
Abstract
Iron is the fundamental element for numerous physiological functions. Plasmalemmal divalent metal ion transporter 1 (DMT1) is responsible for cellular uptake of ferrous (Fe2+), whereas transferrin receptors (TFR) carry transferrin (TF)-bound ferric (Fe3+). In this study we performed detailed analysis of the action of Fe ions on cytoplasmic free calcium ion concentration ([Ca2+]i) in astrocytes. Administration of Fe2+ or Fe3+ in μM concentrations evoked [Ca2+]i in astrocytes in vitro and in vivo. Iron ions trigger increase in [Ca2+]i through two distinct molecular cascades. Uptake of Fe2+ by DMT1 inhibits astroglial Na+-K+-ATPase, which leads to elevation in cytoplasmic Na+ concentration, thus reversing Na+/Ca2+ exchanger and thereby generating Ca2+ influx. Uptake of Fe3+ by TF-TFR stimulates phospholipase C to produce inositol 1,4,5-trisphosphate (InsP3), thus triggering InsP3 receptor-mediated Ca2+ release from endoplasmic reticulum. In summary, these findings reveal the mechanisms of iron-induced astrocytic signalling operational in conditions of iron overload.
Similar content being viewed by others

Introduction
Iron contributes to numerous cellular and biochemical processes and acts as a co-factor in various molecular cascades in the nervous tissue including the synthesis and metabolism of several brain-specific enzymes and neurotransmitters1,2. In biological systems iron is present in either reduced ferrous (Fe2+) or oxidized ferric (Fe3+) state. The brain has the second (after liver) highest quantity of iron in the human body with total non-haem iron in the brain reaching about 60 mg3. The non-haem iron concentration in the serum ranges between 9 and 30 μM, whereas the iron concentration in cerebrospinal fluid (CSF) is much smaller being around 0.3–0.75 μM4,5. Transport of iron across the blood–brain barrier (BBB) is mediated either by transferrin receptor (TFR)-mediated endocytosis of Fe3+-bound to transferrin (holo-TF), or, for non-TF-bound iron, by vesicular and non-vesicular pathways6. Membrane transport of Fe2+ is also mediated by divalent metal ion transporter 1 (DMT1/SLC11A2) which underlies Fe2+ uptake through the plasma membrane or from endosomes6. Under physiological conditions, the intracellular cytosolic ionised iron levels fluctuate around 0.5–1.5 μM7.
In the brain, up to three-fourths of total iron is accumulated within neuroglial cells8. Astrocytes in particular are fundamental elements of ionostatic control over CNS environment9. Ionised Fe2+ enters astrocytes through DMT1/SLC11A2 transporters which are particularly concentrated in the endfeet of cerebral and hippocampal astrocytes10. In physiological condition, DMT1 is widely distributed in the nervous system, being expressed in neurones11,12,13, astrocytes13,14,15 and oligodendrocytes14,16 in vitro and in the brain tissue. In astrocytes, DMT1 mediates non‐transferrin‐bound iron (NTBI) transport, thus contributing to the brain iron homoeostasis in development and adulthood17,18,19. Conditional deletion of DMT1 form oligodendrocyte precursors substantially inhibited both myelination in development and remyelination in pathology16. Evidence for the expression of TFR in astroglial cells remains controversial6,20,21, while iron overload may influence the expression or distribution of TFR in astrocytic compartments21,22. Cellular uptake of Fe3+ requires internalization of TF–TFR complex23. An adaptor protein Disabled-2 (Dab2) plays an essential role in cell signalling, migration and development24. In mice the Dab2 has two isoforms of 96 and 67 kDa (p96 and p6724). In human K562 cells, Dab2 regulates internalization of TFR and uptake of TF25. Dab2 is also widely distributed in immune cells and in neuroglia24, although the functional link between Dab-2 and TFR in astrocytes has not been demonstrated.
Astrocytes possess a special form of intracellular ionic excitability, mediated by temporal and spatial fluctuations in the intracellular ion concentration26,27. Astroglial Ca2+ signalling is mediated by Ca2+ release from the endoplasmic reticulum (ER) following activation of inositol-1,4,5-trisphosphate receptor (InsP3R), or intracellular Ca2+-gated Ca2+ channels known as ryanodine receptors (RyR). Astroglial Ca2+ signals may also be generated by plasmalemmal Ca2+ entry through Ca2+-permeable channels or by sodium-calcium exchanger (NCX) operating in the reverse mode26,28. Astroglial Na+ signalling is shaped by plasmalemmal Na+ entry through cationic channels and numerous Na+-dependent transporters, of which the major role belongs to Na+-dependent glutamate transporters29,30,31; Na+ extrusion is mediated by the sodium-potassium pump (NKA). Both Na+ and Ca2+ signalling systems are closely coordinated, with NKA and NCX accomplishing this coordination at the molecular level32. Astrocytes specifically express α2-subunit containing NKA which is fundamental for astroglial K+ buffering33. Astrocytes express all three isoforms of NCX - NCX1/SLC8A1, NCX2/SLC8A2 and NCX3/SLC8A3, with some evidence indicating higher expression of NCX134. The NKA, the NCX and glutamate transporters are known to be preferentially concentrated in the perisynaptic astroglial membranes indicating intimate relationship between these ion-transporting molecules35,36. The NCX is also known to localise at caveolae rich in caveolin-3 (Cav-3), the latter isoform being predominantly expressed in astrocytes37.
In the present paper we performed an in depth analysis of the action of ferrous and ferric (Fe2+ and Fe3+) on astrocytic Ca2+ and Na+ dynamics. We found that Fe2+ (through DMT1) and Fe3+-TF (through TFR) evoke [Ca2+]i transients in astrocytes in culture and in vivo. Effects of Fe2+ on [Ca2+]i are mediated mainly by the reversed NCX, whereas Fe3+ triggers Ca2+ release from the endoplasmic reticulum by stimulations of InsP3R. In conclusion, we discovered that iron ions trigger astrocytic Ca2+ signalling by acting through two distinct molecular cascades.
Results
Fe2+/Fe3+ trigger [Ca2+]i increase in cortical astrocytes in vitro and in vivo
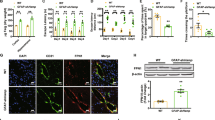
Exposure of cultured astrocytes to Fe2+ leads to a gradual increase in cytoplasmic Fe2+ concentration as revealed by quenching of Fura-2 (Supplementary Figure 1). Washout of Fe2+ leads to a full recovery of Fura-2 fluorescence; these data indicate the existence of plasmalemmal Fe2+ transport system as well as absence of non-specific damaging effect of ionised iron on the cellular membrane. We analysed effects of Fe2+ and Fe3+ Ca2+ dynamics in astrocytes in primary cultures and in vivo in GFAP-eGFP transgenic mice (Fig. 1). In the primary cultured astrocytes, administration of either FeSO4 (Fe2+) or ferric ammonium citrate-TF (Fe3+) increased [Ca2+]i in concentration-dependent manner, albeit with different kinetics. In the presence of Fe2+ an increase in [Ca2+]i demonstrated prominent plateau, whereas Fe3+ triggered transient, relatively rapidly decaying [Ca2+]i response (Fig. 1a).

a Representative images (top panel) and intracellular Ca2+ ([Ca2+]i) recordings from primary cultured astrocytes in response to different concentrations of Fe2+ or Fe3+. Every data point represents mean ± SD, n = 10, p < 0.05, statistically significant difference from the value of baseline in the same group. The experiment was repeated in 10 different cultures. The representative image on the left shows the primary cultured astrocytes co-stained with GLT1 (green), GFAP (red), and DAPI (blue). Representative Fluo-4 images (green) show astrocytes treated by 10 μM Fe2+ or Fe3+ at 0 s and 105 s of the recordings, the astrocytic identity was confirmed by co-staining with SR101. Scale bar, 20 μm. b Images (top panel) and [Ca2+]i recordings from cortical astrocytes in GFAP-eGFP transgenic mice using transcranial confocal microscopy. Every data point represents mean ± SD, n = 10 (different mice), p < 0.05, statistically significant difference from the value of baseline in the same group. Representative Rhod-2 images (red) show astrocytes treated with 100 μM Fe2+ or Fe3+ at 0 s (baseline) and 45 s (peak of the response); GFAP-eGFP images (green) are shown on the right. Scale bar, 10 μm. c Concentration-dependence of the maximal amplitude of [Ca2+]i responses triggered by Fe2+ or Fe3+ in cultured astrocytes.
When imaging cortical astrocytes in vivo (the cells were identified by specific eGFP fluorescence) we found that addition of either Fe2+ or Fe3+ for 30 s induced transient [Ca2+]i increase (Fig. 1b). Administration of Fe2+ increased fluorescent intensity of Rhod-2 to 456.30% ± 18.46% (n = 10, p < 0.0001) whereas Fe3+ increased the peak of fluorescent signal to 308.50% ± 13.01% (n = 10, p < 0.0001) of the basal value. In experiments in vitro we characterised concentration-dependence of [Ca2+]i responses: the EC50 was around 0.4–0.6 μM for both Fe2+ and Fe3+ (Fig. 1c).
DMT1 and TFR mediate Fe2+ and Fe3+ uptake
As mentioned above, Fe2+ uptake could be mediated by plasmalemmal transporter DMT1, whereas Fe3+ is mainly accumulated in TF-bound form by TFRs (Fig. 2a). Immunostaining of cortical tissue preparations and primary cultured astrocytes demonstrated co-localisation of DMT1 and TFR with astroglial GFAP-positive profiles (Fig. 2b). In the cortical tissue both DMT1 and TFR are preferentially localised at perivascular endfeet. Meanwhile, expression of specific DMT1 and TFR mRNA was also detected in the freshly isolated and sorted astrocytes and neurones, as well as and in the cerebral tissue (Fig. 2c).

a Astrocytes accumulate Fe2+ through plasmalemmal divalent metal transporter 1 (DMT1/SLC11A2) whereas Fe3+ is taken up by internalisation of Fe3+-TF-TFR complex. b Images of astrocytes in the somato-sensory cortex preparations or in primary culture double-immunolabeled with antibodies against DMT1 or TFR and against GFAP. Scale bar, 20 μm. c The mRNA expression of DMT1 and TFR measured by qPCR in astrocytes sorted from GFAP-GFP mice, in neurones sorted from Thy1-YFP mice, and in whole cerebral tissue of wild type mice. d Representative western blot bands for DMT1 and TFR in cultured astrocytes treated with sham (Control), siRNA negative control (−) or positive duplex chains (+). The protein levels are shown as the ratio of DMT1 (55 kDa) and β-actin (42 kDa), and TFR (92 kDa) and β-actin. Data represent mean ± SD, n = 6. *Indicates statistically significant (p < 0.05) difference from the value of baseline in the same group. e [Ca2+]i transients evoked by Fe2+ or Fe3+ after RNA interference and down-regulation of protein synthesis. After treatment with DMT1 or TFR siRNA negative control (−) or positive duplex chains (+) for 3 days, [Ca2+]i dynamics in response to Fe2+/Fe3+ was monitored. Every data point represents mean ± SD, n = 10. f Images of astrocytes in primary culture treated with sham (Control) or Fe2+ for 5 min and double-immunolabelled with antibodies against DMT1 and against GFAP. Scale bar, 25 μm. g Redistribution of DMT1 induced by Fe2+ in the extracted proteins of nucleus and cytoplasm. Every data point represents mean ± SD, n = 10. *Indicates statistically significant (p < 0.05) difference from control group.
To reveal the contribution of DMT1 and TFR to Fe2+/Fe3+-induced [Ca2+]i dynamics, we inhibited expression of DMT1 or TFR using siRNA duplex chains. The representative western blots demonstrating the efficacy of knockdown are shown in Fig. 2d. When compared to the control group, the DMT1 siRNA reduced expression of DMT1 to 9.52% ± 2.58% (n = 6, p < 0.0001), whereas treatment with TFR siRNA decreased TFR levels to 7.84% ± 2.10% (n = 6, p < 0.0001) from the control values. Administration of Fe2+ to DMT1-deficient astrocytes failed to induce any changes in [Ca2+]i. At the same time Fe2+ induced robust [Ca2+]i elevation in astrocytes treated with siRNA(−) (Fig. 2e). Similarly, Fe3+ did not produce [Ca2+]i, transients in astrocytes treated with TFR siRNA duplex chains, whereas in cells exposed to negative control siRNA Fe3+ evoked [Ca2+]i elevations (Fig. 2e). At the same time Fe3+-induced [Ca2+]i transients were fully preserved in DMT1-deficient astrocytes (Supplementary Figure 2), hence questioning the role of DMT1 in release of Fe3+ from endosomes.
In unstimulated astrocytes in culture the DMT1 fluorescence was the highest around the nucleus, suggesting its preferred intracellular localisation. After treatment of cultures with Fe2+ for 5 min we observed redistribution of DMT1 from the nuclear region to the plasma membrane (Fig. 2f). The levels of DMT1 were measured in the extracted proteins of the nucleus and cytoplasm (Fig. 2g). After treatment with Fe2+ for 5 min, the level of DMT1 in the nuclei decreased to 36.41% ± 4.33% of control group (n = 10, p < 0.0001), whereas the level of cytoplasmic DMT1 increased to 117.37% ± 5.79% of control group (n = 10, p < 0.0001) (Fig. 2g).
Sources of iron-induced [Ca2+]i mobilisation
The main sources for [Ca2+]i increase in astrocytes are (i) Ca2+ release from the ER following the opening of InsP3Rs or RyRs, or (ii) plasmalemmal Ca2+ influx through either Ca2+ permeable channels (such as L-type Ca2+ channels or TRP channels) or NCX operating in the reverse mode (Fig. 3a) or (iii) combination of some or all of these pathways. To dissect Ca2+ sources we first determined the influence of extracellular Ca2+ on iron-evoked [Ca2+]i transients. Removal of Ca2+ from the extracellular milieu completely abolished Fe2+-induced [Ca2+]i elevations but left Fe3+-evoked [Ca2+]i transients largely intact (Fig. 3b). This highlights the role for plasmalemmal Ca2+ influx in Ca2+ signalling triggered by Fe2+ and ER Ca2+ release for Ca2+ signals triggered by Fe3+.

a Possible pathways mediating Ca2+ influx into the cytosol. b [Ca2+]i recordings performed in the Ca2+-free extracellular medium; removal of external Ca2+ abolished Fe2+ evoked [Ca2+]i increase; conversely [Ca2+]i transient evoked by Fe3+ remains intact. c Pre-treatment with 10 μM nifedipine affects neither Fe2+ nor Fe3+-induced [Ca2+]i responses. d Pre-treatment with NCX inhibitor KB-R7943 at 10 μM suppresses [Ca2+]i response to Fe2+ but not to Fe3+. e Pre-treatment with InsP3 receptor inhibitor XeC at 10 μM suppresses [Ca2+]i response to Fe3+ but not to Fe2+. f Pre-treatment with 10 μM ryanodine (Ry), the inhibitor of ryanodine receptors, affects only plateau phase of Fe2+-induced [Ca2+]i response. In b-f, every data point represents mean ± SD, n = 10.
Incubation of astrocytes with inhibitor of L-type voltage-gated Ca2+ channel nifedipine (10 μM) did not affect [Ca2+]i responses to Fe2+ or to Fe3+ (Fig. 3c). In contrast, inhibition of NCX with 10 μM of selective agonist KB-R7943 completely eliminated [Ca2+]i responses to Fe2+, without affecting Fe3+-induced [Ca2+]i transients (Fig. 3d). Thus, two forms of iron, the ferrous and the ferric, mobilise intracellular Ca2+ through distinct pathways: Fe2+ stimulates Ca2+ influx by NCX, whereas Fe3+ triggers intracellular Ca2+ release. This suggestion was further corroborated by pharmacological inhibition of InsP3 receptors with potent antagonist Xestospongin C (XeC)38, Exposure of cultured astrocytes to 10 μM of XeC effectively suppressed [Ca2+]i response to Fe3+, without much affecting Fe2+-induced [Ca2+]i transient (Fig. 3e). Finally, treatment with 10 μM ryanodine (which at this concentration inhibits RyRs) somewhat decreased the plateau phase of Fe2+-induced [Ca2+]i transient without modifying [Ca2+]i response to Fe3+ (Fig. 3f).
DMT1 transports Fe2+, which inhibits NKA, increases [Na+]i and reverses NCX
Experiments described above have demonstrated that Fe2+, after being transported into the cell by DMT1, leads to a reversal of the NCX, which results in Ca2+ influx. The NCX reversal in astrocytes is triggered by an increase in [Na+]i. Such an increase may originate either from the activation of plasmalemmal Na+ entry or from inhibition of the NKA, which maintains basal [Na+]i28,30. The activity of NKA was suppressed by exposure to 10 μM Fe2+ to 82.40 ± 5.74% (n = 10, p < 0.0001) of the control. Exposure to 100 nM of the specific NKA inhibitor, ouabain, reduced NKA activity to 72.30 ± 5.91% (n = 10, p < 0.0001) of the control (Fig. 4a). When 10 μM Fe2+ and 100 nM ouabain were added together, the NKA activity was reduced to 71.80 ± 7.81% (n = 10, p < 0.0001) (Fig. 4a).

a NKA activity in the presence of Fe2+ and ouabain measured by ELISA; OD value was normalised to controls. Data represent mean ± SD, n = 10. *Indicates statistically significant (p < 0.05) difference from control group; **indicates statistically significant (p < 0.05) difference from any other group. b [Na+]i recordings from cultured astrocytes challenged with Fe2+ and ouabain as indicated on the graph (c) and (d) [Ca2+]i recordings from cultured astrocytes challenged with Fe2+, Fe3+ and ouabain as indicated on the graph. For b, c and d every data point represents mean ± SD, n = 10. e Representative western blot bands for Cav3 in cultured astrocytes treated with sham (Control), siRNA negative control (−), or positive duplex chains (+) to down-regulate Cav3 expression. Protein values are shown as the ratio of Cav3 (22 kDa) to β-actin (42 kDa). Data represent mean ± SD, n = 6. *Indicates statistically significant (p < 0.05) difference from any other group. f Cav3 RNA interference suppresses Fe2+-induced [Ca2+]i response. Every data point represents mean ± SD, n = 10.
Inhibition of NKA in astrocytes results in a substantial elevation in [Na+]i. When monitoring [Na+]i in cultured astrocytes with Na+-sensitive probe SBFI we found that both Fe2+ (10 μM) and ouabain (100 nM) triggered rapid and substantial elevation of [Na+]i (Fig. 4b). These changes in [Na+]i were paralleled by [Ca2+]i dynamics. Exposure of astrocytes to Fe2+, or ouabaine or mixture of Fe2+ and ouabain caused [Ca2+]i elevation (Fig. 4c). When Fe2+ was applied in the presence of ouabain it failed to change [Ca2+]i (Fig. 4d); at the same time application of Fe3+ in the presence of ouabain still triggered additional [Ca2+]i elevation (Fig. 4d).
Fe2+-induced Ca2+ mobilisation is associated with caveolae
Treatment of cultured astrocytes with interfering Cav3 siRNA duplex chains decreased the level of Cav3 to 7.41 ± 4.32% (n = 6, p < 0.0001) of the control (Fig. 4e). An in vitro knock-down of Cav3 significantly reduced amplitudes of Fe2+-induced [Ca2+]i increase; maximal increase in Fluo-4 F/F0 after Cav3 knockdown was 242.00 ± 16.44% (n = 10, p < 0.0001). In control astrocytes treated with siRNA(−) the amplitude of Fe2+-induced [Ca2+]i increase reached 363.34 ± 11.62% (n = 10, p < 0.0001, Fig. 4f). To further analyse the effects of Cav3 on Fe2+-induced [Ca2+]i dynamics the levels of relevant proteins were measured in the extracted caveolae (Fig. 5a). As shown in Fig. 5B, exposure to 10 μM Fe2+ for 5 min significantly increased the level of DMT1 to 293.24 ± 24.89% (n = 10, p < 0.0001) of the control values. After pre-treatment with Cav3 siRNA duplex chains, Fe2+ increased the level of DMT1 only to 195.77 ± 20.19% (n = 10, p < 0.0001) of control group (Fig. 5b). The levels of NCX1 and NKA were similarly affected by Fe2+ and the knocking down of Cav3. Exposure to Fe2+ increased the level of NCX1 and NKA to 248.71 ± 19.58% (n = 10, p < 0.0001) and 263.66 ± 25.93% (n = 10, p < 0.0001) of the controls, after knocking down Cav3, Fe2+ elevated the level of NCX1 and NKA only to 172.96 ± 11.76% (n = 10, p < 0.0001) and 200.86 ± 18.14% (n = 10, p < 0.0001) of control values (Fig. 5b). Of note, Fe2+ did not affect the levels of NCX2 and NCX3 (Fig. 5b).

a Representative protein western blot bands for DMT1, NCX1-3 and NKA-α1/α2 in caveolae membranes extracted from cultured astrocytes. Astrocytes were treated with sham (Control), with siRNA negative control (−) or with Cav3 duplex chains (siRNA(+)) for 3 days. b Protein levels are shown as the ratio of DMT1 (55 kDa) and β-actin (42 kDa), NCX1 (108 kDa) and β-actin, NCX2 (102 kDa) and β-actin, NCX3 (100 kDa) and β-actin, NKA-α1/α2 (100 kDa) and β-actin (42 kDa). Data represent mean ± SD, n = 6. *Indicates statistically significant (p < 0.05) difference from any other group; **indicates statistically significant (p < 0.05) difference from control plus Fe2+ group or Cav3 siRNA(+) plus Ctrl group.
Fe3+ triggers Ca2+ release through stimulation of PLC and increase in InsP3 production
As shown in Fig. 3e, inhibition of InsP3 receptors with XeC suppressed Fe3+-induced [Ca2+]i mobilisation. We therefore analysed effects of Fe3+ on the InsP3 signalling cascade in cultured astrocytes. Incubation of astrocytes with Fe3+ increased the level of InsP3 in cell lysates to 74.10 ± 8.14 ng/ml (n = 10, p < 0.0001), from the control InsP3 level of 24.40 ± 6.35 ng/ml. In cells treated with TF alone the InsP3 level was 28.00 ± 12.62 ng/ml (n = 10, p = 0.4309) (Fig. 6a). Subsequently we analysed the links between scaffolding/signalling protein Dab2 and Fe3+-induced Ca2+ signalling. We suppressed expression of two isoforms of Dab2 by siRNA duplex chains (Fig. 6b). After RNA interfering, expression of 96 kD and 67 kD Dab2 isoforms decreased, respectively, to 5.81 ± 3.56% (n = 6, p < 0.0001) and to 11.88 ± 6.51% (n = 6, p < 0.0001) of the control values (Fig. 6b). The knock-down of Dab2 rendered Fe3+ ineffective: exposure of Dab2-deficient astrocytes to Fe3+ did not affect InsP3 production (control: 31.60 ± 12.51 ng/ml (n = 10); Dab2 knockdown: 30.60 ± 10.42 ng/ml (n = 10, p = 0.1253; Fig. 6c).

a Incubation with Fe3+ increases InsP3 level in cultured astrocytes; InsP3 was measured with ELISA; data are presented as mean ± SD, n = 10. *Indicates statistically significant (p < 0.05) difference from any other group. b Representative protein bands for two isoforms (96 KDa and 67 KDa) of Dab2 in cultured astrocytes treated with sham (Control), siRNA negative control (−) or positive duplex chains (+). Protein values are shown as the ratio of 96 kDa isoform and β-actin (42 kDa), and the ratio of 67 kDa isoform and β-actin. Data represent mean ± SD, n = 6. *Indicates statistically significant (p < 0.05) difference from any other group. c Fe3+-dependent changes in the InsP3 after treatment with Dab2 siRNA duplex chains. After treatment with Dab2 siRNA duplex chains (+) for 3 days, the primary cultured astrocytes were treated with serum-free medium (Control), TF (as negative control) or 10 μM Fe3+-TF (Fe3+) for 5 minutes, the level of InsP3 was measured by ELISA and shown as mean ± SD, n = 10. d Fe3+ -induced [Ca2+]i responses are Dab2 and PLC dependent. Treatment with Dab2 siRNA duplex chains (+) as well as with 10 μM U-73122 (PLC inhibitor) suppressed Fe3+-induced [Ca2+]i responses. Every data point represents mean ± SD, n = 10. e Mechanisms of Fe3+-induced Ca2+ signalling in astrocytes. The uptake of Fe3+ is mediated TFR, TFR internalization requires Dab2; when in the cytosol Fe3+ activated PLC thus stimulating InsP3-induced Ca2+ release from the ER.
When Dab2-deficient astrocytes were challenged with Fe3+, no [Ca2+]i increase was recorded (Fig. 6d). Similarly, after inhibition of the PLC with U-73122, application of Fe3+ did not change [Ca2+]i (Fig. 6d). Hence, we may surmise that uptake of Fe3+ through TFR requires Dab2 protein; after entering the cytosol Fe3+ activates the PLC, which produces InsP3 that triggers InsP3-induced Ca2+ release from the ER (Fig. 6e).
Discussion
In this paper, we describe previously unknown effects of iron ions on cellular [Ca2+]i in astrocytes. Administration of either Fe2+ or Fe3+ triggered a concentration-dependent increase in [Ca2+]i with EC50 of about 0.4–0.6 μM. We performed an in depth analysis of the mechanisms underlying iron transport and iron induced Ca2+ signalling. We also demonstrated that, contrary to the previous beliefs, astrocytes express functional TFR in vitro and in vivo thus allowing accumulation of Fe3+.
In the brain, majority of cells express a full complement of proteins responsible for iron homoeostasis, including TFR and DMT1 for iron uptake, heavy and light chains ferritin for iron sequestration, cytosolic iron exporter ferroportin 1 (FPN1), and iron regulatory protein 1 and 2 (IRP1and IRP2) for regulating intracellular iron homoeostasis39,40. Glial cells, and astrocytes in particular, store up to 75% of ionised iron in the CNS41, and protect the brain against iron overloads42. Transmembrane transport of iron in astrocytes has been identified, but was not studied in details. There is a general agreement on the primary role of plasmalemmal divalent metal transporter 1, DMT1/SLC11A2, which selectively transports Fe2+; the DMT1 was detected in astrocytes in culture and there is evidence indicating its presence in astroglial endfeet in situ43,44,45. In addition, Fe2+ was suggested to enter reactive astrocytes by diffusion through transient receptor potential canonical (TRPC) channels42. Expression of TF-Fe3+-transporting TFR has been noted in astrocytes in culture21,46; it is, nonetheless, generally believed that astrocytes in vivo are not in a possession of TFR and hence cannot accumulate Fe3+ 47,48,49. This conclusion, however, has been made on the basis of rather limited investigations14,46; while expression of TFR-specific mRNA was detected in astrocyte transriptome50. In our study we confirmed expression of DMT1, at mRNA and protein levels as well as by immunostaining, in acutely isolated astrocytes, in astroglial primary culture and in situ in cortical tissue; the DMT1 was particularly enriched in the endfeet (Fig. 2b–d). We also detected astroglial expression of TFR at mRNA level in the transcriptome of acutely isolated and FACS-sorted astrocytes (Fig. 2c). We further confirmed expression of TFR in astrocytes at a protein level and in immunohistochemical analysis of astrocytes in culture and in cortical preparations (Fig. 2b–d). In the cortical tissue TFR labelling was concentrated in perivascular astrocytic endfeet (Fig. 2b–d).
Not much is known about the links between ionised iron and Ca2+ signalling in the cellular elements of the CNS. In the literature, we found only a single example of Fe3+-induced [Ca2+]i transient in cultured hippocampal neurones51. To the best of our knowledge here we present the first recordings of Fe2+/Fe3+ induced Ca2+ signals in astrocytes. Both ions evoked [Ca2+]i elevation in primary cultured astrocytes and in EGFP-labelled astrocytes in the cortices of alive animals studied with transcranial confocal microscopy. Both ions acted in the low μM range of concentrations, however the kinetics of respective [Ca2+]i transients are different. Exposure of cultured astrocytes to Fe2+ triggered rapid [Ca2+]i increase with long-lasting plateau; the [Ca2+]i barely declined in the presence of Fe2+. In contrast, Fe3+-induced transient elevation of [Ca2+]i recovers to the baseline within ~ 200–300 s in the presence of Fe3+ (Figs. 1b and 2e). These distinct kinetics reflect different signalling cascades activated by iron ions.
The Fe2+-induced [Ca2+]i responses require DMT1: in vitro knockdown of DMT1 expression with silencing mRNA completely eliminates Ca2+ signal (Fig. 2). The Fe2+-induced [Ca2+]i changes originate from plasmalemmal Ca2+ entry, because removal of Ca2+ from the extracellular medium inhibits [Ca2+]i response. Finally, Fe2+-induced [Ca2+]i signals require NCX, as pharmacological inhibition of the latter effectively suppresses [Ca2+]i elevation (Fig. 3). These data indicate that Fe2+, after being accumulated in the astrocyte, switches the NCX into the reverse mode of operation thus generating Ca2+ influx into the cell in exchange for Na+. This scenario requires increase in astroglial [Na+]i, which readily reverses the NCX28,52. An increase in [Na+]i is likely to result from the inhibition of NKA, which represents the major Na+ efflux mechanism in astrocytes30. Activity of NKA was effectively suppressed by Fe2+, and probing astrocytes with Na+-sensitive indicator SBFI revealed Fe2+-induced [Na+]i elevation (Fig. 4). In the presence of NKA inhibitor ouabain Fe2+-induced [Ca2+]i responses were eliminated thus corroborating the central role of NKA and NCX in Fe2+-induced Ca2+ signalling (Figs. 4, 7). Of note, exposure to Fe2+ initiated rapid redistribution of DMT1 from nucleus into cytoplasm (Fig. 2g) and arguably to the plasmalemma thus increasing astrocyte capacity for iron uptake.

Uptake of Fe2+ is mediated by DMT1 and uptake of Fe3+ is mediated by TFR. After entering the cytosol, Fe2+ inhibits the activity of NKA, which causes an increase in [Na+]i due to an unopposed influx of Na+ through Na+-dependent transporters or Na+-channels. Increase in [Na+]i switches the NCX into the reverse mode, which results in Ca2+ influx. This influx also activates Ca2+-induced Ca2+ release through RyR that contributes to the plateau phase of [Ca2+]i response. Accumulation of Fe2+ also promotes the formation of the functional unit of DMT1, NCX1 and NKA in the caveolae by recruiting Cav3. Uptake of Fe3+ proceeds through Dab2-assisted internalisation; after entering the cytosol, Fe3+ activates the PLC and stimulates InsP3-dependent Ca2+ release from the ER.
The mechanism of Fe3+-induced Ca2+ signalling is associated with intracellular Ca2+ release. The Fe3+-induced [Ca2+]i responses are preserved in Ca2+-free extracellular solution while being blocked by XeC (inhibitor of InsP3 receptor) and by U-73122 (inhibitor of PLC) thus revealing the central role for InsP3-medaited ER Ca2+ release. Initiation of this signalling cascade requires transmembrane transport of Fe3+: in vitro knockdown of TFR eliminates Fe3+-evoked [Ca2+]i dynamics. The internalisation of TF-Fe3+-TFR complex also requires functional Dab2 protein. This protein is a multi-modular scaffold protein with signalling roles in ion homoeostasis, inflammation and receptors internalization53. Ablation of Dab2 in astrocytes with specific siRNA interrupts signalling chain and blocks Fe3+-dependent Ca2+ signalling (Figs. 6 and 7).
Import of TF-Fe3+-TFR complex involves endocytosis; acidic (pH ~5.6) environment of the endosome achieved through the action of ATP-dependent H+ pumps54, reduces the affinity of TF to Fe3+ and the latter is released55. Subsequently, an endosomal reductase reduces Fe3+ to Fe2+, and Fe2+ can be transported into the cytosol by DMT1 or by ZIP8 or ZIP14 Zn transporters56. Our data question the DMT1 pathway because siRNA knockout of DMT1 did not affect Fe3+-induced Ca2+ signalling; the detailed mechanism of Fe3+ transport in astrocytes remains to be fully characterised.
Caveolae are specific plasmalemmal structures that form functional microdomains involved in various signalling, endocytotic and transporting events57. Caveolae and their main structural and regulatory proteins Caveolin-1,2,3 are present in astrocytes (with predominant expression of Cav3); astrocytic caveolae contribute to signal transduction, formation of signalling protein complexes and are involved in action of various neuroactive substances and drugs58,59. Caveolae are known to form functional Ca2+ signalling units, establish links between Ca2+ channels and various transports and may contribute to formation of plasmalemmal/ER functional domains operational in astrocytes60. We found that down-regulation of expression of Cav3 in cultured astrocytes substantially reduced the amplitude of Fe2+-evoked [Ca2+]i responses. We suggest therefore that Cav3 and caveolae integrate DMT1, NKA and NCX into a single Ca2+ signalling unit (Fig. 7); moreover exposure to iron potentiates formation of such units.
Iron homoeostasis is of fundamental importance for cells, tissues and organisms, as iron contributes to a wide range of vital biological pathways61. The brain contains high concentrations of bound and free iron, which participates in multiple processes from energy production to synaptic transmission41. Iron overload and failures in iron homoeostatic triggers neurotoxicity and is implicated in brain diseases48. Genetic mutations of iron regulatory proteins (the key elements of iron homoeostasis) result in iron deposition in the brain with subsequent neurodegeneration characteristic for aceruloplasminemia62 and neuroferritinopathy63. In aceruloplasminemia patients, iron overload has been observed in astrocytes, particularly in the basal ganglia64. Ceruloplasmin oxidises Fe2+ to Fe3+ in order to generate the oxidised form of iron that can bind to extracellular transferrin; in astrocytes ceruloplasmin is also required for iron export64. Thus, in aceruloplasminemia, iron entering the CNS as ferrous might escape oxidation; hence cells exposed to the excess of Fe2+ may readily become iron overloaded65. Similarly, abnormal iron accumulation has been characterised in neurodegenerative diseases including Alzheimer’s disease, Parkinson disease, amyotrophic lateral sclerosis and Huntington disease to name but a few48. Specific class of neurodegeneration with brain iron accumulation (NBIA) has been also categorised in recent years66.
Based on our data we propose that astrocytes mount the defence against acute iron overload. This defence includes iron accumulation through both DMT1 and TFR, redistribution of DMT1 from intracellular locations to plasmalemma and generation of Ca2+ signals, which control astrocytic reactivity. Iron-induced Ca2+ signalling is activated at low pathological iron concentrations (>1 μM; while physiological iron concentration in the CSF ranges between 0.3 and 0.75 μM). Importantly, two distinct signalling cascades (DMT1 Fe2+ transport, inhibition of NKA and reversal of NCX versus Fe3+-TF-TFR transport, activation of PLC and generation of InsP3-induced Ca2+ release) distinguish between ferric and ferrous. These distinct pathways may define very different outputs: it is known for example that activation of astroglial InsP3 receptors type II is linked to initiation of reactive astrogliosis67,68. In our recent report, exposure to iron was found to stimulate the over-expression of DMT1 in astrocytes and microglia, but not in neurones, which may result in the neuroprotection by glial uptake of excessive iron13. Astrogliosis plays important, if not defining role in evolution of many neurological diseases69. Our previous experiments have shown that formation of brain deposits of iron up-regulates astroglial expression of TFR and instigates reactive astrogliosis70. What characterises the iron-induced reactive phenotype and what is the role of astroglial reactivity in managing excessive iron in the brain remains to be found. In conclusion, our study presented a phenomenon that iron ions (Fe2+ and Fe3+) directly induce intracellular Ca2+ signalling and stimulate astroglial protective mechanisms against iron overload in broad pathological contexts.
Methods
Materials
The culture medium including DMEM and fetal bovine serum were purchased from Gibco Life Technology Invitrogen (Grand Island, NY, USA). Oligo-fectamine, MEMI, fluo-4 AM, sodium-binding benzofuran isophthalate (SBFI) AM, G-agarose bead, TFR antibody, β-actin antibody, GFAP antibody, DMT1 antibody and DMT1 siRNA duplex chains were from Thermo Fisher Scientific (Waltham, MA USA); siRNA duplex chains of TFR, NCX1-3, Cav-3 and Dab2, NCX2 antibody, Na+/K+-ATPase alpha1/2 antibody and the secondary antibodies were bought from Santa Cruz Biotechnology (Santa Cruz, CA, USA). NCX1 antibody, NCX3 antibody and native mouse apo-transferrin (apo-TF; i.e., iron-free) were from Abcam (Cambridge, MA, USA). Donkey serum, xestospongin C (Xe-C), nifedipine, ferrous sulfate heptahydrate (FeSO4), sulforhodamine 101 (SR101) and ferric ammonium citrate were purchase from Sigma-Aldrich (St. Louis, MO, USA). Ryanodine and KB-R7943 were purchased from Calbiochem (La Jolla, CA, USA). Secondary antibody staining with donkey anti-mouse or anti-rabbit Cy-2/3 were from Jackson Immuno-Research (West Grove, PA, USA). Primary antibody of histone H3 was purchased from EarthOx (Millbrae, CA, USA).
Animals
The C57BL/6 mice, FVB/N-Tg(GFAP-eGFP)14Mes/J and B6.Cg-Tg(Thy1-YFP)HJrs/J transgenic mice were all purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The animals were raised in standard housing conditions (22 ± 1 °C; light/dark cycle of 12/12 h), with water and food available ad libitum. All experiments were performed in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 8023) and its 1978 revision, and all experimental protocols were approved by the Institutional Animal Care and Use Committee of China Medical University, No. [2019]059.
Primary culture of astrocytes
Astrocytes were cultured from newborn mice71,72. In brief, the cerebral hemispheres were isolated, dissociated and filtered. Isolated astrocytes were grown in Dulbecco’s Minimum Essential Medium (DMEM) with 7.5 mM glucose supplemented with 10% foetal bovine serum. Astrocytes were incubated at 37 °C in a humidified atmosphere of CO2/air (5:95%). The cultures are highly enriched in astrocytes, the purity is >95% as judged by GFAP staining73.
Iron treatment
For preparing Fe3+-TF solution, ferric ammonium citrate and mouse apo-TF were incubated at a 2:1 ratio in serum-free culture medium for 1 h at 37 °C74,75. The same concentration of apo-TF in the same volume of culture medium but without Fe3+ was used for control treatments. For the Fe2+ solution, FeSO4 was freshly dissociated in serum-free culture medium at 37 °C and used immediately, the same volume of serum-free culture medium was used as the control for Fe2+ group.
RNA interfering
The cultured astrocytes were incubated in DMEM without serum for 12 hours before transfection73,76,77. A transfection solution containing 2 μl oligo-fectamine (Promega, Madison, WI, USA), 40 μl MEMI, and 2.5 μl siRNA (DMT1, TFR, NCX1-3, Cav-3 or Dab2) was added to the culture medium in every well for 8 h. In the siRNA-negative control cultures, transfection solution without siRNA was added. Thereafter, DMEM with three times serum was added to the cultures. These siRNA duplex chains were purchased from Santa Cruz Biotechnology (CA, USA).
Preparation of membrane caveolae
Cell homogenization and the caveolae preparation from astrocytes was made78,79. In brief, primary cultured astrocytes were collected and homogenised in SET (0.315 M sucrose, 20 mM Tris-HCl, and 1 mM EDTA, pH 7.4), and centrifuged for 1 h at 1,000 × g. The pellets were re-solubilised in SET and layered on Percoll (30% in SET) followed centrifugation at 1,000 × g. The pellets were re-homogenised and re-layered to three sucrose density gradient solution (80%, 30% and 5%) with ultra-centrifugation at 175,000 × g. Finally, the purified caveolae were collected and re-suspended in SET.
Co-immunoprecipitation
We used technologies of co-immunoprecipitation and subsequent western blotting to check the conjunction level between NCXs and DMT176. After homogenization, protein content was determined by the Bradford method80 using bovine serum albumin as the standard. For immunoprecipitation of NCX1-3, whole cell lysates (500 μg) were incubated with 20 μg of anti-NCX1, anti-NCX2 or anti-NCX3 antibody for overnight at 4 °C. Then, 200 μl of washed protein G-agarose bead slurry was added, and the mixture was incubated for another 2 hours at 4 °C. The agarose beads were washed three times with cold phosphate buffer solution (PBS) and collected by pulsed centrifugation (5 s in a microcentrifuge at 14,000 × g), the supernatant was drained off, and the beads were boiled for 5 min. Thereafter, the supernatant was collected by pulsed centrifugation, and the entire immunoprecipitates were subjected to 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE).
Cytoplasm and nucleus protein extraction
The subcellular fraction was analyzed using the protein extraction Kit (P0028, Beyotime, Shanghai, China)81, according to the manufacturer’s protocol. For further western blotting assays, cytoplasmic and nucleic protein extractions were kept under −20 °C.
Western blotting
For quantifying expressions of DMT1, TFR, NCX1-3, Cav-3 or Dab2, the samples containing 100 μg of protein were added to slab gels. After transferring to PVDF membranes, the samples were blocked by 10% skimmed milk powder for 1 h, and membranes were incubated overnight with the primary antibodies, specific to either DMT1 at a 1:300 dilution, TFR at a 1:200 dilution, NCX1 at a 1:100 dilution, NCX2 at a 1:200 dilution, NCX3 at a 1:150 dilution, Cav-3 at a 1:200 dilution, Dab2 at a 1:100 dilution, Histone H3 at 1:500 dilution or β-actin at a 1:1000 dilution. After washing, specific binding was detected by horseradish peroxidase-conjugated secondary antibodies. Images were analysed with an Electrophoresis Gel Imaging Analysis System (MF-ChemiBIS 3.2, DNR Bio-Imaging Systems, Israel). Band density was measured with Window AlphaEaseTM FC 32-bit software72,82.
Monitoring of [Ca2+]i
For [Ca2+]i monitoring and imaging in cultured astrocytes83,84, after the pre-treatment with or without inhibitors or siRNA duplex chains, the primarily cultured astrocytes were loaded with 5 μM fluo-4-AM, (Thermo Fisher Scientific (Waltham, MA USA)), for 30 min. Fluo-4 signals were visualised by fluorescent microscopy (Olympus IX71, Japan). The readings from all fluo-4 positive cells in one measured field of view in each culture were included in the statistics, the fluorescence intensity of fluo-4 was normalised to the baseline intensity before stimulation. The measurements were repeated in 10 different cultures. Cultured astrocytes were also stained with specific marker sulforhodamine 101 (SR101) at 1:2000 dilution.
Two-photon in vivo Ca2+ imaging
Adult FVB/N-Tg(GFAP-eGFP)14Mes/J transgenic mice (10–12 weeks old) were anesthetized with ketamine (80 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). Body temperature was monitored using a rectal probe, and the mice were maintained at 37 °C by a heating blanket. A custom made metal plate was glued to the skull with dental acrylic cement and a cranial window was prepared over the right hemisphere at 2.5 mm lateral and 2 mm posterior to bregma. The somato-sensory cortical cells were loaded with Ca2+ indicator Rhod-2 AM (50 μM, 1 h). The transcranial window was superfused with artificial CSF. After a stable baseline recording was obtained, Fe2+ (100 μM) or Fe3+-TF (100 μM) was added for 1 min. Bandpass filters (Chroma) were 540 nm/40 nm for eGFP and 850 nm/70 nm for rhod-2 signals. Time-lapse images of astrocytic Ca2+ signalling were recorded every 5 s using FluoView with a custom-built two-photon laser-scanning setup (Nikon AR1, Japan)84,85.
Intracellular Na+ measurements
For monitoring intracellular ionised Na+ ([Na+]i) in cultured astrocytes, primary cultured astrocytes were loaded with 10 μM of Na+-sensitive indicator SBFI-AM for 30 min in serum-free medium, with subsequent 1 h of washout. SBFI was alternatively excited at 340 nm and 380 nm, and the emission was monitored at 500 nm. The SBFI signals were measured by fluorescent microscopy (Olympus IX71, Japan) and expressed as a ratio (R = F340/F380)52.
Immunofluorescence
The brain tissue was fixed by immersion in 4% paraformaldehyde and cut into 100 μm slices72,82. The cultured cells were fixed with 100% methanol at −20 °C. Brain slices or cells were permeabilised by incubation for 1 h with donkey serum. Primary antibodies against DMT1 or TFR were used at a 1:100 dilution, against glial fibrillary acidic protein (GFAP) was used at 1:200 dilution. And nuclei were stained with marker 4′, 6′-diamidino-2-phenylindole (DAPI) at 1:1000 dilution. The primary antibodies were incubated overnight at 4 °C and then donkey anti-mouse or anti-rabbit Cy-2/3 conjugated secondary antibody for 2 h at room temperature. Images were captured using a confocal scanning microscope (DMi8, Leica, Wetzlar, Germany).
ELISA assays
Astrocytes were incubated at 37 °C in a fresh serum-free culture medium; after the treatment with Fe2+/Fe3+ or inhibitors, the astrocytes were collected and centrifuged at 10,000 × g for 10 min to remove floating cells and/or cell debris at 4 °C. To assay the NKA activity, a commercial ELISA kit (abx255202; Abbexa, Cambridge, UK) was used and operated as the protocols, the sensitivity is 0.19 ng/mL, the optical density (OD) was measured at 450 nm and the OD value was normalized by the control group. To assay the InsP3 concentration86, the supernatant was collected and the concentration of InsP3 assayed using a commercial ELISA kit (E-EL-0059c; Elabscience Biotechnology, Wuhan, China).
Sorting neural cells through fluorescence activated cell sorter (FACS) and quantitative PCR (qPCR)
To measure the mRNA for TFR and DMT1, astrocytes expressing fluorescent marker GFP (GFAP-GFP mice) and neurons expressing fluorescent marker YFP (Thy1-YFP mice) were used; we also extracted the cerebral hemispheres tissues from wild type mice. The cells from transgenic mice were used for specific sorting of astrocytes or neurons with FACS85,87. The RNA of the sorted cells and cerebral tissue was extracted by Trizol. Total RNA was reverse transcribed and PCR amplification was performed in a Robo-cycler thermocycler81,84. The relative quantity of transcripts was assessed using five-folds serial dilutions of RT product (200 ng). RNA quantity was normalised to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and values are expressed as the ratio TFR/GAPDH or DMT1/GAPDH.
Statistics and reproducibility
For statistical analysis, we used one-way analysis of variance (ANOVA) followed by a Tukey’s or Dunnett’s post hoc multiple comparison test for unequal replications using GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA) and SPSS 24 software (International Business Machines Corp., NY, USA). One-way ANOVA for comparisons including more than two groups; unpaired two-tailed t-test for two-group comparisons. All statistical data in the text are presented as the mean ± SD, the value of significance was set at p < 0.05.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Source data underlying the main and supplementary figures are available in Supplementary Data. The data that support the findings of this study are available from the corresponding author Baoman Li upon reasonable request.
References
Youdim, M. B., Ben-Shachar, D., Yehuda, S. & Riederer, P. The role of iron in the basal ganglion. Adv. Neurol. 53, 155–162 (1990).
Wang, Y., Wu, Y., Li, T., Wang, X. & Zhu, C. Iron metabolism and brain development in premature infants. Front. Physiol. 10, 463 (2019).
Schenck, J. F. & Zimmerman, E. A. High-field magnetic resonance imaging of brain iron: birth of a biomarker? Nmr. Biomed. 17, 433–445 (2004).
Gutteridge, J. M. Ferrous ions detected in cerebrospinal fluid by using bleomycin and DNA damage. Clin. Sci. 82, 315–320 (1992).
Bishop, G. M. & Robinson, S. R. Quantitative analysis of cell death and ferritin expression in response to cortical iron: implications for hypoxia-ischemia and stroke. Brain. Res. 907, 175–187 (2001).
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R. & Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060 (2014).
Cabantchik, Z. I. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front. Pharmacol. 5, 45 (2014).
Morris, C. M. et al. Brain iron homeostasis. J. Inorg. Biochem. 47, 257–265 (1992).
Verkhratsky, A. & Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 98, 239–389 (2018).
Wang, X. S., Ong, W. Y. & Connor, J. R. A light and electron microscopic study of the iron transporter protein DMT-1 in the monkey cerebral neocortex and hippocampus. J. Neurocytol. 30, 353–360 (2001).
Moos, T., Trinder, D. & Morgan, E. H. Cellular distribution of ferric iron, ferritin, transferrin and divalent metal transporter 1 (DMT1) in substantia nigra and basal ganglia of normal and beta2-microglobulin deficient mouse brain. Cell Mol. Biol. (Noisy-le.-Gd.) 46, 549–561 (2000).
Skjorringe, T., Burkhart, A., Johnsen, K. B. & Moos, T. Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 8, 19 (2015).
Xia, M. et al. Iatrogenic iron promotes neurodegeneration and activates self-protection of neural cells against exogenous iron attacks. Function 2, zqab003 (2021).
Moos, T. & Morgan, E. H. The significance of the mutated divalent metal transporter (DMT1) on iron transport into the Belgrade rat brain. J. Neurochem. 88, 233–245 (2004).
Cheli, V. T., Correale, J., Paez, P. M. & Pasquini, J. M. Iron metabolism in oligodendrocytes and astrocytes, implications for myelination and remyelination. Asn. Neuro. 12, 1759091420962681 (2020).
Cheli, V. T. et al. The divalent metal transporter 1 (DMT1) is required for iron uptake and normal development of oligodendrocyte progenitor cells. J. Neurosci. 38, 9142–9159 (2018).
Templeton, D. M. & Liu, Y. Genetic regulation of cell function inresponse to iron overload or chelation. Biochim. Biophys. Acta 1619, 113–124 (2003).
Yang, L. et al. Hypoxic preconditioning increases iron transport rate in astrocytes. Biochim. Biophys. Acta 1822, 500–508 (2012).
Chen, Y. et al. Myeloid zinc‐finger 1 (MZF‐1) suppresses prostate tumor growth through enfor-cing ferroportin‐conducted iron egress. Oncogene 34, 3839–3847 (2015).
Xu, J. & Ling, E. A. Studies of the ultrastructure and permeability of the blood-brain barrier in the developing corpus callosum in postnatal rat brain using electron dense tracers. J. Anat. 184, 227–237 (1994).
Hoepken, H. H., Korten, T., Robinson, S. R. & Dringen, R. Iron accumulation, iron-mediated toxicity and altered levels of ferritin and transferrin receptor in cultured astrocytes during incubation with ferric ammonium citrate. J. Neurochem. 88, 1194–1202 (2004).
Wagner, K. R., Sharp, F. R., Ardizzone, T. D., Lu, A. & Clark, J. F. Heme and iron metabolism: role in cerebral hemorrhage. J. Cereb. Blood. Flow. Metab. 23, 629–652 (2003).
Maxfield, F. R. & McGraw, T. E. Endocytic recycling. Nat. Rev. Mol. Cell. Biol. 5, 121–132 (2004).
Ahn, M. et al. Transient activation of an adaptor protein, disabled-2, in rat spinal cord injury. Acta Histochem. 117, 56–61 (2015).
Chu, H. C. et al. Distinct effects of disabled-2 on transferrin uptake in different cell types and culture conditions. Cell. Biol. Int. 38, 1252–1259 (2014).
Verkhratsky, A., Rodríguez, J. J. & Parpura, V. Calcium signalling in astroglia. Mol. Cell. Endocrinol. 353, 45–56 (2012).
Verkhratsky, A., Untiet, V. & Rose, C. R. Ionic signalling in astroglia beyond calcium. J. Physiol. 598, 1655–1670 (2020).
Rose, C. R., Ziemens, D. & Verkhratsky, A. On the special role of NCX in astrocytes: translating Na(+)-transients into intracellular Ca(2+) signals. Cell. Calcium 86, 102154 (2020).
Kirischuk, S., Parpura, V. & Verkhratsky, A. Sodium dynamics: another key to astroglial excitability? Trends Neurosci. 35, 497–506 (2012).
Rose, C. R. & Verkhratsky, A. Principles of sodium homeostasis and sodium signalling in astroglia. Glia 64, 1611–1627 (2016).
Verkhratsky, A. & Rose, C. R. Na(+)-dependent transporters: the backbone of astroglial homeostatic function. Cell. Calcium 85, 102136 (2020).
Verkhratsky, A., Trebak, M., Perocchi, F., Khananshvili, D. & Sekler, I. Crosslink between calcium and sodium signalling. Exp. Physiol. 103, 157–169 (2018).
Larsen, B. R. et al. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 62, 608–622 (2014).
Pappalardo, L. W., Samad, O. A., Black, J. A. & Waxman, S. G. Voltage-gated sodium channel Nav 1.5 contributes to astrogliosis in an in vitro model of glial injury via reverse Na+ /Ca2+ exchange. Glia 62, 1162–1175 (2014).
Minelli, A. et al. Cellular and subcellular localization of Na+-Ca2+ exchanger protein isoforms, NCX1, NCX2, and NCX3 in cerebral cortex and hippocampus of adult rat. Cell. Calcium 41, 221–234 (2007).
Melone, M., Ciriachi, C., Pietrobon, D. & Conti, F. Heterogeneity of astrocytic and neuronal GLT-1 at cortical excitatory synapses, as revealed by its colocalization with Na+/K+-ATPase α Isoforms. Cereb. Cortex. 29, 3331–3350 (2019).
Soares, E. S., Mendonça, M. C., Rocha, T., Kalapothakis, E. & da, Cruz-Höfling, M. A. Are synchronized changes in connexin-43 and caveolin-3 a Bystander effect in a phoneutria nigriventer venom model of blood-brain barrier breakdown? J. Mol. Neurosci. 59, 452–463 (2016).
Gafni, J. et al. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron 19, 723–733 (1997).
Rouault, T. A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2, 406–414 (2006).
Rouault, T. A. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat. Rev. Neurosci. 14, 551–564 (2013).
Gaasch, J. A. et al. Brain iron toxicity: differential responses of astrocytes, neurons, and endothelial cells. Neurochem. Res. 32, 1196–1208 (2007).
Pelizzoni, I., Zacchetti, D., Campanella, A., Grohovaz, F. & Codazzi, F. Iron uptake in quiescent and inflammation-activated astrocytes: a potentially neuroprotective control of iron burden. Biochim. Biophys. Acta 1832, 1326–1333 (2013).
Qian, Z. M., To, Y., Tang, P. L. & Feng, Y. M. Transferrin receptors on the plasma membrane of cultured rat astrocytes. Exp. Brain. Res. 129, 473–476 (1999).
Huang, E., Ong, W. Y. & Connor, J. R. Distribution of divalent metal transporter-1 in the monkey basal ganglia. Neuroscience 128, 487–496 (2004).
Bishop, G. M., Scheiber, I. F., Dringen, R. & Robinson, S. R. Synergistic accumulation of iron and zinc by cultured astrocytes. J. Neural Transm. (Vienna) 117, 809–817 (2010).
Jeong, S. Y. & David, S. Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J. Neurosci. 26, 9810–9819 (2006).
Moos, T., Rosengren, Nielsen, T., Skjørringe, T. & Morgan, E. H. Iron trafficking inside the brain. J. Neurochem. 103, 1730–1740 (2007).
Hare, D., Ayton, S., Bush, A. & Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 5, 34 (2013).
Codazzi, F., Pelizzoni, I., Zacchetti, D. & Grohovaz, F. Iron entry in neurons and astrocytes: a link with synaptic activity. Front. Mol. Neurosci. 8, 18 (2015).
Orre, M. et al. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 35, 1–14 (2014).
Muñoz, P. et al. Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J. Biol. Chem. 286, 13382–13392 (2011).
Unichenko, P., Myakhar, O. & Kirischuk, S. Intracellular Na+ concentration influences short-term plasticity of glutamate transporter-mediated currents in neocortical astrocytes. Glia 60, 605–614 (2012).
Finkielstein, C. V. & Capelluto, D. G. Disabled-2: A modular scaffold protein with multifaceted functions in signaling. Bioessays 38, S45–S55 (2016).
Morgan, E. H. Inhibition of reticulocyte iron uptake by NH4Cl and CH3NH2. Biochim. Biophys. Acta 642, 119–134 (1981).
Luck, A. N. & Mason, A. B. Transferrin-mediated cellular iron delivery. Curr. Top. Membr. 69, 3–35 (2012).
Lambe, T. et al. Identification of a Steap3 endosomal targeting motif essential for normal iron metabolism. Blood 113, 1805–1808 (2009).
Bastiani, M. & Parton, R. G. Caveolae at a glance. J. Cell. Sci. 123, 3831–3836 (2010).
Li, B. et al. Biphasic regulation of caveolin-1 gene expression by fluoxetine in astrocytes: opposite effects of PI3K/AKT and MAPK/ERK signaling pathways on c-fos. Front. Cell. Neurosci. 11, 335 (2017).
Silva, W. I., Maldonado, H. M., Velázquez, G., García, J. O. & González, F. A. Caveolins in glial cell model systems: from detection to significance. J. Neurochem. 103, 101–112 (2007).
Lencesova, L., O’Neill, A., Resneck, W. G., Bloch, R. J. & Blaustein, M. P. Plasma membrane-cytoskeleton-endoplasmic reticulum complexes in neurons and astrocytes. J. Biol. Chem. 279, 2885–2893 (2004).
Aisen, P., Enns, C. & Wessling-Resnick, M. Chemistry and biology of eukaryotic iron metabolism. Int. J. Biochem. Cell. Biol. 33, 940–959 (2001).
Hochstrasser, H. et al. Ceruloplasmin gene variations and substantia nigra hyperechogenicity in Parkinson disease. Neurology 63, 1912–1917 (2004).
Chinnery, P. F. et al. Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation. Brain 130, 110–119 (2007).
Jeong, S. Y. & David, S. Glycosylphosphatidylinositol anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 278, 27144–27148 (2003).
Kaneko, K. et al. Extensive brain pathology in a patient with aceruloplasminemia with a prolonged duration of illness. Hum. Pathol. 43, 451–456 (2012).
Levi, S. & Finazzi, D. Neurodegeneration with brain iron accumulation: update on pathogenic mechanisms. Front. Pharmacol. 5, 99 (2014).
Kanemaru, K. et al. Calcium-dependent N-cadherin up-regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc. Natl Acad. Sci. USA110, 11612–11617 (2013).
Verkhratsky, A. Astroglial calcium signaling in aging and Alzheimer’s disease. Cold. Spring Harb. Perspect. Biol. 11, a035188 (2019).
Verkhratsky, A., Zorec, R. & Parpura, V. Stratification of astrocytes in healthy and diseased brain. Brain. Pathol. 27, 629–644 (2017).
Liang, S. et al. Iron aggravates the depressive phenotype of stressed mice by compromising the glymphatic system. Neurosci. Bull. 36, 1542–1546 (2020).
Li, B., Zhang, S., Zhang, H., Hertz, L. & Peng, L. Fluoxetine affects GluK2 editing, glutamate-evoked Ca(2+) influx and extracellular signal-regulated kinase phosphorylation in mouse astrocytes. J. Psychiatry Neurosci. 36, 322–338 (2011).
Xia, M. et al. The ameliorative effect of fluoxetine on neuroinflammation induced by sleep deprivation. J. Neurochem. Online ahead of print. (2017).
Li, Z. et al. Fluoxetine improves behavioural deficits induced by chronic alcohol treatment by alleviating RNA editing of 5-HT(2C) receptors. Neurochem. Int. 134, 104689 (2020).
Qian, Z. M. & Morgan, E. H. Effect of lead on the transport of transferrin-free and transferrin-bound iron into rabbit reticulocytes. Biochem. Pharmacol. 40, 1049–1054 (1990).
Qian, Z. M. & Morgan, E. H. Effect of metabolic inhibitors on uptake of non-transferrin-bound iron by reticulocytes. Biochim. Biophys. Acta 1073, 456–462 (1991).
Li, B. et al. Lithium inhibits GSK3β activity via two different signaling pathways in neurons after spinal cord injury. Neurochem. Res. 43, 848–856 (2018).
Li, X. et al. Leptin increases expression of 5-HT(2B) receptors in astrocytes thus enhancing action of fluoxetine on the depressive behavior induced by sleep deprivation. Front. Psychiatry 9, 734 (2018).
Waugh, M. G. & Hsuan, J. J. Preparation of membrane rafts. Methods Mol. Biol. 462, 403–414 (2009).
Jia, S., Li, B., Huang, J., Verkhratsky, A. & Peng, L. Regulation of glycogen content in astrocytes via Cav-1/PTEN/AKT/GSK-3β pathway by three anti-bipolar drugs. Neurochem. Res. 43, 1692–1701 (2018).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976).
Li, S. et al. Neddylation promotes protein translocation between the cytoplasm and nucleus. Biochem. Biophys. Res. Commun. 529, 991–997 (2020).
Xia, M., Yang, L., Sun, G., Qi, S. & Li, B. Mechanism of depression as a risk factor in the development of Alzheimer’s disease: the function of AQP4 and the glymphatic system. Psychopharmacology 234, 365–379 (2017).
Li, B. et al. Effects of chronic treatment with fluoxetine on receptor-stimulated increase of [Ca2+]i in astrocytes mimic those of acute inhibition of TRPC1 channel activity. Cell. Calcium 50, 42–53 (2011).
Ren, J. et al. Protective effect of leptin-mediated caveolin-1 expression on neurons after spinal cord injury. Cell. Calcium 76, 122–128 (2018).
Lundgaard, I. et al. Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun. 6, 6807 (2015).
Che, T. et al. Oxytocin hyperpolarizes cultured duodenum myenteric intrinsic primary afferent neurons by opening BK(Ca) channels through IP3 pathway. J. Neurochem. 121, 516–525 (2012).
Zhao, X. et al. Different epidermal growth factor receptor signaling pathways in neurons and astrocytes activated by extracellular matrix after spinal cord injury. Neurochem. Int. 129, 104500 (2019).
Acknowledgements
This work was supported by the National Natural Science Foundation of China, B.L. [Grant number 81871852]; LiaoNing Revitalization Talents Program, B.L. [Grant number XLYC1807137], the Scientific Research Foundation for Returned Scholars of Education Ministry of China, B.L. [grant number 20151098], LiaoNing Thousands Talents Program, B.L. [Grant number 202078], “ChunHui” Program of China Education Ministry, B.L. [grant number 2020703], and the National Natural Science Foundation of China D.G. [Grant number 81671862] and [Grant number 81871529].
Author information
Authors and Affiliations
Contributions
A.V., D.G., M.X. and B.L. designed and supervised the study; M.X., WZ.G., S.L., G.W., BN.C., BJ.C., M.J. and WL.G. collected the data in vitro and analysed the relevant data; WZ.G., SS.L., C.D., X.Z., Y.Z., M.Z., D.Z. and X.L. performed the experiments in vivo and analysed the data; B.L. and A.V. wrote the manuscript. BN.C. and M.Z. collected the images of immunofluorescence, Figs. 1a, b and 2b, f.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guan, W., Xia, M., Ji, M. et al. Iron induces two distinct Ca2+ signalling cascades in astrocytes. Commun Biol 4, 525 (2021). https://doi.org/10.1038/s42003-021-02060-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02060-x
This article is cited by
-
A novel murine model of mania
Molecular Psychiatry (2023)
-
Astrocytes in human central nervous system diseases: a frontier for new therapies
Signal Transduction and Targeted Therapy (2023)
-
Alexei Verkhratsky, the Philosopher of Neuroglia. Preface for the Honorary Issue of Neurochemical Research
Neurochemical Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
