target genes (n = 61) were extracted from PathCards (https://pathcards.genecards.org/; using the “FLT3 signaling” SuperPath52.
Cell stiffness assay
The stiffness of MID-Sens and MID-Res cells was assessed by using FluidFM® (Cytosurge AG, Glattbrugg (Zurich), Switzerland), a new technology adapted to traditional AFM in a Flex-FPM system (Nanosurf GmbH, Langen, Germany). It is mounted on an Axio Observer Z1 inverted microscope (Carl Zeiss, Germany) and a piezoelectric stage of 100 µm retraction range. Micropipette cantilevers (Cytosurge AG, Switzerland) of 8 µm aperture diameter and 0.33 N/m nominal spring constant were used. For the measurements of the stiffness of cells, a cell was immobilized at the aperture of the cantilever by applying a soft underpressure of 30 mbar, then, approaching at 500 nm/s, it was softly pressed against the culture dish until a set point of 3 nN was reached, while acquiring the data points at 3500 Hz. Then the cantilever was retracted, cleaned with NaClO 5% and thoroughly rinsed 4 times with ultrapure water and PBS. The procedure was repeated with 20 cells. The data points corresponding to the approach curves were extracted and processed in MATLAB 2017b (Mathworks, Natick, MA, USA) and fitted with the Hertz Model to obtain the modulus of elasticity, as indicator of the cell stiffness, as explained before53. The cell stiffness was measured in Midostaurin-resistant compared to sensitive cells and resistant cells treated with the combination 7.5 µM Eht1864 + 50 nM Midostaurin versus resistant cells + 50 nM Midostaurin during 24 h.
Adhesion forces assay
In total, 1.106 HS-5 cells were cultured in a 6 well plate for 24 h. Then, 5.105 MID-Sens and MID-Res cells were independently co-cultured with the stromal layer in serum free conditions for 24 h. Afterwards, we quantified and compared the cell adhesion forces of MID-Res and MID-Sens onto HS-5 cells. The quantification of adhesion forces was performed by using the above-described FluidFM® system. Micropipette cantilevers of 4 µm aperture diameter and a nominal spring constant of 0.4 N/m were used. MV4-11 MID-Sens or MID-Res cells were approached with the cantilever at a speed of 5 µm/s until it pressed the surface of a cell with a set point of 3 nN. Next, the cell was immobilized at the cantilever aperture by applying an under pressure of 300 mbar, and retracted at 5 µm/s until complete detachment from the stromal layer. The cantilever was cleaned with NaClO 5% and thoroughly rinsed with water and PBS after measuring each cell. The data acquired during the retraction of the cantilever were extracted with SPIP 6.2.0 software (Image Metrology, Copenhagen, Denmark) and the cell-cell adhesion force was calculated according to Hook’s Law, as explained before53.
siRNAs electroporation in AML cell lines
SiRNAs against FLT3 receptor (Reference: HSS103748) and actin polymerization inducers (ARP2 (Reference: HSS115366), N-WASP (Reference: HSS113263), WAVE2 (Reference: HSS145484) and PFN1 (Reference: HSS107879) were purchased at Thermo Fischer Scientific (Erlangen, Germany). SiRNAs were dispensed into 1.5 ml Eppendorf tubes and mixed with the cell suspension by gentle pipetting. The complete range of electroporation mixes was prepared and electroporation carried out with a Gene Pulser (Bio-Rad Laboratories, Munich, Germany) at a capacity setting of 960 µF and with 300 voltage.
Flow cytometric analysis of FLT3 receptor expression
Cell surface expression of FLT3 (CD135) was analyzed using a PE-conjugated mouse anti-human FLT3 monoclonal antibody (clone 4G8, BD Biosciences, Heidelberg, Germany). In brief, 1 × 106 cells in 100 µl FACS Buffer (1X PBS + 1% FCS + 5 mM HEPES) were stained with 20 µl of PE-FLT3 (the optimum concentration was determined after titrating the antibody) for 30 min at 4 °C. After washing the cells, a viability dye (1:1000) was used (Cat. No. L34960, Thermo Fisher Scientific, Erlangen, Germany) for 30 min at room temperature. Cells were washed and resuspended in 300 µl FACS Buffer and analyzed using the CytoFLEX flow cytometer (Beckman Coulter). 50000 living cells were recorded. The gating strategy consisted of debris removal (FSC/SSC), double discrimination (FSC-A/H), dead cell exclusion (APC-) and FLT3+. Analysis was performed using FlowJo V.10.6 (BD Biosciences).
Immunoblotting
Cells were harvested and lysed using lysis buffer (Thermo Fischer Scientific), reconstituted, and whole-cell lysates were subjected to SDS-PAGE and transferred to Nitrocellulose Blotting Membrane (GEhealthcare Life science). Anti-Phospho Tyrosin 969 FLT3 (working dilution 1:1000), Anti-ARP2 (working dilution 1:1000), anti-N-WASP (working dilution 1:1000), anti-WAVE2 (working dilution 1:1000), anti-PFN1 (working dilution 1:1000), anti–MCL1 (working dilution 1:1000), anti–BCL-2 (working dilution 1:1000), and anti-GAPDH (working dilution 1:2000) antibodies were purchased from Cell Signaling Technology; The HRP-conjugated secondary antibody (working dilution 1:5000) were from Cell Signaling Technology. Moreover, it was used anti-P-N-WASP [Ser484/Ser485] (working dilution 1:1000) and anti-P-WAVE2 [Ser343] (working dilution 1:1000) from Merck. The quantification of western blots were done by ImageJ software: (1) We measure the bands and their backgrounds along with the loading control bands (GAPDH) and their backgrounds, (2) Invert the pixel density for all data, the inverted value is expressed as 255—X, (3) after doing the inversions, it was calculated the net value of our net bands and GAPDH by substracting the inverted background, and (4) was measured fold change of net band/GAPDH relative to the control (scramble in siRNAs experiments and untreated sample in treatments experiment.
Immunofluorescence of actin filaments
In total, 1.5 × 105 cells were seeded on poly-D-lysine (Sigma, P6407-5MG, Munich, Germany) coated Lab-Tek chambers (Lab-Tek II, Nunc, Thermo Fisher Scientific, Erlangen, Germany) and adhered for 1 h at 37 °C and 5% CO2. Cells were fixed using a cytoskeleton buffer (CB, 10 mM MES buffer pH 6.1, 150 mM NaCl, 5 mM EGTA, 5 mM glucose, as previously reported54, with an optimized protocol designed for cytoskeleton staining. Here, the cells were incubated for 1 min in CB1 (CB + 0.25% Triton + 0.3% glutaraldehyde) followed by a 10 min incubation in CB2 (CB + 2% glutaraldehyde). Thereafter autofluorescence was quenched by an incubation of 7 min in 0.1% NaBH4 and unspecific binding was blocked by a 30 min incubation in 5% BSA (Sigma, A3983-100g) after three washing steps. The cells were stained overnight at 4 °C in 0.1 µM Atto643-Phalloidin (ATTO-TEC, AD 643-81) in 5% BSA and stored at 4 °C. Prior to imaging the cells were washed again and imaged in PBS using a Zeiss Elyra S.1 SIM. The z-stackes were rendered and processed with Imaris 8.4.1.
Statistics and reproducibility
All the experiments were repeated three times (Biological triplicates). Data are expressed as means ± standard deviation. Statistical significance between two samples was estimated with Student’s t test (two-tailed). Differences with p values < 0.05 were considered as significant (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ****p < 0.0001). Statistical significance in stiffness and adhesion forces assays was estimated with one way ANOVA test. Statistical analyses were performed utilizing GraphPad Prism 7.0 (GraphPad Software, Inc., San Diego, CA).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available within the manuscripts and its Supplementary files. Source data are provided in Supplementary Data 1. Meta information and gene expression data of 639 AML patients were obtained from ArrayExpress (E-MTAB3444) 51.
References
Ke, Y. Y. et al. Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci. Rep. 5, 11702 (2015).
Smith, C. C. et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 485, 260–263 (2012).
Wodicka, L. M. et al. Activation state-dependent binding of small molecule kinase inhibitors: structural insights from biochemistry. Chem. Biol. 17, 1241–1249 (2010).
Levis, M. Quizartinib in acute myeloid leukemia. Clin. Adv. Hematol. Oncol. 11, 586–588 (2013).
Knapper, S. FLT3 inhibition in acute myeloid leukaemia. Br. J. Haematol. 138, 687–699 (2007).
Weisberg, E. et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 1, 433–443 (2002).
Smith, B. D. et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood 103, 3669–3676 (2004).
Levis, M. et al. Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood 108, 3477–3483 (2006).
Williams, A. B. et al. Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood 120, 3069–3079 (2012).
Wu, M., Li, L., Hamaker, M., Small, D. & Duffield, A. S. FLT3-ITD cooperates with Rac1 to modulate the sensitivity of leukemic cells to chemotherapeutic agents via regulation of DNA repair pathways. Haematologica 104, 2418–2428. (2019).
Lawson, C. D. & Burridge, K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 5, e27958 (2014).
Tomasevic, N. et al. Differential regulation of WASP and N-WASP by Cdc42, Rac1, Nck, and PI(4,5)P2. Biochemistry 46, 3494–3502 (2007).
Takenawa, T. & Miki, H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 114, 1801–1809 (2001).
Yang, C. et al. Profilin enhances Cdc42-induced nucleation of actin polymerization. J. Cell Biol. 150, 1001–1012 (2000).
Castagnino, A. et al. Coronin 1C promotes triple-negative breast cancer invasiveness through regulation of MT1-MMP traffic and invadopodia function. Oncogene 37, 6425–6441. (2018).
Salvi, A. & Thanabalu, T. WIP promotes in-vitro invasion ability, anchorage independent growth and EMT progression of A549 lung adenocarcinoma cells by regulating RhoA levels. Biochem. Biophys. Res. Commun. 482, 1353–1359 (2017).
Velaithan, R. et al. The small GTPase Rac1 is a novel binding partner of Bcl-2 and stabilizes its antiapoptotic activity. Blood 117, 6214–6226 (2011).
Li, G. et al. Rac1b enhances cell survival through activation of the JNK2/c-JUN/Cyclin-D1 and AKT2/MCL1 pathways. Oncotarget 7, 17970–17985 (2016).
Cardama, G. A. et al. Relevance of small GTPase Rac1 pathway in drug and radio-resistance mechanisms: Opportunities in cancer therapeutics. Crit. Rev. Oncol. Hematol. 124, 29–36 (2018).
Shutes, A. et al. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 282, 35666–35678 (2007).
Padrick, S. B., Doolittle, L. K., Brautigam, C. A., King, D. S. & Rosen, M. K. Arp2/3 complex is bound and activated by two WASP proteins. Proc. Natl Acad. Sci. USA 108, E472–E479 (2011).
Kunschmann, T. et al. The Small GTPase Rac1 Increases Cell Surface Stiffness and Enhances 3D Migration Into Extracellular Matrices. Sci. Rep. 9, 7675 (2019).
Fletcher, D. A. & Mullins, R. D. Cell mechanics and the cytoskeleton. Nature 463, 485–492 (2010).
Janmey, P. A. et al. Effect of ATP on actin filament stiffness. Nature 347, 95–99 (1990).
Meister, A. et al. FluidFM: combining atomic force microscopy and nanofluidics in a universal liquid delivery system for single cell applications and beyond. Nano Lett. 9, 2501–2507 (2009).
Azab, A. K. et al. RhoA and Rac1 GTPases play major and differential roles in stromal cell-derived factor-1-induced cell adhesion and chemotaxis in multiple myeloma. Blood 114, 619–629 (2009).
Cheng, E. H. et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8, 705–711 (2001).
Minami, Y. et al. Different antiapoptotic pathways between wild-type and mutated FLT3: insights into therapeutic targets in leukemia. Blood 102, 2969–2975 (2003).
Spiekermann, K., Bagrintseva, K., Schwab, R., Schmieja, K. & Hiddemann, W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin. Cancer Res. 9, 2140–2150 (2003).
Zhou, J., Goh, B. C., Albert, D. H. & Chen, C. S. ABT-869, a promising multi-targeted tyrosine kinase inhibitor: from bench to bedside. J. Hematol. Oncol. 2, 33 (2009).
Broekman, F., Giovannetti, E. & Peters, G. J. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2, 80–93 (2011).
Campa, C. C., Ciraolo, E., Ghigo, A., Germena, G. & Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 6, 71–80 (2015).
Chatterjee, A. et al. Regulation of Stat5 by FAK and PAK1 in Oncogenic FLT3- and KIT-Driven Leukemogenesis. Cell Rep. 9, 1333–1348 (2014).
Ma, J. et al. Inhibition of Bcl-2 synergistically enhances the antileukemic activity of midostaurin and gilteritinib in preclinical models of FLT3-mutated acute myeloid leukemia. Clin. Cancer Res. 25, 6815–6826. (2019).
Van Aelst, L. & D’Souza-Schorey, C. Rho GTPases and signaling networks. Genes Dev. 11, 2295–2322 (1997).
Cory, G. O., Cramer, R., Blanchoin, L. & Ridley, A. J. Phosphorylation of the WASP-VCA domain increases its affinity for the Arp2/3 complex and enhances actin polymerization by WASP. Mol. Cell 11, 1229–1239 (2003).
Nakanishi, O., Suetsugu, S., Yamazaki, D. & Takenawa, T. Effect of WAVE2 phosphorylation on activation of the Arp2/3 complex. J. Biochem 141, 319–325 (2007).
Kazazian, K. et al. Plk4 promotes cancer invasion and metastasis through Arp2/3 complex regulation of the actin cytoskeleton. Cancer Res. 77, 434–447. (2017).
Lu, Y. et al. Profilin 1 induces drug resistance through Beclin1 complex-mediated autophagy in multiple myeloma. Cancer Sci. 109, 2706–2716. (2018).
Holmes, K. C., Popp, D., Gebhard, W. & Kabsch, W. Atomic model of the actin filament. Nature 347, 44–49 (1990).
Nguyen, T. V., Sleiman, M., Moriarty, T., Herrick, W. G. & Peyton, S. R. Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials 35, 5749–5759 (2014).
Zustiak, S., Nossal, R. & Sackett, D. L. Multiwell stiffness assay for the study of cell responsiveness to cytotoxic drugs. Biotechnol. Bioeng. 111, 396–403 (2014).
Sharma, S. et al. Correlative nanomechanical profiling with super-resolution F-actin imaging reveals novel insights into mechanisms of cisplatin resistance in ovarian cancer cells. Nanomedicine 8, 757–766 (2012).
Al Absi, A. et al. Actin cytoskeleton remodeling drives breast cancer cell escape from natural killer-mediated cytotoxicity. Cancer Res. 78, 5631–5643. (2018).
Desouza, M., Gunning, P. W. & Stehn, J. R. The actin cytoskeleton as a sensor and mediator of apoptosis. Bioarchitecture 2, 75–87 (2012).
Moujalled, D. M. et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 33, 905–917. (2019).
Siu, K. T. et al. BCL2 blockade overcomes MCL1 resistance in multiple myeloma. Leukemia 33, 2098–2102. (2019).
Schubert, E. K. A., Emrich, T., Züfle, A., Schmid, K. A. & Zimek, A. A framework for clustering uncertain data. 8 (Proceedings of the VLDB Endowment, 2015) https://dl.acm.org/doi/10.14778/2824032.2824115.
Wolter, S. et al. rapidSTORM: accurate, fast open-source software for localization microscopy. Nat. Methods 9, 1040–1041 (2012).
Garitano-Trojaola, A. et al. Deregulation of linc-PINT in acute lymphoblastic leukemia is implicated in abnormal proliferation of leukemic cells. Oncotarget 9, 12842–12852. (2018).
Stavropoulou, V. et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell 30, 43–58 (2016).
Belinky, F. et al. PathCards: multi-source consolidation of human biological pathways. Database (Oxford). 2015, bav006 (2015).
Sancho, A., Vandersmissen, I., Craps, S., Luttun, A., & Groll, J. A new strategy to measure intercellular adhesion forces in mature cell-cell contacts. Sci. Rep. 7, 46152 (2017).
Michie, M. S. et al. Cyanine conformational restraint in the far-red range. J. Am. Chem. Soc. 139, 12406–12409 (2017).
Acknowledgements
This research was supported by the IZKF grant (B-334). As well, we thank Patricia Maiso Laboratory (Marta Lasa and Sarai Sarvide) for providing HS-5 cell line and the protocol for co-culturing mesenchymal stromal cells and tumor cells.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conception and design: A.G.T., A.S., J.G., H.E., S.K., and K.M.K. Development of methodology: A.G.T., A.S., R.G., P.E., S.W., H.J., and J.G.P. Acquisition of data (provided reagents, facilities, etc.): A.G.T., A.S., H.J., S.W., M.S., and J.G. Reagents and materials: E.T. and N.R. Analysis and interpretation of data (statistical analysis, biostatistics analysis): A.G.T., A.S., E.A., R.G., and P.E. Provided and managed patients: M.C.D., S.K., and R.T. Writing, review, and/or revision of the manuscript: A.G.T., A.S., R.G., P.E., L.H., A.R., M.S., L.R., M.H., J.G., H.E., S.K., and K.M.K. Study supervision: A.G.T., S.K., and K.M.K. All authors critically reviewed and approved this paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Garitano-Trojaola, A., Sancho, A., Götz, R. et al. Actin cytoskeleton deregulation confers midostaurin resistance in FLT3-mutant acute myeloid leukemia. Commun Biol 4, 799 (2021). https://doi.org/10.1038/s42003-021-02215-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02215-w
This article is cited by
-
Cysteine- and glycine-rich protein 1 predicts prognosis and therapy response in patients with acute myeloid leukemia
Clinical and Experimental Medicine (2024)
-
ACTN1 promotes HNSCC tumorigenesis and cisplatin resistance by enhancing MYH9-dependent degradation of GSK-3β and integrin β1-mediated phosphorylation of FAK
Journal of Experimental & Clinical Cancer Research (2023)
-
HIV-1 diverts cortical actin for particle assembly and release
Nature Communications (2023)
-
Population distributions of single-cell adhesion parameters during the cell cycle from high-throughput robotic fluidic force microscopy
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
