
Abstract
N-terminal acetylation is one of the most common protein modifications in eukaryotes and is carried out by N-terminal acetyltransferases (NATs). It plays important roles in protein homeostasis, localization, and interactions and is linked to various human diseases. NatB, one of the major co-translationally active NATs, is composed of the catalytic subunit Naa20 and the auxiliary subunit Naa25, and acetylates about 20% of the proteome. Here we show that NatB substrate specificity and catalytic mechanism are conserved among eukaryotes, and that Naa20 alone is able to acetylate NatB substrates in vitro. We show that Naa25 increases the Naa20 substrate affinity, and identify residues important for peptide binding and acetylation activity. We present the first Naa20 crystal structure in complex with the competitive inhibitor CoA-Ac-MDEL. Our findings demonstrate how Naa20 binds its substrates in the absence of Naa25 and support prospective endeavors to derive specific NAT inhibitors for drug development.
Similar content being viewed by others


Introduction
Nα-acetylation is the most common protein modification in eukaryotes. Approximately 60% of all soluble proteins in yeast, more than 70% in plants, and 80–90% in humans are N-terminally acetylated1,2,3. This modification is involved in many cellular processes affecting the stability, folding and degradation of proteins, protein interactions, subcellular localization and it is linked with several human diseases like cancer, Parkinson or Huntington disease4,5,6. During N-terminal acetylation, an acetyl group is transferred from acetyl coenzyme A (AcCoA) to the α-amino group of a polypeptide. This modification is mostly carried out co-translationally7 by Nα-acetyltransferase complexes (NATs), which differ in subunit composition and substrate specificity, but comprise at least one catalytic subunit1,8. Eight different eukaryotic NATs have been identified so far (NatA to NatH)9. The NatB-complex, one of the major NATs, is composed of two subunits, the catalytic subunit Naa20 and the auxiliary subunit Naa25 (formerly known as Nat3 and Mdm20, respectively)10. Both subunits are conserved within eukaryotic model organisms and the NatB complex is found associated with the ribosome11,12,13. Deletion mutants of NAA25 and NAA20 in Saccharomyces cerevisiae show a slow growth phenotype, are unable to form actin cables, have a defect in vacuolar and mitochondrial inheritance and are sensitive to DNA-damage causing agents3,14,15,16. Recently, ScNaa20 dependent acetylation was suggested to have a protective function in regard to protein degradation and a role in protein synthesis17,18,19. An involvement of the NatB complex in vacuolar protein sorting and cell wall maintenance, as well as in influencing the shutoff activity of influenza A virus was also suggested20,21. Additionally, NatB is involved in the regulation of plant development, abiotic stress response and is linked to the microRNA pathway22,23 and in human cell proliferation, cell survival and liver cancer progression13,24,25,26. Noteworthy, as NatB subunits exhibit partly divergent phenotypes, it was speculated that both subunits may have functions independent from each other24,27, and Naa25 was found to not always coexpress with Naa20 in mouse neurons28. Importantly, Naa25 was shown to be essential for the activity of Naa2015,16,24 and localizes in the cytoplasm, while Naa20 is found in both, the nucleus13 and cytoplasm independent of Naa2511,13.
NatB acts on substrates presenting N-termini with the initial methionine, which is retained and directly acetylated, followed by an acidic residue (MD-, ME-, MN- or MQ-)3,13,16,23,29,30. Recently, crystal structures of Candida albicans NatB in complex with the bisubstrate inhibitor CoA-Ac-MDSEVA and in the free state were reported30. They show that Naa20 adopts the canonical Gcn5-related N-acetyltransferase (GNAT) fold and accommodates a peptide at its substrate binding pocket. Naa25 forms a horse-shoe-like structure holding Naa20, and overall the CaNatB structures resemble the human NatB cryo-EM structure visualizing the high evolutionary conservation31. However, while for the isolated catalytic subunits Naa10 and Naa50 crystal structures were reported32,33, structural information on Naa20 were not available so far.
In order to dissect the molecular mechanism of NatB and its use as a potential therapeutic target, we studied Chaetomium thermophilum NatB complex and its individual subunits. Our results show that CtNaa20 is active towards a canonical NatB substrate without CtNaa25 in vitro, however less efficient than in NatB. We designed, synthesized and characterized the NatB inhibitor CoA-Ac-MDEL and solved the crystal structure of the catalytic subunit Naa20 in complex with this inhibitor. The structure reveals the basis of CtNaa20 substrate binding and activity towards the MDEL peptide.
Results
CtNaa20 binds with high affinity to CtNaa25
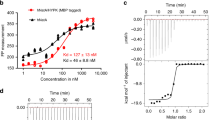
To functionally and structurally characterize the NatB complex and the subunits Naa20 and Naa25, we used the conserved orthologous proteins of the thermophilic model organism Chaetomium thermophilum (Ct) to benefit from their often superior properties in biochemical and structural studies34,35. A BLAST search using the Candida albicans (Ca) NAA20 and NAA25 sequences as query revealed two candidates with 42% and 20% amino acids sequence identity, respectively (Supplementary Fig. 1). Next, full length CtNaa25, CtNaa20 and a truncated CtNaa201-166 variant were cloned and expressed in E. coli. The CtNaa201-166 was based on secondary structure prediction and comprises the predicted GNAT-fold. The auxiliary subunit CtNaa25 and the catalytic subunit CtNaa20 were individually expressed and purified to homogeneity (Fig. 1a, b). Co-expression of CtNaa25 and CtNaa20 led to the formation of the NatB complex, which could also be purified to homogeneity (Fig. 1c). In order to characterize the NatB complex, size exclusion chromatography coupled to multi angle light scattering (SEC-MALS) was carried out. The individual subunits CtNaa25 and CtNaa20 and NatB eluted as single symmetric peaks and analyses of the molecular mass showed that CtNaa20 and CtNaa25 are monomers in solution (Fig. 1a, b). A molecular mass of 136.5 kDa determined for NatB confirms a 1:1 stoichiometry of its subunits (Fig. 1c). To further characterize NatB complex formation, we performed isothermal titration calorimetry experiments. Titration of CtNaa25 into CtNaa20 was endothermic (ΔH = 5.4 kcal/mol) and resulted in the formation of a stable complex with a dissociation constant Kd of 17.8 ± 11.9 nM and a molar ratio of one (Fig. 1d).

a–c SEC-MALS analysis of CtNaa20 (a), CtNaa25 (b), and CtNatB (c). The experimentally determined molecular weight of CtNatB, CtNaa25, and CtNaa20 is 136.5 kDa (theoretical molecular is 136.1 kDa), 110.7 kDa (theoretical Mw is 115.1 kDa), and 23.6 kDa (theoretical Mw is 23.1 kDa), respectively. The UV-signals (black) of the corresponding SEC chromatograms are shown together with the light scattering signals (gray) and the mass distributions (black dots). d Isothermal titration calorimetry measurement of CtNatB complex formation. CtNaa20 was titrated by CtNaa25. The signal of one representative measurement is given in differential points (DP) and the dissociation constant Kd and binding enthalpy are given in the table. The heats of dilution of the buffer to CtNaa20 (triangle) and CtNaa25 to buffer (cross) control runs are represented. Measurements were performed in triplicate and the values represent the means and standard deviations. e Electrophoretic mobility shift assay with the tip of CtES27 (expansion segment 27) RNA, CtNaa20, and CtNatB. CtNaa20 and CtNatB were mixed in different ratios with the RNA and free or bound RNA is indicated.
As NatB acts co-translationally at the ribosome, we wanted to address ribosome binding of CtNa20 and CtNaB. Electrophoretic mobility shift assays (EMSA) were performed to analyze CtNaa20 or CtNatB binding to rRNA as indicator for putative ribosome interaction. CtNaa20 and CtNatB were incubated with a C. thermophilum expansion segment 27 RNA (CtES27) fragment. ES27 was shown to be involved in the ribosome binding of the ribosome-associated factors NatA, Arx1, and Ebp136,37,38. A shift of the RNA band was observed upon addition of CtNatB but not for CtNaa20, indicating that CtNatB but not CtNaa20 alone can bind to CtES27 (Fig. 1e). To analyze the specificity of the interaction, hammerhead ribozyme RNA was used as control (Supplementary Fig. 2). No RNA binding for CtNaa20 could be detected, while CtNatB binding to hammerhead RNA was observed, showing that CtNatB binds nonspecifically to RNA. This suggests that Naa20 does not associate with the ribosome on its own, but only when in complex with Naa25.
CtNatB acetylates specifically the MDEL peptide
We then investigated CtNatB substrate specificity and enzymatic activity by in vitro acetylation assays. Canonical NatA (SESS)1, Naa80/Naa10 (EEEI)39,40, NatB (MDEL)13, and NatC/E/F (MVNALE and MLGTE)3 substrates were tested. CtNatB acetylates only the MDEL peptide, highlighting that NatB specificity is conserved (Fig. 2a). The MDEL peptide was then used to determine the NatB enzymatic parameters. CtNatB showed a Michaelis constant (Km) of 45.6 ± 4.8 μM for AcCoA and a turnover number (kcat) of 68.8 ± 2.0 min−1 (Fig. 2b). These values are in good agreement with the values observed for CaNatB and AtNatB (Supplementary Fig. 3a)23,30. Based on these results, we designed and synthesized a bisubstrate analog, CoA-Ac-MDEL (Fig. 2c and Supplementary Fig. 3b). This bisubstrate is a potent competitive NatB inhibitor with a half-maximum inhibitor concentration (IC50) of 1.56 ± 0.24 μM and an inhibitor constant Ki of 0.41 ± 0.14 μM (Fig. 2d and Supplementary Fig. 3c). The potency of CoA-Ac-MDEL in inhibiting CtNatB is in the same range as CoA-Ac-MVNAL inhibiting NatF and CoA-SASEA inhibiting NatA (Fig. 2e)32,41. Taken together, our results show that NatB substrate specificity is evolutionarily conserved, and indicate that all NATs bind their specific inhibitors with similar affinities.

a Substrate specificity of CtNatB tested with five different peptides. SESS, EEEI, MDEL, MLGTE and MVNALE were previously identified as NatA, Naa10/Naa80, NatB and NatC/E/F substrates. b Michaelis–Menten curve of the CtNatB mediated acetylation of MDEL. c Structure of the bisubstrate analog CoA-Ac-MDEL. d Dose-response curve of the NatB mediated acetylation, inhibited by CoA-Ac-MDEL. e Inhibitor characteristics of CoA-Ac-MDEL compared to other bisubstrate analogs32,41. All reactions were performed in triplicates and error bars represent the standard deviation.
CtNaa20 is active and specifically acetylates the NatB substrate MDEL
To further characterize CtNatB, we investigated the activity of the catalytic subunit CtNaa20 alone. In contrast to previous reports on NatB, CtNaa20 shows a clearly detectable and specific activity towards the MDEL peptide (Fig. 3a) with a Km value for AcCoA of 12.0 ± 1.0 μM and a kcat of 9.0 ± 0.2 min−1 (Supplementary Fig. 4a). These data show that the catalytic subunit alone is active, but less efficient than in complex with CtNaa25. The bisubstrate analog CoA-Ac-MDEL is also a potent inhibitor of CtNaa20, with an IC50 of 6.5 ± 2.5 μM (Supplementary Fig. 4b). Compared to NatB the higher IC50 indicates a lower affinity of CtNaa20 to this inhibitor. The difference between the enzymatic activities of CtNaa20 alone and as part of NatB might be explained by different affinities for the MDEL substrate. To test this, we performed kinetic experiments with constant AcCoA, but varying MDEL concentrations. CtNatB shows a Km of 232 ± 28 μM for MDEL, which is significantly lower than the one of CtNaa20 alone (4.4 ± 0.9 mM), while the kcat is in a similar range (Supplementary Fig. 4c, d). These data suggest that CtNaa25 increases the affinity of CtNaa20 for NatB substrates.

a Substrate specificity of CtNaa20 tested with five different peptides. b Melting temperatures of CtNatB in the presence of varying concentrations of AcCoA and CoA-Ac-MDEL. c Melting temperatures of CtNaa20 in the presence of varying concentrations of AcCoA and CoA-Ac-MDEL. d Melting temperatures of CtNatB and CtNaa20 in the presence of varying concentrations of CoA-Ac-MDEL, CoA-Ac-SESS, or CoA-Ac-MVNAL. All measurements were performed in triplicates and error bars represent the standard deviation.
To further investigate the CtNatB and CtNaa20 ligand interactions, we performed nanoDSF based binding assays using CoA-Ac-MDEL and AcCoA. Binding of both ligands to CtNatB and CtNaa20 is indicated by protein stabilization (Fig. 3b, c). When AcCoA is added to CtNatB, a mild stabilizing effect was observed by an increase of the unfolding transition temperature from 58 °C to 62 °C (using a 1/64 ratio of protein/ligand). Upon addition of the bisubstrate analog, CtNatB melting temperatures increased drastically from 58 °C to 71 °C. This illustrates a major contribution of the NatB specific peptide to the stabilization of the protein, compared to AcCoA alone (Fig. 3b). Noteworthy, the bisubstrate has a stronger effect on CtNaa20 melting temperature increase than AcCoA. These data confirm that CtNaa20 alone is able to bind MDEL and support the observation that it can acetylate MDEL without CtNaa25 (Fig. 3c). As a control we used two similar bisubstrate analogs with different peptide moieties in the nanoDSF assay (Fig. 3d). CoA-Ac-SESS and CoA-Ac-MVNAL were described as NatA and NatF inhibitors, respectively41,42,43. As they contain a CoA moiety, they exhibit a mild stabilizing effect on CtNatB, which is significantly lower than the effect of CoA-Ac-MDEL. The difference in stabilization highlights that MDEL binds to CtNaa20, but not the other peptides. Noteworthy, when only the MDEL peptide was added in a saturating amount to CtNatB or CtNaa20, no significant stabilizing effect was detected (Supplementary Fig. 4e). This is in accordance with the mechanism reported for Naa50, where AcCoA needs to bind before a substrate can bind44. Taken together, our data clearly show that CtNaa20 is active towards a canonical NatB substrate without CtNaa25. However, CtNaa25 increases the CtNaa20 affinity for this substrate, allowing for a more efficient acetylation.
CtNaa20 crystal structure in complex with CoA-Ac-MDEL
So far, structural information on Naa20 in the absence of the adaptor subunit Naa25 has not been available. In order to characterize Naa20 on an atomic level, we crystallized CtNaa20 in complex with the bisubstrate analog CoA-Ac-MDEL. The structure was solved in space group P 21 with two molecules per asymmetric unit. The initial phases were obtained by molecular replacement with the CaNaa20 structure part of the CaNatB complex (pdb: 5k18)30. The structure could be built at 1.57 Å resolution, revealing the expected GNAT-fold (Table 1; Fig. 4a). The high quality of the electron density map allowed building residues 2–190 together with one CoA-Ac-MDEL ligand (with the peptide part M1pD2pE3pL4p) for both protein chains. The root mean square deviation (rmsd) between the two protein molecules is 0.2 Å (for 190 Cα), indicating a very low level of flexibility.

a Overall structure of CtNaa20 with CoA-Ac-MDEL. CtNaa20 is shown in red as a cartoon and CoA-Ac-MDEL as gray sticks with nitrogen, oxygen, phosphorus, and sulfur shown in blue, red, orange, and yellow, respectively. The 2mFobs-DFcal electron density around the bisubstrate is shown at a contour level of 1σ (gray mesh). b The CtNaa20 structure is superimposed to CaNaa20 as part of CaNatB (pdb: 5k1830). Parts of the CaNaa25 structure are shown transparent to clarify the orientation. c Hydrogen bonds between CtNaa20 and CoA-Ac-MDEL are visualized with blue dotted lines and the corresponding atom distances.
The CtNaa20 structure consists of 4 α-helices and 8 β-strands and the bisubstrate is bound in the known V-shaped binding groove typical for NATs30,33,41,42. The β8-strand is a short additional strand compared to other NATs and shows that CtNaa20 C-terminal residues fold back along the β6 strand (Fig. 4a). This interaction stabilizes the enzyme compared to a C-terminally truncated version CtNaa201-166 (Supplementary Fig. 4f).
Comparison of our structure with the CaNaa20 part of CaNatB shows that both catalytic subunits superimpose very well with a rmsd of 1.0 Å (for 158 Cα)30, although one is in complex with its auxiliary subunit and the other not (Fig. 4b). The nicely resolved CtNaa20 C-terminal region superimposes well with the only partially built CaNaa20 C-terminal region (Fig. 4b). The comparison shows that the Naa20 substrate peptide binding site does not undergo major conformational changes upon complex formation, and explains the ability of CtNaa20 to bind and acetylate MDEL without CtNaa25. Nevertheless, some loop regions differ. For example, the β3-β4 loop is longer in CtNaa20 and does not superimpose well with the CaNaa20 β3-β4 loop. The CtNaa20 β6-β7 loop folds over the substrate peptide and contacts the α1-α2 loop, while the CaNaa20 β6-β7 loop turns away from the peptide (Fig. 4a, b). This results in a narrower peptide binding site in CtNaa20 compared to CaNaa20. Taken together, we obtained a high-resolution crystal structure of CtNaa20 alone, which superimposes well with CaNaa20 as part of the CaNatB complex, but also shows minor differences.
CtNaa20 structure explains the substrate specificity
The CtNaa20 structure demonstrates how the catalytic subunit binds a substrate peptide in the absence of CtNaa25. The loops α1-α2 and β6-β7 and the elongated β3-β4 loop confine the size of the substrate entry site. Loops α1-α2 and β6-β7 fold over the peptide, whereas loop α3-α4 is positioned underneath the peptide, but does not contact it directly (Fig. 4a). There are specific interactions between the ligand and CtNaa20, which were also observed in the CaNatB structure30. The acetyl bridge of the bisubstrate is coordinated by the amide backbone of V84. The amide nitrogen of M1p, the target of acetylation, is binding to the carbonyl oxygen of F118 (Fig. 4c and Supplementary Fig. 5a). The sidechain of M1p is situated in a pocket built by Y27 and A83 with a contribution of T24 and E25 sidechains (Fig. 4c and Supplementary Fig. 5a). Noteworthy, this pocket is less hydrophobic compared to methionine pockets of other NATs, like Naa50 or Naa60, which also act on the initiator methionine33,41,45. The M1p backbone carbonyl is bound by the hydroxyl group of Y145 and the D2p amide is coordinated by the T82 carbonyl group (Supplementary Fig. 5a). The specificity for acidic residues in peptide position two is caused by the H80 and T28 sidechains, which both form hydrogen bonds to the D2p sidechain (Fig. 4c). The D2p backbone carbonyl binds to the Y27 sidechain and the amide of E3p hydrogen bonds to Y144 sidechain (Supplementary Fig. 5a). The E3p peptide sidechain hydrogen bonds to the G146 backbone (Supplementary Fig. 5a), but no sidechain specific protein-ligand interactions are found for E3p and L4p, highlighting that the substrate specificity is mainly determined by the first two positions. Noteworthy, a well-ordered water can be found in the active site, which may be involved in catalysis. This water is coordinated by the backbone of F118 and I81, the M1p amide and the D2p sidechain, (Supplementary Fig. 5b).
Besides, the electrostatic surface potential of CtNaa20 reveals a positive area at the conserved AcCoA binding site, but no further exposed positive patches (Supplementary Fig. 5c), corroborating the lack of CtNaa20 binding to RNAs (Fig. 1e and Supplementary Fig. 2). In summary, the structural data support the observation that CtNaa20 acetylates canonical NatB substrates in vitro and show that H80 is important for specific peptide binding.
Specific residues are crucial for CtNatB activity
To further investigate the enzymatic mechanism of CtNatB, we mutated residues in CtNaa20 which are suggested to be important for acetylation efficiency30. Single mutations in the CtNaa20 substrate-binding pocket and active site (Y27A, H80A, H80Y, F118A, and Y145A) do not affect protein stability (Fig. 5a), but impair CtNatB acetylation efficiency (Fig. 5b). When CtNaa20 Y27 was replaced by F, which is the corresponding residue in CaNaa20 (Supplementary Fig. 1b), or F118 replaced by H, the corresponding residue in Naa10, Naa50 and Naa60 (Supplementary Fig. 6a), the catalytic efficiency of the resulting CtNatB complex does not change (Fig. 5b). Noteworthy, the H80Y and F118H CtNatB mutants and the corresponding double mutant, which were created to mimic the Naa10, Naa50 or Naa60 sequences at these positions (Supplementary Fig. 6a), are not sufficient to alter the substrate specificity of NatB to accept SESS, EEEI or MVNAL peptides (Supplementary Fig. 6b). To test whether CtNaa20 H80 is the major determinant for Naa20 and NatB substrate specificity (Fig. 4c), we created the Arabidopsis thaliana Naa60 Y115H mutant, to mimic the Naa20 sequence at the corresponding position (Supplementary Figs. 1b and 6a). This Naa60 mutant acetylated the NatB substrate MDEL in addition to its canonical substrates (Supplementary Fig. 6c), while the AtNaa60 wild-type is not active towards MDEL41. This confirms that H80 is indeed a key residue for Naa20 substrate specificity.

a The stability of different NatB mutants (mutations in the catalytic subunit CtNaa201-166) was assessed by nanoDSF in triplicates and error bars represent the standard deviations. b The wild-type efficiencies of different point mutants were calculated from kcat and Km values, determined in individuals experiments in triplicates. The data represent the mean values with standard deviations of these experiments, normalized to the wild-type value and considering the propagation of uncertainty. c Superimposition of mutated residues with the corresponding CaNaa20 residues. CtNaa20 is shown as transparent cartoon with catalytically important residues shown as red sticks. The corresponding CaNaa20 residues (pdb: 5k1830) are shown as cyan sticks and CoA-Ac-MDEL is shown as gray sticks.
The inactive CtNaa20 mutants (Y27A, H80A, H80Y, F118A, and Y145A) were also tested for CoA-Ac-MDEL binding using nanoDSF (Supplementary Fig. 7). The stabilizing effect of CoA-Ac-MDEL is higher than that of AcCoA for the Y27A and F118H mutant, indicating that they still bind MDEL (Supplementary Fig. S7a, e). In contrast, the H80A, H80Y, F118A, and Y145A mutants are not able to bind MDEL (Supplementary Fig. 7b–f) explaining their acetylation deficiency. Comparison with CaNaa20 shows that the corresponding residues superimpose well (Fig. 5c), which implies that their role in peptide binding and substrate acetylation is conserved between Naa20 proteins. Taken together, our data show that CtNaa20 alone is active in substrate acetylation and highlight the importance of specific residues for peptide binding and acetylation activity, and suggest that Naa25 binding does not induce conformational changes in Naa20.
Discussion
The majority of the proteome is N-terminally acetylated with around 20% being acetylated by NatB in yeast, plants, and human46. However, compared to the major NatA/NatE complexes, NatB has been less studied. For NatA and NatE, structures of the complexes32,42,47 and of the individual catalytic subunits Naa10 and Naa5032,33 were reported and analyzed in depth. Recently, the CaNatB crystal structure and the HsNatB cryo-EM structure were determined30,31. This showed how Naa25 binds to Naa20, and provided the first molecular basis of NatB substrate specificity. However, the Naa20 subunit could not be purified alone in previous CaNatB and Arabidopsis thaliana (At) NatB studies23,30. Therefore, to characterize NatB in more detail, we aimed to investigate CtNaa20 structure and function in the absence of CtNaa25.
Both CtNatB subunits were expressed and purified independently to homogeneity, and were shown to be monomers in solution. A stable CtNatB complex was formed with a 1:1 stoichiometry and a Kd of 17.8 ± 11.9 nM. Comparison with NatA binding to Naa50 (Kd = 46 ± 8.8 nM)48 shows that the binding affinities are in the same range. CtNatB acetylates specifically the MDEL peptide with enzymatic parameters similar to the ones described for other organisms. This emphasizes a high degree of conservation of NatB substrate specificity and mode of action3,23,30,49. Surprisingly, we also observed that CtNaa20 alone specifically acetylates the MDEL peptide in vitro, but with a lower efficiency than CtNatB. So far, Naa20 was considered to be inactive without Naa25 in vivo15,16,24 and the in vitro activity was not tested due to the lack of stable Naa20. In order to understand the seeming discrepancy between in vivo inactivity and the in vitro activity described in this study, we determined the CtNaa20 RNA binding capability and analyzed the CtNaa20 electrostatic surface potential. CtNaa20 does not bind RNA and does not present exposed positive patches that would allow for a direct ribosome interaction (Fig. 1e and Supplementary Fig. 5c). Therefore, Naa20 alone most likely does not bind to the ribosome and might have limited access to its substrates, which could explain the in vivo inactivity of Naa20. For comparison, Naa10 does not expose positive patches and is not involved in NatE-ribosome interactions36. Noteworthy, Naa20 is also found in the nucleus as a single subunit13 and thereby might acetylate a subset of presumed NatB substrates post-translationally. Interestingly, NatB-type substrates are found overrepresented in the nucleus compared to the whole worm lysate in C. elegans and a higher acetylation rate is observed in the nuclear fraction12.
The observed differences in CtNatB and CtNaa20 acetylation efficiency is reminiscent of differences in the activity towards the MLGP peptide between human Naa50 alone and as part of the human NatE complex48. In this case, HsNaa50 in complex with HsNatA (forming HsNatE) increased its affinity for the peptide, which lead to a more efficient acetylation. Similarly, we showed by enzymatic measurements that one function of Naa25 is to increase the affinity of Naa20 for NatB substrates. Probably, this is due to a stabilizing effect of CtNaa25 on CtNaa20, which leads to a more efficient acetylation.
Based on our results, showing that CtNaa20 specifically acetylates the MDEL peptide, we designed and synthesized the CoA-Ac-MDEL bisubstrate analog. This ligand was used for co-crystallization with CtNaa20 and structure determination at 1.57 Å resolution. The structure with the CoA-Ac-MDEL ligand allowed understanding the observed substrate specificity for the MDEL peptide. The M1p sidechain of the ligand is located in a wide pocket and importantly the D2p sidechain is coordinated by the H80 sidechain. Compared to other catalytic subunits acting on the initiator methionine, like Naa50 or Naa60, this specific sidechain interaction with the substrate residue in position two is a unique feature of Naa20 and of NatB30. The responsible histidine sidechain is conserved in all Naa20 subunits (Supplementary Fig. 1b) and explains the specificity for acidic residues at substrate position two. Compared to other NATs, Naa10 also has a conserved corresponding histidine, which is however not involved in substrate binding42. Interestingly, an AtNaa60 Y115H mutant was sufficient to broaden Naa60 substrate specificity towards NatB substrates, highlighting that this histidine plays a central role in recognition of an acidic residue in substrate position two. Naa30, Naa50, or Naa60 have a tyrosine at this position (Supplementary Fig. 6a). Nevertheless, a single H80Y mutation in CtNaa20 was not sufficient to change the substrate specificity to NatC/E/F like substrates. The CtNaa20 methionine binding pocket is more hydrophilic compared to Naa50 and Naa6033,41, consistent with Naa20 specificity for polar residues in position two. The substrate peptide residues E3p and L4p are not involved in specific protein-ligand interactions and may play only a minor role in substrate recognition. Accordingly, the third and fourth position of NatB substrates was found to be highly variable3,23,49.
When compared with the CaNaa20 subunit in complex with CaNaa25, CtNaa20 alone superimposes very well and only differs in several loop regions. The CtNaa20 β3-β4 loop is longer compared to Ca, and the β6-β7 loop folds over the MDEL peptide and is in closer contact to loop α1-α2 than in Ca. The high similarity of both structures suggests that Naa25 does not induce major rearrangements in Naa20 and that the mechanism of substrate binding and acetylation is very similar between NatB and the isolated Naa20 subunit. Similarly, binding of CaNaa20 seems not to induce conformational changes in the CaNaa25 subunit30. Here, NatB differs distinctly from NatA, as the peptide-binding pocket of Naa20 remains unchanged upon NatB complex formation and Naa20 is catalytically active on its own. Formation of NatA induces rearrangements of catalytically important residues in Naa10 and alters its substrate specificity as Naa10 alone is not active towards NatA substrates32. The CtNaa20/CoA-Ac-MDEL structure supports the observation that CtNaa20 is active towards canonical NatB substrates in vitro. Nevertheless, we cannot exclude that CtNaa20 is also active towards other substrates in the absence of CtNaa25.
Our structure unravels the Naa20 peptide binding mode, but does not allow to deduce an exact catalytic mechanism. In general, NATs were shown to use a base-mediated mechanism. Naa10 uses a distinct glutamate as general base and the corresponding E25 of CtNaa20 is conserved in Naa20 proteins (Supplementary Figs. 1b and 6a)32. However, in the CtNaa20 structure, E25 is not positioned in a way to serve as a base and the corresponding E25A mutation in the CaNatB complex even increased the catalytic efficiency30. Naa50 and Naa60 were shown to use a dual-base mechanism with conserved tyrosine and histidine residues and a well-ordered water molecule33,41. A well-ordered water was also found in the active site of the CtNaa20 structure. This water is likely to be involved in the catalysis, however, a basic residue is not involved in the coordination of the water and therefore a definite catalytic mechanism cannot be deduced. In CtNaa20, H80 corresponds to the catalytically important tyrosine of Naa50/60 and is important for binding D2p, but H80 cannot additionally hydrogen bond to the active site water or the M1p amide. The CtNaa20 residue corresponding to the catalytically important histidine of Naa50/60 is F118, which again is crucial for peptide binding and involved in coordinating the active site water, but is not a basic residue and therefore cannot be involved in proton transfer. Additionally, the F118H mutant showed no change in acetylation efficiency, whereas the corresponding H154F mutation in Naa60 impaired activity41. However, one has to consider that the bisubstrate analogs used here and in previous structures do not reflect the accurate transition state geometry of N-terminal acetylation, which proceeds via a tetrahedral conformation of the transferred acetyl-group carbon. This different geometry may hinder the identification of the CtNaa20 catalytic base, or the exact role of the active site water. Nevertheless, the CoA-Ac-MDEL bisubstrate analog is a potent competitive NatB inhibitor and a useful tool to understand the binding mechanism, structural features, and function of NATs. As NAT dysregulation is linked to multiple human diseases9, it is momentous to design NAT inhibitors for therapeutic purposes, based on substrate specificities and available structures, and to design ways to synthesize bio-available inhibitors in the future. NatB is an interesting therapeutic target because its depletion leads to the most severe phenotypes among all NATs50.
Taken together, our data contribute to a detailed understanding of NAT structures and functions, by providing the first Naa20 crystal structure. We show how Naa20 binds and specifically acetylates its substrates, which indicates that Naa20 may have a function without Naa25. Therefore, our study provides the structural and mechanistic framework to fully integrate NatB into the landscape of N-terminal protein acetylation.
Methods
Cloning of NatB constructs
CtNAA25 was amplified by PCR from cDNA and an internal NcoI-site was abolished by introducing a silent mutation in the forward primer. The resulting fragment was digested with the NcoI and BamHI enzymes and ligated into the pETNHis-vector (G. Stier, BZH) to obtain the pETNHis::CtNAA25 construct with a TEV-site cleavable His6-tag. CtNAA20 was amplified from cDNA by PCR and transferred into the TOPO-vector (Thermo Fischer). One internal NcoI-cutting site was removed by site-directed mutagenesis and a C-terminal His6-tag was introduced. The CtNAA20 fragment was ligated into the pET21d vector, leading to the pET21d::CtNAA20-His construct. Finally, the C-terminally truncated construct pET21d::CtNAA201-166-His was obtained by PCR using the full-length construct as template and cloning into the pET21d vector. Different CtNAA201-166 point mutants were obtained by site-directed mutagenesis using the quikchange lightning kit according to the manufacturer protocol (Agilent). All used primers are listed in Supplementary Table 1.
Expression and purification of native proteins
The pET21d::CtNAA20His construct was expressed in Rosetta II (DE3) E. coli strain (Novagen). Cells were grown at 18 °C in auto-induction-media, supplemented with chloramphenicol (34 μg ml−1) and ampicillin (50 μg ml−1). For purification, the cells were resuspended in lysate buffer (20 mM HEPES pH 7.5, 200 mM NaCl, 40 mM Imidazole), supplemented with a protease inhibitor mix, lysed using a microfluidizer (M-110L, Microfluidics) and the lysate was cleared by centrifugation (50000 × g, 25 min, 4 °C). For Ni-IMAC (immobilized metal affinity chromatography), the supernatants were loaded on a 1 ml HisTrap HP column (GE Healthcare). The tagged protein was eluted by adding 250 mM imidazole to the lysis buffer and further purified by size exclusion chromatography (SEC) using a S75 16/60 gel-filtration column (GE Healthcare) and buffer G (20 mM HEPES pH 7.5, 200 mM NaCl).
For the purification of the CtNaa25 subunit, the CtNatB complex or the complex with Naa201-166 point mutants, a three-step purification was performed. For this, the pETNHis::CtNAA25 construct, was transformed alone or cotransformed with the pET21d::CtNAA20-His, or one of the CtNAA201-166-His point mutants, with Rosetta II (DE3) E. coli cells (Novagen). Cells were grown at 18 °C for 18 h in auto-induction-media, supplemented with chloramphenicol (34 μg ml−1) and kanamycin (50 μg ml−1) for the CtNaa25 expression and additionally with carbenicillin (50 µg ml−1) for the different CtNatB complex species. For purification, the cells were resuspended in lysate buffer, supplemented with a protease inhibitor mix, lysed using a microfluidizer (M-110L, Microfluidics) and the lysate was cleared by centrifugation (50000 g, 25 min, 4 °C). The proteins were purified with Ni-IMAC by loading the supernatant on two 1 ml HisTrap HP column (GE Healthcare). Elution was performed with 250 mM imidazole. Afterward, the samples were dialyzed against IEX buffer A (100 mM NaCl and 50 mM sodium citrate pH 5.5) and loaded on a 5 ml HiTrap SP column for cation-exchange chromatography. The proteins were eluted using IEX buffer B (50 mM sodium citrate pH 5.5 and 1100 mM NaCl) by applying a step gradient of 18% buffer B. Afterwards the buffer was exchanged to buffer G via dialysis and the sample was loaded on a Superdex 200 26/60 gel-filtration column (GE Healthcare) for SEC. The AtNaa60_Y115H mutant was expressed and purified as described recently41.
Crystallization of CtNaa20
Crystallization was performed at 18 °C using the sitting drop vapor diffusion method. CtNaa20 was concentrated after gel-filtration to 20 mg/ml and mixed in a 1:3 molar ratio with CoA-Ac-MDEL and incubated on ice for 18 h. The crystallization drops contained 200 nl protein solution and 200 nl precipitant solution (15% (v/v) propanol, 0.2 M ammonium acetate, and 0.1 M TRIS pH 8.5). Crystals appeared after 3 days and were cryo-protected with 20% glycerol and flash-frozen in liquid nitrogen.
Data collection and structure determination
Data sets for the CtNaa20 crystals were collected at beamline P14 (DESY) at cryogenic temperature. The images were integrated with XDS51. Afterwards the images were scaled using AIMLESS52. Phases were obtained by molecular replacement with PHASER-MR53 implemented in the PHENIX package54. The CaNaa20 part of the CaNatB complex (30 pdb:5k18) was used as an initial search model. Finally, iterative model building and refinement were performed with Coot55 and Phenix.refine56. The CoA-Ac-MDEL ligand was parametrized with the PHENIX eLBOW module in AM1 QM mode57. Model quality was analyzed with MolProbity58. Interfaces and crystal packing were analyzed with PISA59. Structure figures were prepared with PyMOL60. Crystallographic data are summarized in Table 1. Coordinates and structure factors are deposited at the Protein Data Bank PDB with accession code 6ZMP.
SEC-MALS analyses
The CtNatB complex (0.12 mg) and its subunits CtNaa25 (0.13 mg) and CtNaa20 (0.1 mg) were successively injected onto a Superdex 200 10/300 gel-filtration column (GE Healthcare) in buffer G. The column was connected to a MALS system (Dawn Heleos II 8+ and Optilab T-rEX, Wyatt Technology). Data were analyzed using the Astra 6 software (Wyatt Technology).
RNA preparation and electrophoretic mobility shift assay
The 32 nucleotides long (5′-GGGCCTCTAGCCGGGCAACCGGCCGGCGGCTC-3′) CtES27 fragment of the 25 S rRNA fused to the 3′-hammerhead ribozyme was amplified by primer extension PCR and cloned under the control of the T7 RNA polymerase promotor into the pUC18 vector (Promega) digested EcoRI/ Hind III. In vitro transcription of the CtES27 fragment and hammerhead ribozyme RNA construct was performed as described previously61. In brief, the CtES27 fragment RNA and hammerhead ribozyme RNA were transcribed in vitro and purified by urea-polyacrylamide gel electrophoresis, extracted by crush-and-soak followed by isopropanol precipitation and desalting.
For the EMSA, CtNaa20 and CtNatB (5–20 µM) were incubated with 5 µM CtES27 or hammerhead ribozyme RNA in EMSA buffer (20 mM Tris pH 8, 200 mM NaCl, 10 mM MgCl2, 10 mM KCl and 20% v/v glycerol) at 20 °C for 30 min. The samples were separated electrophoretically with 0.8% (w/v) agarose gels (supplemented with ethidium bromide) in 0.5x TB buffer (45 mM boric acid and 45 mM Tris pH 8). RNA-free and protein-free samples were used as controls. The RNA migration was visualized under UV light.
Synthesis of CoA-Ac-MDEL inhibitor
For the synthesis of the bisubstrate analog CoA-Ac-MDEL, the Foyn et al. protocol was modified43. The MDEL peptide was synthesized with a MultiPep RSi peptide synthesizer (Intavis) on solid support. A leucine preloaded 2-chlorotrityl resin (50 µmol, 1 equiv.) was used with 9-Fluorenylmethoxycarbonyl (Fmoc)-amino acids (250 µmol, 5 equiv), HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluoro-phosphate; 250 µmol, 5 equiv), HOBt (1-Hydroxybenzotriazol; 0.2 mol/l) and DIPEA (Diisopropylethylamine; 500 µmol, 10 equiv) in DMF (N,N-Dimethylformamide) doing double couplings for 40 min. Fmoc deprotection was carried out applying 40% piperidine in DMF for 3 min and then 20% piperidine in DMF for 14 min. 70 mg Bromoacetic acid (500 µmol, 10 equiv), dissolved in DMF and mixed with 155 µl DIC (N,N’-diisopropylcarbodiimide; 126 mg, 1 mmol, 20 equiv) was added to the peptide-linked resin. The suspension was shaken for 24 h at room temperature and then the resin was washed three times with DMF and DCM (Dichloromethane), respectively. The bromo-acetylated peptide was cleaved from the resin by gently shaking in a cleavage cocktail (Trifluoroacetic acid:Triisopropylsilane:water 95:2.5:2.5) for 3 h and was precipitated in 40 ml cold diethylether and dried under vacuum. The precipitate was dissolved in 2 ml water/acetonitrile mixture (80:20) and purified by reverse phase HPLC. The solvent was removed using a rotary evaporator revealing a white solid (3.5 mg, 5.6 µmol, 11.2% yield). Subsequently, the intermediate was dissolved in 200 µl of 1 M triethylammonium bicarbonate buffer at pH 8.5 and a 200 µl solution of 6.4 mg Coenzyme A trilithium salt (8.49 µmol, 1.5 eq.), in the same buffer, was added. The mixture was stirred at room temperature for 24 h protected from light. The solvent was removed under vacuum and the crude product was dissolved in water and purified by reverse phase HPLC (water/acetonitrile 10–30%) to a purity of >95%. The solvent was removed by lyophilization to give 0.9 mg of CoA-Ac-MDEL (0.7 µmol, 12.5% yield for CoA coupling; MALDI/TOF (pos): m/z calc. for C43H71N11O26P3S2+: 1314.32 [M + H]+; found: 1314.2).
CtNatB activity and CoA-Ac-MDEL inhibition assays
All enzymatic assays (the substrate specificity tests of CtNatB and CtNaa20, the Michaelis–Menten analysis of the CtNatB complex and the complex with the mutated catalytic subunits, the inhibitor assays to determine the IC50 value of CoA-Ac-MDEL and the mode of inhibition) were performed using microplate assays described earlier62 and used as modified recently41. For all assays a protein concentration of 500 nM was used. For all assays, either a constant peptide concentration of 1.5 mM or a constant AcCoA concentration of 370 µM with varying concentrations of 6–500 µM AcCoA or 39–2500 µM MDEL were used. Background control reactions were performed in the absence of the enzyme, or of the peptides and all reactions were performed in triplicates. Data were evaluated using the GraphPad Prism software.
ITC Measurements
ITC binding measurements between CtNaa25 and CtNaa201-166 were performed using a PEAQ-ITC microcalorimeter (Malvern Instrument GmbH). Prior to the measurements, the protein samples were dialyzed against buffer G overnight.
CtNaa201-166 concentrations of 25–35 µM in the cell were titrated with CtNaa25 concentrations of 250–400 µM in the syringe at 20 °C. The data were fitted and analyzed using a single-site binding model in the MicroCal PEAQ-ITC analysis software. Measurements were performed in triplicates. In addition, buffer to buffer, buffer to CtNaa201-166 and CtNaa25 to buffer ITC runs were performed as control reactions.
NanoDSF Measurements
To determine melting temperatures Tm of different protein samples, nano differential scanning fluorimetry (nanoDSF) was used. Intrinsic tyrosine and tryptophan fluorescence at emission wavelengths of 330 nm and 350 nm were measured continuously applying a temperature gradient of 20–90 °C in the Prometheus NT.48 nanoDSF system. The Tm was calculated by the supplied software (NanoTemper Technologies GmbH). To assess the stability of the different CtNatB mutants, 1 mg/ml samples were measured in buffer G. The stability changes of CtNatB, CtNaa20, and its mutants upon addition of different ligands, were measured using 15–30 µM protein in buffer G, after incubation with a varying excess of AcCoA, CoA-Ac-MDEL, CoA-Ac-SESS, or CoA-Ac-MVNAL for 10 min on ice.
Statistics and Reproducibility
All kinetic experiments were performed in triplicates. Error bars in figures represent the standard deviations. All nanoDSF assays were performed in triplicates or quadruplicates and error bars represent the standard deviations. Individual data points are depicted in all figures, apart for Fig. 5b. For Fig. 5b, kcat and Km values of each mutant were determined in individuals Michaelis-Menten experiments in triplicates, which were further used to calculate the enzymatic efficiency, normalized to the wild-type efficiency. The data represent the mean values with standard deviations considering the propagation of uncertainty. The ITC measurements were performed in triplicates and Kd and ΔH represent the mean values with corresponding standard deviations.
Data availability
Coordinates and structure factors have been deposited at the Protein Data Bank under the accession code 6ZMP. All source data underlying graphs and charts are presented in Supplementary Data 1. Further data supporting the findings of this study are available from the corresponding author upon reasonable request.
References
Arnesen, T. et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl Acad. Sci. USA 106, 8157–8162 (2009).
Bienvenut, W. V. et al. Comparative large scale characterization of plant versus mammal proteins reveals similar and idiosyncratic N-α-acetylation features. Mol. Cell. Proteom. 11, M111.015131 (2012).
Van Damme, P. et al. N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc. Natl Acad. Sci. USA 109, 12449–12454 (2012).
Arnesen, T. Towards a functional understanding of protein N-terminal acetylation. PLoS Biol. 9, e1001074 (2011).
Dörfel, M. J. & Lyon, G. J. The biological functions of Naa10—from amino-terminal acetylation to human disease. Gene 567, 103–131 (2015).
Starheim, K. K., Gevaert, K. & Arnesen, T. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem. Sci. 37, 152–161 (2012).
Gautschi, M. et al. The yeast Nα-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol. Cell. Biol. 23, 7403–7414 (2003).
Arnesen, T. et al. Identification and characterization of the human ARD1–NATH protein acetyltransferase complex. Biochem. J. 386, 433–443 (2005).
Aksnes, H., Ree, R. & Arnesen, T. Co-translational, Post-translational, and Non-catalytic Roles of N-Terminal Acetyltransferases. Mol. Cell 73.6, 1097–1114 (2019).
Polevoda, B., Arnesen, T. & Sherman, F. A synopsis of eukaryotic N α-terminal acetyltransferases: nomenclature, subunits and substrates. in BMC proceedings 3 S2 (BioMed Central, 2009).
Polevoda, B., Brown, S., Cardillo, T. S., Rigby, S. & Sherman, F. Yeast Nα‐terminal acetyltransferases are associated with ribosomes. J. Cell. Biochem. 103, 492–508 (2008).
Gao, J. et al. N-terminal acetylation promotes synaptonemal complex assembly in C. elegans. Genes Dev. 30, 2404–2416 (2016).
Starheim, K. K. et al. Identification of the human Nα-acetyltransferase complex B (hNatB): a complex important for cell-cycle progression. Biochem. J. 415, 325–331 (2008).
Singer, J. M., Hermann, G. J. & Shaw, J. M. Suppressors of mdm20 in yeast identify new alleles of ACT1 and TPM1 predicted to enhance actin-tropomyosin interactions. Genetics 156, 523–534 (2000).
Singer, J. M. & Shaw, J. M. Mdm20 protein functions with Nat3 protein to acetylate Tpm1 protein and regulate tropomyosin–actin interactions in budding yeast. Proc. Natl Acad. Sci. 100, 7644–7649 (2003).
Polevoda, B., Cardillo, T. S., Doyle, T. C., Bedi, G. S. & Sherman, F. Nat3p and Mdm20p are required for function of yeast NatB Nα-terminal acetyltransferase and of actin and tropomyosin. J. Biol. Chem. 278, 30686–30697 (2003).
Kats, I. et al. Mapping degradation signals and pathways in a eukaryotic N-terminome. Mol. Cell 70, 488–501 (2018).
Nguyen, K. T., Kim, J.-M., Park, S.-E. & Hwang, C.-S. N-terminal methionine excision of proteins creates tertiary destabilizing N-degrons of the Arg/N-end rule pathway. J. Biol. Chem. 294, 4464–4476 (2019).
Kamita, M. et al. Nα-Acetylation of yeast ribosomal proteins and its effect on protein synthesis. J. Proteom. 74, 431–441 (2011).
Lee, K.-E., Ahn, J.-Y., Kim, J.-M. & Hwang, C.-S. Synthetic lethal screen of NAA20, a catalytic subunit gene of NatB N-terminal acetylase in Saccharomyces cerevisiae. J. Microbiol. 52, 842–848 (2014).
Oishi, K., Yamayoshi, S., Kozuka-Hata, H., Oyama, M. & Kawaoka, Y. N-terminal acetylation by NatB is required for the shutoff activity of influenza A virus PA-X. Cell Rep. 24, 851–860 (2018).
Ferrandez-Ayela, A. et al. Mutation of an Arabidopsis NatB N-alpha-terminal acetylation complex component causes pleiotropic developmental defects. PLoS ONE 8, e80697 (2013).
Huber, M. et al. NatB-mediated N-terminal acetylation affects growth and abiotic stress responses. Plant Physiol. 182, 792–806 (2019).
Caesar, R., Warringer, J. & Blomberg, A. Physiological importance and identification of novel targets for the N-terminal acetyltransferase NatB. Eukaryot. Cell 5, 368–378 (2006).
Ametzazurra, A., Larrea, E., Civeira, M., Prieto, J. & Aldabe, R. Implication of human N-α-acetyltransferase 5 in cellular proliferation and carcinogenesis. Oncogene 27, 7296 (2008).
Neri, L. et al. NatB-mediated protein N-α-terminal acetylation is a potential therapeutic target in hepatocellular carcinoma. Oncotarget 8, 40967 (2017).
Yasuda, K., Takahashi, M. & Mori, N. Mdm20 modulates actin remodeling through the mTORC2 pathway via its effect on rictor expression. PLoS ONE 10, e0142943 (2015).
Ohyama, K., Yasuda, K., Onga, K., Kakizuka, A. & Mori, N. Spatio-temporal expression pattern of the NatB complex, Nat5/Mdm20 in the developing mouse brain: implications for co-operative versus non-co-operative actions of Mdm20 and Nat5. Gene Expr. Patterns 12, 36–45 (2012).
Ametzazurra, A. et al. Characterization of the human N α-terminal acetyltransferase B enzymatic complex. in BMC proceedings 3 S4 (BioMed Central, 2009).
Hong, H. et al. Molecular basis of substrate specific acetylation by N-terminal acetyltransferase NatB. Structure 25, 641–649 (2017). e3.
Deng, S., Pan, B., Gottlieb, L., Petersson, J. & Marmorstein, R. Molecular basis for N-terminal alpha-synuclein acetylation by human NatB. eLife 9, e57491 (2020).
Liszczak, G. et al. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat. Struct. Mol. Biol. 20, 1098–1105 (2013).
Liszczak, G., Arnesen, T. & Marmorstein, R. Structure of a ternary Naa50p (NAT5/SAN) N-terminal acetyltransferase complex reveals the molecular basis for substrate-specific acetylation. J. Biol. Chem. 286, 37002–37010 (2011).
van Noort, V. et al. Consistent mutational paths predict eukaryotic thermostability. BMC Evolut. Biol. 13, 7 (2013).
Amlacher, S. et al. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 146, 277–289 (2011).
Knorr, A. G. et al. Ribosome–NatA architecture reveals that rRNA expansion segments coordinate N-terminal acetylation. Nature structural &. Mol. Biol. 26, 35–39 (2019).
Wild, K. et al. MetAP-like Ebp1 occupies the human ribosomal tunnel exit and recruits flexible rRNA expansion segments. Nat. Commun. 11, 1–10 (2020).
Bradatsch, B. et al. Structure of the pre-60S ribosomal subunit with nuclear export factor Arx1 bound at the exit tunnel. Nat. Struct. Mol. Biol. 19, 1234 (2012).
Van Damme, P. et al. Proteome-derived peptide libraries allow detailed analysis of the substrate specificities of Nα-acetyltransferases and point to hNaa10p as the post-translational actin Nα-acetyltransferase. Mol. Cell. Proteom. 10, M110.004580 (2011).
Drazic, A. et al. NAA80 is actin’s N-terminal acetyltransferase and regulates cytoskeleton assembly and cell motility. Proc. Natl Acad. Sci. USA 115, 4399–4404 (2018).
Linster, E. et al. The Arabidopsis Nα‐acetyltransferase NAA60 locates to the plasma membrane and is vital for the high salt stress response. N. Phytologist 228, 554–569 (2020).
Weyer, F. A. et al. Structural basis of HypK regulating N-terminal acetylation by the NatA complex. Nat. Commun. 8, 1–10 (2017).
Foyn, H. V. et al. Design, synthesis, and kinetic characterization of protein N-terminal acetyltransferase inhibitors. ACS Chem. Biol. 8, 1121–1127 (2013).
Evjenth, R. H. et al. Human protein N-terminal acetyltransferase hNaa50p (hNAT5/hSAN) follows ordered sequential catalytic mechanism combined kinetic and NMR study. J. Biol. Chem. 287, 10081–10088 (2012).
Støve, S. I. et al. Crystal structure of the Golgi-associated human Nα-acetyltransferase 60 reveals the molecular determinants for substrate-specific acetylation. Structure 24, 1044–1056 (2016).
Linster, E. & Wirtz, M. N-terminal acetylation: an essential protein modification emerges as an important regulator of stress responses. J. Exp. Bot. 69, 4555–4568 (2018).
Deng, S. et al. Structure and mechanism of acetylation by the N-terminal dual enzyme NatA/Naa50 complex. Structure 27, 1057–1070 (2019).
Deng, S., McTiernan, N., Wei, X., Arnesen, T. & Marmorstein, R. Molecular basis for N-terminal acetylation by human NatE and its modulation by HYPK. Nat. Commun. 11, 1–14 (2020).
Helbig, A. O. et al. Perturbation of the yeast N-acetyltransferase NatB induces elevation of protein phosphorylation levels. BMC genomics 11, 685 (2010).
Aksnes, H., Hole, K. & Arnesen, T. Molecular, cellular, and physiological significance of N-terminal acetylation. in International review of cell and molecular biology, Vol. 316, 267–305 (Elsevier, 2015).
Kabsch, W. XDS. Acta Crystallogr D. Biol. Crystallogr. 66, 125–132 (2010).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D: Biol. Crystallogr. 69, 1204–1214 (2013).
Bunkóczi, G. et al. Phaser. MRage: automated molecular replacement. Acta Crystallogr. Sect. D: Biol. Crystallogr. 69, 2276–2286 (2013).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D: Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 66, 486–501 (2010).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D. Biol. Crystallogr. 68, 352–367 (2012).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. Sect. D: Biol. Crystallogr. 65, 1074–1080 (2009).
Richardson, D. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 66, 12–21 (2010).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Schrödinger, L. L. C. The PyMOL molecular graphics system. Version 1, 8 (2015).
Becker, M. M., Lapouge, K., Segnitz, B., Wild, K. & Sinning, I. Structures of human SRP72 complexes provide insights into SRP RNA remodeling and ribosome interaction. Nucleic Acids Res. 45, 470–481 (2017).
Andrew Skaff, D. & Miziorko, H. M. A visible wavelength spectrophotometric assay suitable for high-throughput screening of 3-hydroxy-3-methylglutaryl-CoA synthase. Anal. Biochem. 396, 96–102 (2010).
Acknowledgements
We thank F.A. Weyer for initial cloning and for fruitful input, and N. Höfflin for support in the bisubstrate synthesis. We thank C. Siegmann from the BZH crystallization platform for protein crystallization, S. Adrian for assistance in cloning, and G. Stier and E. Hurt for providing cloning vectors and CtcDNA, respectively. X-ray data collections were performed at beamlines X06DA-PXIII (SLS, Paul Scherrer Institut, Villigen, Switzerland) and beamline P14 (EMBL Hamburg at the PETRA III storage ring, DESY, Hamburg, Germany), respectively. We acknowledge the data storage service SDS@hd supported by the Ministry of Science, Research and the Arts Baden-Württemberg (MWK) and the German Research Foundation (DFG) through grant INST 35/1314-1 and INST 35/1503-1 FUGG. I.S. is an investigator of the Cluster of Excellence: CellNetworks. This work was funded by the German Research Foundation (DFG) through the Leibniz program (SI 586/6-1) and Project-ID 201348542 – SFB 1036 (TP22) to I.S., and by an ERC Starting Grant (#336567) to M.K.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
D.L. designed, performed, and analyzed experiments, collected and processed X-ray diffraction data, analyzed all data, synthesized the bisubstrate analog, wrote the manuscript. J.K. performed crystallization screening, collected X-ray diffraction data sets and analyzed X-ray data. M.F. designed and synthesized the bisubstrate analog. M.K. supervised and planned the synthesis of the bisubstrate analog. K.L. designed and analyzed experiments, wrote manuscript. I.S. conceived the study, designed and analyzed experiments, acquired funding, wrote manuscript. All authors read and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Layer, D., Kopp, J., Fontanillo, M. et al. Structural basis of Naa20 activity towards a canonical NatB substrate. Commun Biol 4, 2 (2021). https://doi.org/10.1038/s42003-020-01546-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-01546-4
This article is cited by
-
Diverging co-translational protein complex assembly pathways are governed by interface energy distribution
Nature Communications (2024)
-
Metabolic regulation of proteome stability via N-terminal acetylation controls male germline stem cell differentiation and reproduction
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
