
Abstract
Although the important roles of glycolysis in T cells have been demonstrated, the regulatory mechanism of glycolysis in activated T cells has not been fully elucidated. Furthermore, the influences of glycolytic failure on the T cell-dependent immune response in vivo remain unclear. We therefore assessed the role of glycolysis in the T cell-dependent immune response using T cell-specific Pgam1-deficient mice. Both CD8 and CD4 T cell-dependent immune responses were attenuated by Pgam1 deficiency. The helper T cell-dependent inflammation was ameliorated in Pgam1-deficient mice. Glycolysis augments the activation of mTOR complex 1 (mTORC1) and the T-cell receptor (TCR) signals. Glutamine acts as a metabolic hub in activated T cells, since the TCR-dependent increase in intracellular glutamine is required to augment glycolysis, increase mTORC1 activity and augment TCR signals. These findings suggest that mTORC1, glycolysis and glutamine affect each other and cooperate to induce T cell proliferation and differentiation.
Similar content being viewed by others
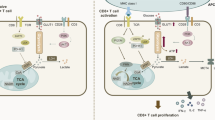


Introduction
On antigen recognition, naïve T cells initiate a cell-intrinsic program that induces a proliferative response and differentiation into effector T cells. Recent evidence suggests that T cells dramatically alter their metabolic activity during T cell receptor (TCR)-mediated activation1,2,3. Non-proliferating naïve T cells use fatty acid oxidization and/or a low rate of glycolysis and subsequently oxidize glucose-derived pyruvate via oxidative phosphorylation (OXOPHOS) to generate ATP4,5. In contrast, the activated T cells rely on an increasing rate of glycolysis despite the aerobic condition which is required for rapid cellular proliferation and acquisition of the effector function4,6, known as the Warburg effect7. Although effector T cells dominantly use aerobic glycolysis for glucose metabolism, OXPHOS also continues to occur6,8. This change in the metabolic status is termed “metabolic reprogramming” and is believed to play an important role in the regulation of T cell-mediated immune responses9.
Phosphoglycerate mutase 1 (Pgam1) is a glycolytic enzyme that catalyzes the conversion of 3-phosphoglycerate (3-PG) to 2-phosphoglycerate (2-PG). Pgam1 is thought to regulate both glycolysis and biosynthesis, since 3-PG inhibits 6-phosphogluconate dehydrogenase in the pentose phosphate pathway (PPP) while 2-PG activates 3-phosphoglycerate dehydrogenase in serine synthesis10. Recent studies have revealed that Pgam1 is upregulated in several human cancers and linked to the tumor growth and survival11,12. As described above, the metabolic profiles of activated T cells are similar to the profile in tumors, represented by aerobic glycolysis. Hence, Pgam1 also plays an important role in regulating the metabolic reprogramming of T cells.
The mechanistic target of rapamycin (mTOR) is a crucial regulator of cellular metabolism13,14. mTOR signaling is essential for immune signals and alteration of cellular metabolism for proper differentiation and function of T cells15,16,17. mTOR exists in two complexes, mTOR complex 1 (mTORC1) and mTORC2. mTORC1 and mTORC2 orchestrate metabolic reprograming to exit the quiescent state of T cells18. mTORC1 regulates protein synthesis via phosphorylation of the S6 kinases and the inhibitory eukaryotic initiation factor 4E (eIF4E)-binding proteins. It was reported that the mTORC1-dependent induction of Myc promotes glucose and glutamine metabolism in activated T cells8,19. Myc-dependent metabolic reprograming promotes the T-cell proliferative response and differentiation18,20. Thus, a consequence of increased mTORC1 activity during quiescence exit is the increased translation of pro-anabolic enzymes and transcription factors, such as Myc21.
Proliferating cells vigorously import extracellular glutamine which supply carbon and nitrogen for the biomass accumulation22,23. T cells also increase glutamine take up after TCR-stimulation, and glutamine promote T-cell activation and proliferation8,24. In the glutaminolysis, glutamine is converted to glutamate, and then, glutamate is catabolized to α-ketoglutarate (α-KG), which is consumed through tricarboxylic acid (TCA) cycle25,26. Glutamine is thought to activate T cells through several mechanisms. Recently, we reported that glutamine activates mTORC1 signaling, partly via supplementation of α-KG27.
In the present study, we examined the role of glycolysis using T-cell-specific Pgam1 knockout (KO) mice (Pgam1flox/flox with CD4-Cre transgenic) and found that glycolysis, mTORC1 and glutamine affect each other and cooperate to induce T-cell proliferation and differentiation.
Results
Glycolysis controls TCR-mediated signal transduction
Upon antigen recognition, T cells show a dramatic increase in glucose metabolism1,2,3. However, the influence of glycolytic failure on the T-cell-dependent immune response in vivo is poorly understood. The mRNA expression of pgam1 was induced by TCR-stimulation in CD8 T cells, whereas the level of pgam2 mRNA, an isozyme of Pgam1, was decreased (Supplementary Fig. 1a). We therefore generated T cell-specific Pgam1 KO mice to clarify the roles of glycolysis during TCR-mediated activation and the T-cell-dependent immune response. The reduction in Pgam1 protein in Pgam1 KO CD8 T cells was confirmed by immunoblotting (Supplementary Fig. 1b). Pgam1 deficiency showed no effect on thymic T cell development or the T cell number in the spleen (Supplementary Fig. 2a). The memory/activated phenotype CD4 and CD8 T cells were marginally decreased in Pgam1 KO mice compared with wild-type mice (Supplementary Fig. 2b). The numbers of Foxp-positive CD4 T cells and invariant NKT cells were decreased in the spleen, whereas the numbers of these cells in the thymus and mesenteric lymph node were comparable (Supplementary Fig. 2c, d). Interleukin (IL)-2 production was significantly lower in Pgam1 KO CD4 T cells than in wild-type CD4 T cells. (Supplementary Fig. 2e).
We first assessed the metabolic profile in Pgam1 KO T cells using an extracellular flux analyzer. The glycolysis assessed by the extracellular acidification rate (ECAR) at 24 h after TCR-stimulation was lower in Pgam1 KO activated CD8 T cells than in wild-type cells (Fig. 1a). Pgam1 KO activated CD4 T cells also showed reduced ECAR compared with wild-type (Supplementary Fig. 3a). The basal oxygen consumption rate (OCR) and spare respiratory capacity (SRC) at 24 h after TCR-stimulation showed a significant reduction under conditions of Pgam1 deficiency in CD8 T cells (Fig. 1b). The basal OCR in Pgam1 KO activated CD4 T cells was comparable to that in wild-type CD4 T cells, whereas the SRC was decreased in Pgam1 KO CD4 T cells (Supplementary Fig. 3b). The ECAR in Pgam1 KO activated CD8 T cells at 8 h was comparable to that in wild-type CD8 T cells (Supplementary Fig. 4a). In addition, the basal OCR at 8 h in Pgam1 KO activated CD8 T cells was comparable to that in wild-type CD8 T cells, whereas the SRC was decreased in Pgam1 KO CD8 T cells (Supplementary Fig. 4b). The intracellular concentration of glycolytic intermediates before the Pgam-dependent catabolizing step (G6P, F6P, F1-6P DHAP, and 3PG) at 24 h after TCR-mediated activation was increased in Pgam1 KO CD8 T cells in comparison to wild-type CD8 T cells (Fig. 1c). In contrast, the intracellular level of lactate, an end product of anaerobic glycolysis, was decreased in Pgam1 KO cells (Fig. 1c). Pgam1-deficiency only showed a marginal effect on the intracellular concentrations of glycolytic products at 6 h after stimulation (Fig. 1c). These results were consistent with the expression pattern of pgam1 mRNAs that demonstrated the shift from pgam2 to pgam1 upon the TCR-mediated activation of CD8 T cells (Supplementary Fig. 1a). The concentrations of TCA cycle intermediates, succinate, fumarate, and malate were decreased in Pgam1-deficient CD8 T cells at 24 h after stimulation, whereas the levels of citrate and cis-aconitate were not affected by Pgam1 deficiency (Supplementary Fig. 4c). The intracellular concentrations of TCA cycle intermediates at 6 h were moderately increased in Pgam1 KO CD8 T cells in comparison to wild-type CD8 T cells (Supplementary Fig. 4c). The intracellular amounts of both NAD+ and NADH at 24 h were significantly decreased in Pgam1 KO CD8 T cells in comparison to wild-type cells, although these concentrations were comparable at 6 h (Supplementary Fig. 4d). The intracellular concentration of intermediates of the pentose phosphate pathway (PPP) at 24 h was moderately increased in Pgam1 KO CD8 T cells in comparison to wild-type cells (Supplementary Fig. 4e), and the intracellular levels of IMP, AMP, GMP, and UMP were reduced by Pgam1 deficiency (Supplementary Fig. 4f). These results suggest that nucleotide synthesis, but not PPP, is inhibited by Pgam1 deficiency in activated CD8 T cells. The intracellular concentration of ATP in Pgam1 KO CD8 T cells was equivalent to that in wild-type CD8 T cells at 6 h after TCR stimulation (Fig. 1d). While the level of ATP was further increased in wild-type CD8 T cells at 24 h, it was not increased but instead decreased in Pgam1 KO CD8 T cells (Fig. 1d).

a, b WT and Pgam1 KO naive CD8 T cells were stimulated for 24 h, and then a the extracellular acidification rate (ECAR) and b oxygen consumption rate (OCR) were determined (n = 3, technical replicate). Error bars represent the standard deviation. The results of the analyses are representative of at least three independent experiments with similar results. c WT and Pgam1 KO naïve CD8 T cells were stimulated with anti-TCR-β and anti-CD28 mAbs for 6 or 24 h, and the intracellular amounts of glycolytic intermediates were determined by metabolic profiling (n = 3–4, biological replicates). d The intracellular amount of ATP of the cells in c. e The phospho-tyrosine and β-actin levels in WT and Pgam1 KO CD8 T cells stimulated for indicated periods were determined by an immunoblot analysis. The protein amount of β-actin was used as a loading control. The numbers below the bands indicate the densitometry ratio with WT stimulated for 24 h. f The results of the immunoblot analysis of phospho-Zap70 (Tyr319), phosphor-Plcγ1 (Tyr783), and β-actin (control) in cells in e are shown. The numbers below the bands indicate the densitometry ratio with WT stimulated for 24 h. The results of the immunoblot analyses are representative of at least three independent experiments with similar results and are presented as cropped images. The full-length blots are presented in Supplementary Fig. 18. *P < 0.05, **P < 0.01 (Student’s t test).
ATP is well known to act as a phosphate donor and is required for the protein kinase activity28. We therefore assessed the protein-tyrosine phosphorylation status of Pgam1 KO activated CD8 T cells. The protein tyrosine phosphorylation level was lower in Pgam1 KO activated CD8 T cells than in wild-type cells at 24 h after TCR stimulation, whereas the level at 6 h was comparable in both cell types (Fig. 1e). The tyrosine phosphorylation status of the TCR signaling molecules Zap70 (Tyr319) and Plcγ1 (Tyr783) was also decreased in Pgam1 KO activated CD8 T cells at 24 h after stimulation and the level at 6 h was equivalent to that in wild-type cells. (Fig. 1f). These results indicate that the glycolytic activity and the intracellular concentration of ATP are correlated with the signal strength in activated CD8 T cells.
Glycolysis is required for the development of effector CD8 T cells
Wild-type and Pgam1 KO CD8 T cells were stimulated with anti-TCRβ mAb plus anti-CD28 mAb for 36 h, and the expression of activation makers was assessed by FACS. The expression of CD25, IL-2 receptor α chain, was decreased in Pgam1 KO CD8 T cells compared to wild-type CD8 T cells, whereas the CD44 and CD69 levels were unaffected by Pgam1 deficiency (Fig. 2a). Consistent with the decreased expression of CD25, the IL-2-dependent phosphorylation of Stat5 was reduced in Pgam1 KO CD8 T cells in comparison to the wild-type CD8 T cells (Fig. 2b), indicating that the reactivity of activated CD8 T cells to IL-2 was dependent on glycolytic activation. The TCR-dependent proliferative response was decreased in Pgam1 KO CD8 T cells compared to wild-type CD8 T cells in vitro (Fig. 2c). Furthermore, Pgam1 KO mice showed a lower expansion of OVA-specific CD8 T cells in the spleen after OVA-peptide-expressing Listeria monocytogenes (Lm-OVA) infection (Fig. 2d and Supplementary Fig. 5). Thus, the T-cell expansion was attenuated by Pgam1 deficiency both in vitro and in vivo. The essential role of de novo nucleotide synthesis in the cell cycle progression of activated T cells has been reported29. We demonstrated the decrease in the intracellular nucleotides concentration in Pgam1 KO activated CD8 T cells (Supplementary Fig. 4e), which would result in an impaired T-cell proliferative response.

a Representative staining profile of CD25, CD44, and CD69 in WT and Pgam1 KO CD8 T cells stimulated with anti-TCR-β plus anti-CD28 mAbs for 36 h. b Representative staining profiles of phospho-STAT5 (Tyr694) in activated CD8 T cells. WT and Pgam1 KO naïve CD8 T cells were stimulated with anti-TCR-β mAb plus anti-CD28 mAb for 48 h, and then the cells were cultured with or without IL-2 for 24 h. c WT and Pgam1 KO naive CD8 T cells were labeled with eFluor670 and stimulated with anti-TCR-β mAb plus anti-CD28 mAb. The cell division was detected by flow cytometry at the indicated number of hours after the initial stimulation. d The immune response of antigen-specific CD8 T cells after Lm-OVA infection was analyzed by staining with an OVA-specific tetramer (Tet). A representative staining profile of Tet/CD8 gated on the CD8-positive cells in the spleen at 7 days post infection (upper). The percentages of cells are indicated in the circle. The absolute number of Tet+ CD8 T cells in the spleen is shown (lower). Each point represents an individual mouse. e Representative results of the intracellular FACS analysis of IFN-γ/IL-2 in the WT and Pgam1 KO CD8 T cells cultured under IL-2 conditions on day 5. The percentages of cells are indicated in each quadrant. f The results of an ELISA for IL-2, IFN-γ, and TNF-α in the supernatants of the cells in e (n = 3, biological replicate). The results of the analyses are representative of at least three independent experiments with similar results. The results are indicated with the standard deviation. *P < 0.05, **P < 0.01 (Student’s t-test).
We next assessed the effector differentiation in Pgam1 KO CD8 T cells in vitro. The production of IL-2, interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) in Pgam1 KO CD8 T cells was significantly lower than in wild-type CD8 T cells (Fig. 2e, f). The impairment of effector differentiation in Pgam1 KO CD8 T cells was not restored by the supplementation of pyruvate, the end product of glycolysis (Supplementary Fig. 6a, b). These results suggest that glycolysis is required for maximal proliferation and effector differentiation of CD8 T cells.
Impaired TH2 and TH17 cell differentiation in Pgam1 KO naïve CD4 T cells
We next focused on the role of glycolysis in effector CD4 T cell development. In contrast to Pgam1-deficient CD8 T cells, Pgam1 KO CD4 T cells showed only a moderate reduction in CD25 (Supplementary Fig. 7a). The expression of CD44 and CD69 in Pgam1 KO CD4 T cells was comparable to that in wild-type CD4 T cells, whereas the expression of CD40L showed a marked reduction (Supplementary Fig. 7a). The TCR-dependent proliferative response was decreased in Pgam1 KO CD4 T cells compared to wild-type CD4 T cells in vitro (Supplementary Fig. 7b). Consistent with the decreased expression of CD40L in Pgam1 KO activated CD4 T cells, the serum levels of dinitrophenyl (DNP)-specific IgM, IgG1, and IgG2c but not IgA in 2,4-dinitrophenylated ovalbumin (DNP-OVA)-immunized Pgam1 KO mice were lower than in wild-type mice (Supplementary Fig. 8).
We next assessed the differentiation to effector helper T (Th) cell subsets in Pgam1 KO naïve CD4 T cells in vitro. The generation of IFNγ-producing TH1 cells was moderately decreased in the Pgam1 KO naïve CD4 T cells compared to the wild-type naïve CD4 T cells (Fig. 3a). The production of IFN-γ and TNF-α in in vitro differentiated Pgam1 KO TH1 cells was decreased to half, whereas the production of IL-2 was equivalent to that in wild-type TH1 cells (Fig. 3b). The generation of IL-4-producing TH2 cells was severely decreased, and the induction of IFN-γ-producing cells was detected in Pgam1 KO naïve CD4 T cells cultured under TH2 conditions (Fig. 3c). Although the decreased IL-4 production was confirmed by an enzyme-linked immunosorbent assay (ELISA), the production of IL-5 and IL-13 showed only a moderate reduction in Pgam1 KO TH2 cells (Fig. 3d). Pgam1 KO naïve CD4 T cells cultured under TH17 conditions showed a striking reduction in the generation of IL-17A-producing cells compared with wild-type naive CD4 T cells (Fig. 3e). The generation of Foxp3-positive regulatory T (Treg) cells was slightly decreased, so the ratio of Foxp3-postive cells and IL-17A-producing cells was increased (Fig. 3e). The decreased production of IL-17A and IL-17F was confirmed by an ELISA (Fig. 3f). Although the production of TH1 cytokine (IFN-γ and TNF-α) in Pgam1 KO CD4 T cells was restored by the supplementation of pyruvate (Fig. 3b), TH2 and TH17 cell differentiation in Pgam1 KO CD4 T cells was insensitive to pyruvate supplementation (Fig. 3d, f). Interestingly, the production of IL-2 was suppressed by pyruvate supplementation in both wild-type and Pgam1 KO Th1 cells (Fig. 3b).

a Representative results of the intracellular FACS analysis of IFN-γ/IL-4 in the WT and Pgam1 KO CD4 T cells cultured under Th1 conditions for 5 days. b WT and Pgam1 KO CD4 T cells cultured under Th1 conditions in the presence or absence of pyruvate for 5 days. Then the cells were restimulated with immobilized ani-TCRβ mAb for 16 h. The amounts of IL-2, IFN-γ, and TNF-α in the supernatants was determined by an ELISA (n = 3, biological replicate). c Representative results of the intracellular FACS analysis of IFN-γ/IL-4 in the WT and Pgam1 KO CD4 T cells cultured under Th2 conditions for 5 days. d WT and Pgam1 KO CD4 T cells cultured under Th2 conditions in the presence or absence of pyruvate for 5 days. Then the cells were restimulated with immobilized ani-TCRβ mAb for 16 h. The amounts of IL-4, IL-5, and IL-13 in the supernatants was determined by an ELISA (n = 3, biological replicate). e Representative results of the intracellular FACS analysis of Foxp3/IL-17A in the WT and Pgam1 KO CD4 T cells cultured under Th17 condition for 3 days (left panel). The ratio of Foxp3-positive cells/IL-17A-positive cells is shown in the right panel. f WT and Pgam1 KO CD4 T cells cultured under Th17 conditions in the presence or absence of pyruvate for 3 days. Then the cells were restimulated with immobilized ani-TCRβ mAb for 16 h. The amounts of IL-17A and IL-17F in the supernatants was determined by an ELISA (n = 3, biological replicate). The results of the FACS analyses are representative of at least three independent experiments with similar results. The percentages of cells are indicated in each quadrant. The results are indicated with the standard deviation. *P < 0.05, **P < 0.01 (Student’s t-test).
Attenuated helper T-cell-dependent inflammation in Pgam1 KO mice
Next, we assessed TH cell-dependent inflammatory response in Pgam1 KO mice. To assess the impact of glycolytic inhibition on the in vivo TH2 immune-response, allergic airway inflammation was induced in wild-type and Pgam1 KO mice by the intranasal administration of OVA following immunization with OVA. A significant decrease in mononuclear cells infiltrating the peribronchiolar regions of the lungs was observed in the Pgam1 KO mice (Fig. 4a). The bronchioles of the Pgam1-deficient mice showed less mucus production and goblet cell neoplasia than wild-type mice, as assessed using staining with periodic acid-Schiff reagent (Fig. 4a). Decreased mucus hyper-production was confirmed by the reduction in muc5ac and mclca3 mRNA in the lungs of Pgam1 KO mice (Fig. 4b). The infiltration of inflammatory cells, including eosinophils, neutrophils, and lymphocytes, in the bronchoalveolar lavage (BAL) fluid was significantly reduced in OVA-challenged Pgam1 KO mice compared to OVA-challenged wild-type mice (Fig. 4c and Supplementary Fig. 9a). The effect of Pgam1 deficiency on the TH1/TH17-mediated autoimmune responses was determined using an experimental autoimmune encephalitis (EAE) model in vivo. As shown in Fig. 4d, Pgam1 KO mice did not develop any signs of disease, while all wild-type mice developed such signs. A reduction in the body weight was observed in wild-type mice but not in Pgam1 KO mice (Fig. 4d). In addition, the infiltration of T cells (CD3ε-positive cells) into the spinal cord was not detected, and the number of infiltrated cells was decreased in Pgam1 KO mice (Fig. 4e). The reduction of infiltrating cells was also confirmed by hematoxylin and eosin staining of spinal code (Fig. 4f and Supplementary Fig. 9b).

a–c WT and T-cell-specific Pgam1 KO mice were immunized intraperitoneally with 100 μg OVA in 2 mg of aluminum hydroxide gel. The immunized mice were intranasally challenged with OVA in saline (100 μg per mouse) on days 7, 8, and 9 to induce allergic airway inflammation. Twenty-four hours after the last OVA challenge, the lung samples (a) and BAL fluid samples (c) were prepared. a The microscopic appearance of the lungs fixed and stained with hematoxylin and eosin (H&E; left panel) or periodic acid-Schiff (PAS) reagent (right panel). Original magnification ×200 (Scale bars = 100 μm). b The results of the quantitative RT-PCR analysis of the muc5ac and clca3 mRNA in the lungs. The results are presented relative to the expression of 18s rRNA. Each point represents an individual mouse. c Quantification of eosinophils, neutrophils, lymphocytes, macrophages, and total cells in the BAL fluid (n = 8 per group). Each point represents an individual mouse. The results are presented relative to the expression of 18s rRNA. Each point represents an individual mouse. d–f WT and Pgam1 KO mice were immunized with MOG35–55 peptides in CFA and pertussis toxin to induce experimental autoimmune encephalomyelitis. d The clinical scores (upper panel) and weight change (lower panel) were indicated (n = 6–7 per group). e Mice were killed on day 17, and the number of CD3+ and total cells in the spinal cord were analyzed by flow cytometry (n = 7 per group). f The microscopic appearance of the spinal cords stained with H&E (left panels; arrows highlight inflammatory foci) or Luxol fast blue (LFB; right panels; arrows highlight demyelinated foci). Scale bars = 100 μm. The results are indicated with the standard deviation. *P < 0.05, **P < 0.01 (Student’s t test).
To assess demyelination and axonal damage within the central nervous system of diseased mice, histological analyses of spinal cord sections were carried out. The extent of demyelination and loss of axons were detected by luxol fast blue staining. Immunized Pgam1 KO mice showed fewer regions of demyelination than wild-type mice (Fig. 4f and Supplementary Fig. 9b). These results demonstrate that TH-mediated inflammation is dependent on the glycolytic activation of CD4 T cells.
Glycolysis supports sustained the activation of mTORC1 signaling
To further investigate the regulatory role of glycolysis in T-cell activation and differentiation, we assessed the activation status of mTORC1 signaling. The phosphorylation status of S6 (Ser235/236) and 4E-BP1 (Thr37/46) proteins, which are downstream targets of mTORC1, in Pgam1 KO naïve CD8 T cells at 8 h after TCR stimulation was equivalent to that in wild-type CD8 T cells (Fig. 5a). However, the phosphorylation levels of both proteins were lower in Pgam1 KO CD8 T cells at 24 h than in wild-type cells (Fig. 5a). Treatment with 2-DG showed similar effects on the phosphorylation of S6 (Ser235/236) protein in wild-type CD8 T cells (Supplementary Fig. 10). mTORC1 controls the protein expression of c-Myc, Hif1α and Srebp1 through post-transcriptional regulation18. As expected, the protein expression of c-Myc, Hif1α, and Srebp1 was decreased in Pgam1 KO CD8 T cells 24 h after TCR stimulation (Fig. 5b). The levels of c-Myc, Hif1α, and Srebp1 mRNA were marginally affected by Pgam1 deficiency (Supplementary Fig. 11a). mTORC1 provokes the expressions of enzymes and transporters that activate glycolysis, glutaminolysis, and fatty acid synthesis via the induction of c-Myc, Hif1α, and Srebp1. The induction of a facilitative glucose transporter slc2a1, hk2, and gapdh was lower in Pgam1 KO activated CD8 T cells than in wild-type activated CD8 T cells (Fig. 5c). Furthermore, the decreased expression of ldha was detected in Pgam1 KO activated CD8 T cells, while the levels of ldhb mRNA were increased at 24 and 48 h, but not 6 h, after TCR stimulation (Supplementary Fig. 11b). The reduction in the ldha/ldhb ratio indicates the attenuation of anaerobic glycolysis in Pgam1 KO activated CD8 T cells (Supplementary Fig. 11b). The mRNA expression of the regulatory targets of c-Myc (gls2) and Srebp1 (acly, elovl6, and scd1) was also attenuated in Pgam1 KO activated CD8 T cells (Fig. 5d, e). These results suggest that TCR-mediated activation of glucose metabolism supports the sustained mTORC1 activation and induces the metabolic reprograming of T cells that is required for clonal expansion and differentiation.

a Representative results of the intracellular FACS analysis of phospho-ribosomal S6 (S6; Ser235/236) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1; Thr37/46) in WT and Pgam1 KO CD8 T cells stimulated with anti-TCR-β mAb plus anti-CD28 mAb for the indicated number of hours. The results of the FACS analyses are representative of at least three independent experiments with similar results. b The results of the immunoblot analysis of c-myc, Hif1α, SREBP1 and β-actin in WT and Pgam1 KO CD8 T cells stimulated with anti-TCR-β mAb plus anti-CD28 mAb for 24 h. The protein amount of β-actin was used as a loading control. The numbers below the bands indicate the densitometry ratio with WT. The results of the immunoblot analyses are representative of at least three independent experiments with similar results and are presented as cropped images. The full-length blots are presented in Supplementary Figure 18. c–e WT and Pgam1 KO CD8 T cells were stimulated with anti-TCR-β mAb plus anti-CD28 mAb for the indicated number of hours, and the mRNA expression of the enzymes for glycolysis (c), glutaminolysis (d), and lipid synthesis (e) were determined by qRT-PCR. The results are presented relative to the expression of 18s rRNA with the standard deviation (n = 3, technical replicates). *P < 0.05, **P < 0.01 (Student’s t test).
Glutamine is required for the sustained activation of mTORC1
Next, we explored the molecular mechanism by which glycolysis induces the sustained activation of mTORC1 signaling. We previously reported that glutamine activates mTORC1 via the glutaminolysis-dependent supplementation of α-KG27. The levels of glutamine and glutamate were lower in Pgam1 KO activated CD8 T cells in comparison to wild-type activated CD8 T cells at 24 h after TCR-stimulation, although those levels at 6 h were equivalent (Fig. 6a). In addition, the glutamine level in the culture medium was higher in Pgam1 KO activated CD8 T cells in comparison to wild-type activated CD8 T cells at 24 h after TCR-stimulation, although those levels were equivalent at 6 h (Supplementary Fig. 12). The intracellular concentrations of several amino acids (His, Ile, Leu, Lys, and Tyr) were moderately affected by Pgam1 deficiency (Supplementary Fig. 13). The reduction of intracellular glutamine and glutamate levels at 24 h after TCR stimulation was also observed by 2-DG treatment (Fig. 6b). These results indicate that glycolysis supports the increased glutamine uptake and subsequent glutaminolysis at 24 h after TCR stimulation in CD8 T cells.

a The intracellular amounts of glutamine and glutamate in WT and Pgam1 KO CD8 T cells stimulated with anti-TCR-β mAb plus anti-CD28 mAb for 6 or 24 h (n = 3–4, biological replicate). b The intracellular amounts of glutamine and glutamate in WT CD8 T cells stimulated with anti-TCR-β mAb plus anti-CD28 mAb in the presence or absence of 10 mM 2-DG for 24 h (n = 3, biological replicate). c WT naïve CD8 T cells were stimulated in the presence or absence of glutamine for 36 h, and then the ECAR was determined. The results are indicated with the standard deviation (right panel; n = 3, technical replicate). d The intracellular levels of lactate in activated CD8 T cells in the presence or absence of glutamine for 24 h are indicated with the standard deviation (n = 3, biological replicate). e Representative results of the intracellular FACS analysis of phospho-S6 (Ser235/236) in CD8 T cells stimulated in the presence or absence of glutamine for the indicated number of hours. f The results of the immunoblot analysis of c-Myc, Hif1α, Srebp1, and β-actin in CD8 T cells cultured in the presence or absence of glutamine and rapamycin for 24 h. The protein amount of β-actin was used as a loading control. The results are presented as cropped images. The numbers below the bands indicate the densitometry ratio with CD8 T cells cultured under Gln (+), Rapa (−) conditions. The results of the immunoblot analyses are representative of at least three independent experiments with similar results. The full-length blots are presented in Supplementary Fig. 18. f The levels of Pgam1, Gapdh, Scd1, and Elovl6 mRNA in CD8 T cells stimulated in the presence or absence of glutamine for the indicated number of hours were determined by the quantitative RT-PCR analysis. The effect of rapamycin on glutamine-dependent alteration was also determined. The results are presented relative to the expression of 18s rRNA with the standard deviations (n = 3, technical replicates). The results of the FACS analyses are representative of at least three independent experiments with similar results. *p < 0.05, **p < 0.01 (Student’s t test).
Increases in the intracellular concentration of glutamine and glutamate in activated CD8 T cells were dependent on extracellular glutamine, since the levels of both amino acids were markedly decreased by the withdrawal of extracellular glutamine (Supplementary Fig. 14a). Glycolysis assessed by ECAR was decreased by the restriction of extracellular glutamine (Fig. 6c). The intracellular concentrations of glycolytic intermediates (F16P, DHAP, glycerophosphate, 3PG, and lactate), acetyl-CoA, and intermediates of TCA cycle (citrate, succinate, fumarate, and malate) were decreased by the depletion of extracellular glutamine in the culture medium (glutamine-deprived conditions; Fig. 6d, Supplementary Fig. 14b, c). NADH/NAD+ ratio in activated CD8 T cells was severely reduced by extracellular glutamine deprivation (Supplementary Fig. 14d). The intracellular levels of nucleotides were also decreased by extracellular glutamine deprivation (Supplementary Fig. 14e). These results indicate that both the glucose and glutamine metabolisms were impaired by glutamine deprivation in activated CD8 T cells in vitro.
Next, the effect of glutamine deprivation on mTORC1 activation was assessed by the phosphorylation status of S6 and 4E-BP1 proteins as an index. The phosphorylation levels of S6 protein (Fig. 6e) and 4E-BP1 (Supplementary Fig. 15a) in activated CD8 T cells cultured under glutamine-deprived conditions were comparable to those cultured under glutamine-sufficient conditions at 2 and 8 h after TCR stimulation. However, the phosphorylation levels of these proteins at 24 h after stimulation were lower in activated CD8 T cells cultured under glutamine-deprived conditions than in those cultured under glutamine-sufficient conditions (Fig. 6e and Supplementary Fig. 15a). Furthermore, the phosphorylation of S6 and 4E-BP1 proteins at 24 h after TCR stimulation was induced in a glutamine dose-dependent manner (Supplementary Fig. 15b). Consistent with the mTORC1 activity, the protein expression of c-Myc, Hif1α, and Srebp1 at 24 h after TCR stimulation was induced by glutamine and inhibited by rapamycin (Fig. 6f). In addition, the mRNA expression of pgam1, gapdh, scd1, and elovl6 was induced in a glutamine-dependent manner in activated CD8 T cells and inhibited by rapamycin treatment (Fig. 6g). These results suggest that glutamine is required for the sustained activation of the mTORC1 signaling pathway in activated CD8 T cells.
Glutamine controls TCR- and IL-2-mediated signals
Finally, we examined whether or not glutamine regulates TCR- and IL-2-mediated signals in activated CD8 T cells, since these signals are impaired in Pgam1 KO activated CD8 T cells. The intracellular concentration of ATP was increased in a glutamine-dependent manner at 24 h after TCR stimulation. In contrast, the intracellular ATP concentration at 6 h after activation was less affected by glutamine (Fig. 7a). The protein-tyrosine phosphorylation in activated CD8 T cells was increased by extracellular glutamine at 24 h after TCR stimulation (Fig. 7b), and the glutamine-dependent upregulation of tyrosine-phosphorylation was inhibited by rapamycin (Fig. 7c). The tyrosine phosphorylation at 6 h after TCR stimulation was glutamine-independent (Fig. 7b) and rapamycin-insensitive (Fig. 7c). The tyrosine phosphorylation of the TCR signaling molecules Zap70 (Tyr319) and Plcγ1 (Tyr783) at 24 h after TCR stimulation was also enhanced by glutamine, and the increased tyrosine phosphorylation was inhibited by rapamycin (Fig. 7d, e). The phosphorylation of Zap70 and Plcγ1 at 6 h after TCR stimulation was induced in a glutamine-independent and rapamycin-insensitive manner (Fig. 7d, e).

a The intracellular amounts of ATP in CD8 T cells stimulated with anti-TCR-β mAb plus anti-CD28 mAb in the presence or absence of glutamine for the indicated number of hours. The results are presented with the standard deviations (n = 3, biological replicate). b The results of the immunoblot analysis of protein-tyrosine phosphorylation and β-actin in naïve CD8 T cells cultured with or without glutamine for the indicated number of hours. The protein amount of β-actin was used as a loading control. The numbers below the bands indicate the densitometry ratio with CD8 T cells cultured with glutamine. c The effect of rapamycin on glutamine-induced protein-tyrosine phosphorylation. Naïve CD8 T cells were stimulated under glutamine-sufficient conditions in the presence or absence of rapamycin for the indicated number of hours. The numbers below the bands indicate the densitometry ratio with CD8 T cells cultured without rapamycin. d The results of the immunoblot analysis of phospho-Zap70 (Tyr319), phosphor-Plcγ1 (Tyr783) and β-actin (control) in cells in (b). The numbers below the bands indicate the densitometry ratio with CD8 T cells cultured with glutamine. e The results of the immunoblot analysis of phospho-Zap70 (Tyr319), phosphor-Plcγ1 (Tyr783) and β-actin (control) in cells in (c). The numbers below the bands indicate the densitometry ratio with CD8 T cells cultured without rapamycin. f Representative results of the FACS analysis of CD25 and CD69 in CD8 T cells stimulated under the indicated conditions for 48 h. g Representative results of the intracellular FACS analysis of phosphor-Stat5 in CD8 T cells cultured with or without IL-2 stimulation. Naïve CD8 T cells were stimulated with anti-TCR-β mAb plus anti-CD28 mAb for 48 h, and then the cells were cultured with or without IL-2 for 24 h. The results of the FACS and immunoblot analyses are representative of at least three independent experiments with similar results. The results of the immunoblot analyses are presented as cropped images. The full-length blots are presented in Supplementary Fig. 18. *p < 0.05, **p < 0.01 (Student’s t test).
We showed that glucose metabolism controls IL-2 signaling by regulating the CD25 expression (Fig. 2a, b). Therefore, we investigated the glutamine dependency of IL-2 signaling in activated CD8 T cells. The expression of CD25 was decreased under glutamine-deprived conditions, whereas the CD69 expression was not influenced (Fig. 7f). Furthermore, the IL-2-induced tyrosine phosphorylation of Stat5 was impaired in activated CD8 T cells cultured under glutamine-deprived conditions (Fig. 7g). Consistent with these results, the effector cytokine production in CD8 T cells cultured under glutamine-deprived conditions was markedly decreased compared with that in cells cultured under glutamine-sufficient conditions (Supplementary Fig. 16a, b). These results suggest that glutamine is required to sustain TCR- and IL-2-mediated signaling as well as glycolysis (Supplementary Fig. 17).
Discussion
The mTORC1-mediated regulation of c-Myc, Hif1α, and Srebp1 protein expression has been reported19,30. In this study, we demonstrated that glutamine is required for c-Myc, Hif1α, and Srebp1 protein expression and sustained activation of mTORC1 signaling. Glutamine-induced c-Myc, Hif1α, and Srebp1 protein expression and the subsequent mRNA expression of glycolytic enzymes in activated CD8 T cells was inhibited by rapamycin, indicating that glutamine controls glycolysis through the regulation of the mTORC1 activity. The uptake of l-glutamine is the rate-limiting step in the essential amino acid- and growth factor-dependent activation of mTORC1. The counter transport of l-glutamine and l-leucine through the solute carrier family 7 member 5 (SLC7A5)/SLC3A2 complex controls mTORC1 activity31. Furthermore, we and others have reported that glutamine activates mTORC1 signaling via the supplementation of αKG27,32. We also found that the TCR-mediated glycolytic activation is required to sustain mTORC1 activity and to regulate the glutamine uptake in activated CD8 T cells. The inhibitions of glycolysis, glutamine uptake resulted in the impaired proliferation and effector differentiation of T cells. A previous report revealed that c-Myc sorting occurred asymmetrically during the first division33. c-Myclow cells do not show metabolic reprogramming or proliferative burst. The c-Myc-dependent regulation of the SLC1A5 expression and sustained mTORC1 activity was also reported8,33. Conversely, the transport of glutamine through SLC7A5, but not glutaminolysis, regulates the c-Myc expression in NK cells34. We demonstrated that the c-Myc protein expression was reduced by both the deletion of the Pgam1 and glutamine-depletion in activated CD8 T cells. The glutamine-dependent expression of the c-Myc protein was rapamycin-sensitive. Thus, these findings suggested that mTORC1, glycolysis, glutamine, and c-Myc affect each other and cooperate to induce proliferation, differentiation, and a T-cell-dependent immune response. Thus, the loss of Pgam1 influences the T-cell-dependent immune response through multiple pathways.
We showed in the present study that TCR signaling assessed by the tyrosine phosphorylation levels of Zap70 and Plcγ was not augmented in Pgam1 KO activated CD8 T cells. TCR-mediated protein-tyrosine phosphorylation did not show a time-dependent increase in Pgam1 KO naïve CD8 T cells. Glutamine was required for the augmentation of the TCR signaling and protein-tyrosine phosphorylation in activated CD8 T cells, and the effect of glutamine was antagonized by rapamycin. The upregulation of the intracellular ATP was also impaired by Pgam1 deficiency and glutamine restriction. These results suggest that both glycolysis and glutamine are required to activate signal transduction in activated T cells via the supplementation of metabolic products, such as ATP. Furthermore, ATP regulates the activity of ATP-dependent chromatin remodelers that control cellular differentiation35. ATP-dependent chromatin alteration is accomplished by chromatin-remodeling complexes that use the energy derived from ATP hydrolysis to alter nucleosome structure and conformation. Changes in nucleosomes control transcription by regulating the accessibility of transcription factors36. Metabolic products, such as acetyl-CoA, αKG, S-adenosylmethionine, NAD+, succinate, and fumarate, are known to be used as substrates and cofactors of enzymes that regulate epigenetic modifications37. It is possible that mTORC1, glycolysis, and glutamine also control T cell differentiation by regulating cellular signals and the epigenetic status.
Pgam1 is reported to mediate PPP and serine synthesis through 3-PG and 2-PG in cancer cells10, however, Pgam1 KO CD8 T cells did not show the reduction of PPP intermediates and serine. Although the causes of these inconsistency need further elucidation, the accumulation of upstream metabolites, glucose-6-phosphate and 3-PG, may diminish those effects. On the other hand, the intracellular nucleotide concentration strikingly decreased in Pgam1 KO CD8 T cells after 24 h from stimulation, despite the normal PPP activity. This is thought to be caused by the reduction of intracellular glutamine and ATP, which are essential for de novo nucleotide synthesis from ribose-5-phosphate. To support this observation, glutamine-deprivation also reduced the intracellular nucleotide concentration in CD8 T cells.
Pgam1 deficiency reduced OXPHOS in both CD4 and CD8 T cells at 24 h after TCR-stimulation. Although activated T cells dominantly rely on aerobic glycolysis to generate ATP, OXPHOS also increases activity during metabolic reprograming after TCR-stimulation in an mTOR-dependent manner38,39. Thus, it is hypothesized that the decreased mTORC1 activity, due to glycolytic failure, attenuated OXPHOS in Pgam1 KO T cells. Interestingly, Pagm1 KO T cells did not respond to proton ionophore FCCP at 24 h after stimulation. NADH and succinate are required to generate a proton gradient across the mitochondrial membrane via mitochondria respiratory complex I and II, respectively. The NADH and NAD+ levels were both decreased in Pgam1 KO activated CD8 T cells. The decreased purine nucleotide level possibly resulted in the reduction of the NADH and NAD+ levels. In addition, the intracellular levels of succinate and fumarate were also decreased in Pgam1 KO activated CD8 T cells. It is possible that the reduction of NADH and succinate, which are required to generate a proton gradient across the mitochondrial membrane, limits electron transport chain activity in Pgam1 KO CD8 T cells and causes poor responsiveness to FCCP.
The TCR-dependent expression of CD25 was inhibited by Pgam1 deficiency. The IL-2 responsiveness assessed by tyrosine phosphorylation of Stat5 was impaired in Pgam1 KO activated CD8 T cells. Furthermore, the expression of CD25 was low in activated CD8 T cells cultured under glutamine-restricted conditions compared to that in cells cultured under glutamine-sufficient conditions. The decreased expression of CD25 in Rptor, the scaffolding protein of mTORC1 complex, -deficient TH2 cells was previously reported19. Similar to that observed in Pgam1 KO activated T cells, the expression of CD44 and CD69 was unchanged in Rptor KO T cells. In addition to the Rptor and Pgam1 KO activated CD8 T cells, the mTORC1 activity was also abrogated in activated CD8 T cells under glutamine-restricted conditions. These results suggest that the responsiveness to IL-2 and the proliferative response of activated CD8 T cells are dependent on mTORC1, glycolysis, and glutamine, all of which are indispensable.
The differentiation of TH1, TH2, and TH17 was impaired in Pgam1 KO naïve CD4 T cells, whereas the Foxp3-positive Treg cell generation was only marginally affected. The TH17/Treg ratio was reduced in Pgam1 KO CD4 T cell under TH17 conditions compared to wild-type CD4 T cells. These results were consistent with a previous observation in which effector T cell development was found to depend on anaerobic glycolysis, while Treg cells preferentially used β-oxidation40,41,42,43. However, the reduction of IFN-γ-producing TH1 cells in Pgam1-deficient CD4 T cells was milder than that in IL-4-producing TH2 cells and IL-17-producing TH17 cells in our experiments. In addition, the reduced generation of IFN-γ-producing TH1 cells in Pgam1-deficient CD4 T cells was restored by the supplementation of pyruvate in vitro, whereas the development of TH2 and TH17 cells was not. We demonstrated that OXHOS was reduced in Pgam1-deficient activated CD4 T cells; thus, it is possible that OXPHOS also plays an important role in TH1 differentiation. These results suggest that glycolysis induces the differentiation of TH subsets through various different pathways.
Several studies have shown that the generation of Treg can be enhanced by rapamycin in vitro and in vivo44,45,46. Furthermore, the genetic deletion of components of the mTORC signaling pathway in T cells results in the enhanced generation of Tregs and decreased effector T cell development47,48. However, Treg-specific Rptor KO mice developed systemic autoimmunity, suggesting that the mTORC1 activity is required for the Treg function49. The number of Tregs was decreased in the spleen of Pgam1 KO mice, and we did not examine the function of Pgam-deficient Treg cells. Recently, the mTORC2-dependent TH1, TH2 and Treg differentiation and functions have been reported50,51. In the present study, we did not assess the effect of Pgam1 deficiency or glutamine-deprivation on the mTORC and Akt activity in CD4 T cells. It is likely that glycolysis and glutamine also regulate the mTORC2 activity via the supplementation of ATP. Thus, the interaction between mTOR, glycolysis, and glutamine in CD4 T cells should be further clarified in the future.
Methods
Mice
CD4-Cre transgenic (TG) mice, OT-1 Tg mice, Thy1.1+ mice, and Thy1.2+ mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Pgam1flox/flox mice were kindly provided by Drs. Hiroshi Kondoh and Takumi Mikawa (Kyoto University). In brief, the exon 2–4 of pgam1 was pinched by two loxP sequences, and FLP-flanked neo-cassette was inserted in the 5′ upstream region of exon 2 to generate the pgam1 targeting vector. Targeting vector was injected into C57BL/6 mouse embryonic stem cells. The founder mice were crossed with FLP transgenic mice to remove the neo-cassette gene to generate Pgam1-floxed mice. Pgam1-floxed mice were crossed with CD4-Cre TG mice to generate T-cell-specific Pgam1-deficient mice. Gene-manipulated mice with a C57BL/6 background were used in all experiments. These mice were purchased from Clea Japan, Inc. (Tokyo, Japan). Both male and female mice were used in the experiments. All mice were used at 8–12 weeks of age.
All of the animal experiments received approval from the Ehime University Administrative Panel for Animal Care. All animal care was conducted in accordance with the guidelines of Ehime University. All experiments using Lm-OVA were performed in accordance with the protocols approved by the Ehime University Institution Biosafety Committee.
Reagents and antibodies
Rapamycin was purchased from Wako Chemicals (cat# R0161; Osaka, Japan). Antibodies used for intracellular and cell-surface staining were as follows: anti-CD8-violetFluor 450 mAb (0.066 μg/106 cells, 2.43; TONBO Biosciences, San Diego, CA, USA), anti-CD4-fluorescein isothiocyanate (FITC), mAb (0.04 μg/106 cells, cat# 553047; BD Biosciences, San Jose, CA, USA), anti-TNF-α-phycoerythrin (PE), mAb (0.08 μg/106 cells, cat# 554419; BD Biosciences), anti-IFN-γ-FITC mAb (0.025 μg/106 cells, cat# 554411; BD Biosciences), IL-2- allophycocyanin (APC), mAb (0.01 μg/106 cells, cat# JES6-5H4; TONBO Biosciences), anti-Phospho-S6 (Ser235/236)-Alexa Fluor 647 mAb (0.125 μg/106 cells, cat# 4851; Cell Signaling Technology, Danvers, MA, USA), anti-4E-BP1 (Thr37/46)-Alexa Fluor 488 mAb (0.05 μg/106 cells, 236B4; cat# 2846 S; Cell Signaling Technology), anti-phospho-STAT5 (Tyr694)-Alexa Fluor 647 mAb (20 μl/test/106 cells, cat# 612599; BD Biosciences), anti-CD44-PE mAb (0.02 μg/106 cells, IM7; TONBO Biosciences), anti-CD62L-APC mAb (0.02 μg/106 cells, cat# 20-0621; TONBO Biosciences), anti-CD25-PE mAb (0.066 μg/106 cells, 3C7; BD Biosciences), anti-CD40L-PE mAb (0.02 μg/106 cells, cat# 106505; BioLegend), anti-Thy1.1-Alexa Fluor 647 mAb (0.005 μg/106 cells, OX-7; BioLegend), anti-Thy1.2-APC-Cy7 mAb (0.02 μg/106 cells, 30-H12; BioLegend), anti-IL-4-PE mAb (0.066 μg/106 cells, cat# 554435; BD Bioscience), anti-IL17A-PE mAb (0.066 μg/106 cells, cat# 506904; BioLegend), and anti-Foxp3- Alexa Fluor 647 mAb (0.25 μg/106 cells, cat# 320014; BioLegend).
The antibodies for immunoblotting were as follows: anti-phospho-tyrosin mAb (1:1000, cat# 8954 S; Cell Signaling Technology), anti-phospho-zap70 (Y319/352) mAb (1:1000, cat# 2717 T; Cell Signaling Technology), anti-phospho-PLCγ1 (Y783) mAb (1:1000, cat# 14008 T; Cell Signaling Technology), anti-β-actin Ab (1:1000, cat# 4970SS; Cell Signaling Technology), anti-c-Myc mAb (1:1000, cat# 5605 S; Cell Signaling Technology), anti-Hif1α Ab (1:2000, A300-286A; Bethyl Laboratories, Montgomery, AL, USA), and anti-Srebp1 mAb (1:200, cat# 13551; Santa Cruz Biotechnology, Dallas, TX, USA). All antibodies were diluted and used according to the manufacturer’s instructions.
T-cell stimulation and differentiation in vitro
Naive CD8 T (CD44lowCD62Lhigh) cells and naive CD4 T (CD44lowCD62LhighCD25negative) cells were prepared using a Naive CD8+ T-cell Isolation kit (cat# 130-096-543; Miltenyi Biotec, San Diego, CA, USA) and a Naive CD4+ T-cell Isolation kit (cat# 130-093-227; Miltenyi Biotec), respectively. Naive T cells (7.5 × 105) were stimulated with immobilized anti-TCR-β mAb (3 μg/ml, H57-597; BioLegend) and anti-CD28 mAb (1 μg/ml, 37.5; BioLegend) for 2 days. The cells were then transferred to a new plate and further cultured.
The cytokine conditions were as follows: IL-2 conditions, IL-2 (10 ng/ml); Th1 conditions, IL-2 (10 ng/ml), IL-12 (1 ng/ml), and anti-IL-4 mAb (5 mg/ml, 11B11; BioLegend); Th2 conditions, IL-2 (10 ng/ml), IL-4 (1 ng/ml), and anti-IFN-γ mAb (5 mg/ml, R4-6A2; BioLegend); Th17 conditions, IL-6 (10 ng/ml), IL-1β (10 ng/ml), TGF-β1 (1 ng/ml), anti-IL-4 mAb (5 mg/ml), and anti-IFN-γ mAb (5 mg/ml). The cells were cultured in RPMI 1640 with l-glutamine (cat# 189-02025; Wako Chemicals) supplemented with 10% fetal bovine serum, 2 mM l-glutamine (16948-04; Nacalai Tesque, Kyoto, Japan), 1% MEM nonessential amino acids (cat# 06344-56; Nacalai Tesque), 10 mM HEPES (cat# 15630-080; Thermo Fisher Scientific, Waltham, MA, USA), 55 μM 2-Mercaptoethanol (cat# 21985-023; Thermo Fisher Scientific), and 1% penicillin-streptomycin (cat# 26253-84; Nacalai Tesque).
For glutamine-deprived conditions, the cells were cultured in RPMI 1640 without l-glutamine (cat# 183-02165; Wako Chemicals) supplemented with 10% fetal bovine serum, 1% MEM nonessential amino acids, 10 mM HEPES, 55 μM 2-mercaptoethanol, and 1% penicillin-streptomycin. The medium was basically deprived of pyruvate, an end product of the glycolytic pathway.
Intracellular staining of cytokines
The cells were differentiated in vitro and stimulated with an immobilized anti-TCR-β mAb (3 μg/ml, H57-597; BioLegend) for 6 h with monensin (2 μM, cat# M5273; Sigma-Aldrich, St. Louis, MO, USA) for the intracellular staining of cytokines. Intracellular staining was then performed as described previously27,52. For the intracellular staining of phosphorylated S6 ribosomal proteins, the CD8 T cells were fixed and permeabilized with BD Phosflow Lyse/Fix Buffer (cat# 558049; BD Biosciences) and BD Phosflow Perm III (cat# 558050; BD Biosciences) in accordance with the manufacturer’s instructions. Flow cytometry (FACS) was performed using a FACS Caliber instrument (BD Biosciences) and Gallios instrument (Beckman Coulter, CA, USA), and the results were analyzed using the FlowJo software program (Tree Star, Ashland, OR, USA).
ELISA
The cells were stimulated with an immobilized anti-TCR-β mAb (3 μl/ml) for 16 h. The concentrations of TNF-α, IL-13, IL-17A, and IL-17F were determined using the DuoSet ELISA kits (R&D Systems, Minneapolis, MN, USA) in accordance with the manufacturer’s instructions. The concentrations of IL-2, IFN-γ, IL-4, and IL-5 in the supernatants were determined using ELISAs, as described previously52.
Quantitative reverse transcriptase polymerase chain reaction
Total RNA was isolated using TRIzol regent, and complementary DNA (cDNA) was synthesized using the superscript VILO cDNA synthesis kit (cat# 11754; Thermo Fisher Scientific). Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was performed using the Step One Plus Real-Time PCR System (Thermo Fisher Scientific).
Immunoblot analyses
Nuclear and cytoplasmic extracts were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (cat# 78833; Thermo Fisher Scientific). The lysates were separated on an SDS polyacrylamide gel and then subjected to immunoblotting with specific antibodies. The western blot bands were quantified using the Image J software program (http://rsb.info.nih.gov/ij).
Metabolic profiling
Metabolome measurements and data processing were performed through a facility service at Human Metabolome Technology, Inc. (Yamagata, Japan) as we described previously27.
In brief, WT and Pgam1 KO naïve CD8 T cells were stimulated with plate-bound anti-TCR-β mAb plus anti-CD28 mAb in the presence or absence of glutamine for 24 h. The cells (5 × 106 cells) were washed with 5% (w/w) mannitol and then lysed with 800 μl of methanol and 500 μl of Milli-Q water containing internal standards (H3304-1002; Human Metabolome Technology, Inc.) and left to rest for another 30 s. The extract was obtained and centrifuged at 2300 × g at 4 °C for 5 min, and then 800 μl of the upper aqueous layer was centrifugally filtered through a Millipore 5-kDa cut-off filter at 9100 × g at 4 °C for 120 min to remove proteins. The filtrate was centrifugally concentrated and re-suspended in 50 μl of Milli-Q water for capillary electrophoresis-mass spectrometry (CE-MS). Cationic compounds were measured in the positive mode of capillary electrophoresis-time of flight-mass spectrometry (CE-TOFMS), and anionic compounds were measured in the positive and negative modes of capillary electrophoresis-tandem mass spectrometry (CE-MS/MS), in accordance with the methods developed by Soga et al53,54.
The ECAR and OCR were measured using an Extracellular Flux Analyzer XFp (Agilent Technologies, Santa Clara, CA, USA). The culture medium was changed to Seahorse XF RPMI Base Medium (Cat# 103336-100; Agilent Technologies) before the analysis. Activated CD8 T cells (1.5 × 105 at 8 h and 1 × 105 at 24 h after activation) or CD4 T cells (1.5 × 105) were adhered to Cell Tak coated seahorse eight-well plates and pre-incubated at 37 °C for 45 min in the absence of CO2. The ECAR was then measured at the baseline and in response to 10 mM glucose, 1 μM oligomycin, and 50 mM 2-DG (XFp glycolysis stress test kit; cat# 103017-100; Agilent Technologies). The OCR was measured under basal conditions and in response to 1 μM oligomycin, 2 μM FCCP and 0.5 μM rotenone/antimycin A (XFp mito stress test kit; cat# 103010-100; Agilent Technologies).
Adoptive transfer of CD8 T cells and Listeria infection
Naïve CD8 T cells were prepared from the spleens of wild-type OT-1 Tg (Thy1.1+) or Pgam1 KO OT-1 Tg mice (Thy1.2+). The purified cells were mixed at a 1:1 ratio (wild-type/Pgam1 KO) and intravenously transferred into naive C57BL/6 (Thy1.1+Thy1.2+) mice (1 × 104 cells per mouse). The mice were then infected with the Listeria monocytogenes-expressing OVA (Lm-OVA) strain at 5 × 103 colony-forming units (CFU) intravenously 18–24 h later. The donor cells in the spleen were gated, and the expansion was assessed on days 5 and 7 after Lm-OVA infection.
EAE induction in mice
Wild-type and T cell-specific Pgam1 KO mice were immunized subcutaneously with 200 μg MOG35-55 peptide in CFA. The mice were infected with pertussis toxin (100 ng) intraperitoneally on days 0 and 2, and monitored for the EAE clinical score and weight changes. Possible EAE scores were 0 = normal, 1 = flaccid tail, 2 = hind limb weakness, 3 = partial hind limb paralysis, 4 = complete hind limb paralysis, and 5 = moribund state. For FACS and histological analyses, mice were killed on day 17.
OVA-induced allergic inflammation in mice
Wild-type and T-cell-specific Pgam1 KO mice were immunized intraperitoneally with 100 µg OVA in 2 mg of aluminum hydroxide gel on day 0. Next, the mice were intranasally challenged with OVA in saline (100 µg per mouse) on days 7, 8, and 9. Twenty-four hours after the last OVA challenge, BAL-fluid cells and lung samples were prepared for a histological examination.
Primers and probes for qRT-PCR
The primer and TaqMan probes used for the detection of Gapdh were purchased from Thermo Fisher Scientific. The specific primers and Roche Universal Probes were as follows: acly: 5′-AAGAAGGAGGGGAAGCTGAT-3′ (forward), 5′-TCGCATGTCTGGGTTGTTTA-3′ (reverse), probe#105 c-myc: 5′-CCTAGTGCTGCATGAGGAGA-3′ (forward), 5′-TCCACAGACACCACATCAATTT-3′ (reverse), probe#77, elovl6: 5′-GCAAAGCACCCGAACTAGG-3′ (forward), 5′-GAGACACAGTGATGTGGTGGT-3′ (reverse), probe #4, gls2: 5′-GTATGACTTCTCGGGCCAGT-3′ (forward), 5′-TCCTGACCCAGCTGACTTGG-3′ (reverse), probe #97, hif1a: 5′-GGCTATCCACATCAAAGCAA-3′ (forward), 5′-GCACTAGACAAAGTTCACCTGAGA-3′ (reverse), probe #95, hk2: 5′-CAACTCCGGATGGGACAG-3′ (forward), 5′-CACACGGAAGTTGGTTCCTC-3′ (reverse), probe #21, ldha: 5′-GGCACTGACGCAGACAAG-3′ (forward), 5′-TGATCACCTCGTAGGCACTG-3′ (reverse), probe #12, ldhb: 5′-GCCCTGAGTCCAGGAAGTAAG-3′ (forward), 5′-CGATGGCTCCTGTGAGTTTT-3′ (reverse), probe #63, pgam1: 5′-AGTTCCTGGGAGACGACGAG-3′ (forward), 5′-CTGCCTCTTCACCTTCACTTC-3′ (reverse), probe #5, scd1: 5′-TTCCCTCCTGCAAGCTCTAC-3′ (forward), 5′-CAGAGCGCTGGTCATGTAGT-3′ (reverse), probe #34, pgam2: 5′-AACCTCATGGGCTTGG-3′ (forward), 5′-GGCATTGTGAAACATC -3′ (reverse), probe #93, scd1: 5′-TTCCCTCCTGCAAGCTCTAC-3′ (forward), 5′-CAGAGCGCTGGTCATGTAGT-3′ (reverse), probe #34, srebf1: 5′-GGYYYGAACGACATCGAAGA-3′ (forward), 5′-CGGGAAGTCACTGTCTTGGT-3′ (reverse), probe #78, slc2a1: 5′-GACCCTGCACCTCATTGGT-3′ (forward), 5′-GATGCTCAGATAGGACATCCAAG-3′ (reverse), probe #99, and 18s: 5′-GCAATTATTCCCCATGAACG-3′ (forward), and 5′-GGGACTTAATCAACGCAAGC-3′ (reverse), probe #48.
Statistics and reproducibility
Student’s t test was used for the statistical analyses. Experiments were independently repeated at least three times in general as mentioned in figure legends.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. Source data can be found in Supplementary Data 1.
References
Ganeshan, K. & Chawla, A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 32, 609–634 (2014).
Pearce, E. L., Poffenberger, M. C., Chang, C. H. & Jones, R. G. Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454 (2013).
Pollizzi, K. N. & Powell, J. D. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 14, 435–446 (2014).
MacIver, N. J., Michalek, R. D. & Rathmell, J. C. Metabolic regulation of T lymphocytes. Annu Rev. Immunol. 31, 259–283 (2013).
Rathmell, J. C., Vander Heiden, M. G., Harris, M. H., Frauwirth, K. A. & Thompson, C. B. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol. Cell 6, 683–692 (2000).
Buck, M. D., O’Sullivan, D. & Pearce, E. L. T cell metabolism drives immunity. J. Exp. Med. 212, 1345–1360 (2015).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Wang, R. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Assmann, N. & Finlay, D. K. Metabolic regulation of immune responses: therapeutic opportunities. J. Clin. Invest. 126, 2031–2039 (2016).
Hitosugi, T. et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 22, 585–600 (2012).
Li, C. et al. Expression of PGAM1 in renal clear cell carcinoma and its clinical significance. Int. J. Clin. Exp. Pathol. 8, 9410–9415 (2015).
Peng, X. C. et al. Proteomics identification of PGAM1 as a potential therapeutic target for urothelial bladder cancer. J. Proteom. 132, 85–92 (2016).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Kim, J. & Guan, K. L. Amino acid signaling in TOR activation. Annu. Rev. Biochem. 80, 1001–1032 (2011).
Waickman, A. T. & Powell, J. D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 249, 43–58 (2012).
Yang, K. & Chi, H. mTOR and metabolic pathways in T cell quiescence and functional activation. Semin. Immunol. 24, 421–428 (2012).
Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 12, 325–338 (2012).
Chapman N. M., Boothby M. R., Chi H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 20, 55–70 (2019).
Yang, K. et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity 39, 1043–1056 (2013).
Wang, R. & Green, D. R. Metabolic checkpoints in activated T cells. Nat. Immunol. 13, 907–915 (2012).
Piccirillo, C. A., Bjur, E., Topisirovic, I., Sonenberg, N. & Larsson, O. Translational control of immune responses: from transcripts to translatomes. Nat. Immunol. 15, 503–511 (2014).
DeBerardinis, R. J. & Cheng, T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–324 (2010).
DeBerardinis, R. J. et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl Acad. Sci. USA 104, 19345–19350 (2007).
Carr, E. L. et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 185, 1037–1044 (2010).
DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G. & Thompson, C. B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 (2008).
Mullen, A. R. et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388 (2012).
Suzuki, J. et al. The tumor suppressor menin prevents effector CD8 T-cell dysfunction by targeting mTORC1-dependent metabolic activation. Nat. Commun. 9, 3296 (2018).
Wang, Z. & Cole, P. A. Catalytic mechanisms and regulation of protein kinases. Methods Enzymol. 548, 1–21 (2014).
Quemeneur, L. et al. Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J. Immunol. 170, 4986–4995 (2003).
Duvel, K. et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183 (2010).
Nicklin, P. et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534 (2009).
Carbonneau, M. et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 7, 12700 (2016).
Verbist, K. C. et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 532, 389–393 (2016).
Loftus, R. M. et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 9, 2341 (2018).
Narlikar, G. J., Sundaramoorthy, R. & Owen-Hughes, T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154, 490–503 (2013).
Hota, S. K. & Bruneau, B. G. ATP-dependent chromatin remodeling during mammalian development. Development 143, 2882–2897 (2016).
Reid, M. A., Dai, Z. & Locasale, J. W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 19, 1298–1306 (2017).
Tan, H. et al. Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46, 488–503 (2017).
Ron-Harel, N. et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 24, 104–117 (2016).
Finlay, D. K. et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 209, 2441–2453 (2012).
Michalek, R. D. et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011).
Macintyre, A. N. et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72 (2014).
Jacobs, S. R. et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol. 180, 4476–4486 (2008).
Zeiser, R. et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood 111, 453–462 (2008).
Valmori, D. et al. Rapamycin-mediated enrichment of T cells with regulatory activity in stimulated CD4+ T cell cultures is not due to the selective expansion of naturally occurring regulatory T cells but to the induction of regulatory functions in conventional CD4+ T cells. J. Immunol. 177, 944–949 (2006).
Battaglia, M. et al. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J. Immunol. 177, 8338–8347 (2006).
Delgoffe, G. M. et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844 (2009).
Haxhinasto, S., Mathis, D. & Benoist, C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 205, 565–574 (2008).
Zeng, H. et al. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490 (2013).
Lee, K. et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32, 743–753 (2010).
Charbonnier, L. M. et al. Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat. Immunol. 20, 1208–1219 (2019).
Kuwahara, M. et al. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-beta and suppresses T(H)2 differentiation. Nat. Immunol. 13, 778–786 (2012).
Soga, T. et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 281, 16768–16776 (2006).
Soga, T. et al. Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal. Chem. 81, 6165–6174 (2009).
Acknowledgements
We thank A. Tamai and D. Shimizu for their excellent technical assistance. This work was supported by JSPS KAKENHI Grants (18K08409, 18H05036, 17H04086, 18K05364, 20H035040, and 20H049480), the Kanae Foundation for the Promotion of Medical Science, the Naito Foundation Natural Science Scholarship, the Uehara Memorial Foundation, and the TAKEDA Science Foundation.
Author information
Authors and Affiliations
Contributions
K.T. and M.K. performed the experiments, analyzed the data and wrote the manuscript, N.T., A.K., T.Yorozuya, T.Yamada, T.S., A.S. and M.Y. performed and supported the experiments, H.K. and T.M. established and provided the Pgam1flox/flox mice, and M.Y. conceptualized the research, directed the study, and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Toriyama, K., Kuwahara, M., Kondoh, H. et al. T cell-specific deletion of Pgam1 reveals a critical role for glycolysis in T cell responses. Commun Biol 3, 394 (2020). https://doi.org/10.1038/s42003-020-01122-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-01122-w
This article is cited by
-
CD8+ T cell metabolic flexibility elicited by CD28-ARS2 axis-driven alternative splicing of PKM supports antitumor immunity
Cellular & Molecular Immunology (2024)
-
PRSS37 deficiency leads to impaired energy metabolism in testis and sperm revealed by DIA-based quantitative proteomic analysis
Reproductive Sciences (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
