
Abstract
Periodical cicadas comprise three species groups containing three pairs of 13- and 17-year life cycle species showing parallel divergence, along with a more anciently diverged 13-year species (Magicicda tredecim). The mechanism and genetic basis of this parallel divergence is unknown. Here we use orthologous transcriptome sequences to explore the demographic processes and genomic evolution associated with parallel life cycle divergence. The three 13- and 17-year species pairs have similar demographic histories, and the two life cycles diverged 200,000–100,000 years ago. Interestingly, these life cycle differences have been maintained despite substantial gene flow between 13- and 17-year species within species groups, which is possible during co-emergences. Sequence divergence between 13- and 17-year species in each species group (excluding M. tredecim) is minimal, and we find no shared divergent single-nucleotide polymorphisms (SNPs) or loci associated with all instances of life cycle divergence. The two life cycles may be controlled by highly limited genomic differences.
Similar content being viewed by others


Introduction
Life history diversity is a remarkable feature of living organisms and underlies fundamental evolutionary questions1. Periodical cicadas of the genus Magicicada are found only in the eastern United States and are well known for their unusual life history patterns, characterised by prolonged juvenile periods of 13 or 17 years, followed by synchronised mass emergence of adults within local populations2. Only one cohort, or ‘brood’, of periodical cicadas emerges every 13 or 17 years in any given location. There are three co-occurring species groups of periodical cicadas, Decim, Decula and Cassini. Each has one species with a 17-year life cycle and one or two species with a 13-year cycle, and there are seven described species (four 13-year and three 17-year) in total3,4,5 (Fig. 1). Although the species groups clearly differ in morphology, male songs and female song preferences, the 13-year and 17-year species within each species group are extremely similar or indistinguishable in these characters4,5; thus, the difference in life cycle length is one of the only diagnostic characters for their identification.

Species and species groups of Magicicada periodical cicadas. a Seven Magicicada species in three species groups, with relationships and divergence times for four lineages estimated in our previous study6. Individuals in photographs are all males. Photographs were taken by T. Sota. b Distribution ranges of 13- and 17-year cicadas. The dark pink area in the range of 13-year cicadas indicates the contact zone with M. neotredecim (northern species) and M. tredecim (southern species) in the Decim group5
The three species groups are estimated to have diverged 3.9–2.5 million years ago (mya), and subsequent divergence of the present 13-year (mostly southern) and 17-year (mostly northern) life cycles has occurred in parallel in the three species groups during the Quaternary, except for the first split of the 13-year species, M. tredecim, in the Decim group (0.5 mya) (Fig. 1)6. The synchronisation of prolonged life cycles among species groups is thought to have evolved for a predation-avoidance strategy7, an ecological problem shared among co-occurring species. The divergence of 13-year and 17-year life cycles may have been related to adaptation to climatic changes across glacial cycles; the 4-year extension of juvenile stages may have been advantageous for surviving in cooler northern environments8,9.
The genetic basis of life cycle length has not been studied because the long life cycles complicate genetic crosses. An early explanation for life cycle control in periodical cicadas proposed a one-locus, two-allele system in which either the 13- or the 17-year cycle is dominant10,11. Differences between the two life cycle lengths may be attributable to differences in juvenile developmental rate12,13, which may be regulated by one locus or a small number of loci. However, life cycle regulation in periodical cicadas may not always be strict, because 4-year acceleration and/or deceleration of emergences have been observed in both groups of cicadas, events unlikely to have resulted from fortuitous mass mutation14. These observations have led to the hypothesis that all periodical cicadas possess monomorphic developmental plasticity14 and that this common plasticity underlies the switching of life cycle lengths triggered by environmental cues (e.g., a drastic change in temperature during juvenile development), followed by a genetic change in a life-cycle control locus (genetic accommodation15), which enables a permanent life cycle shift4.
In general, parallelism in adaptive character divergence among closely related species results from parallel mutation or selection, ancestral polymorphism with balancing selection, or adaptive introgression16,17. In periodical cicadas, an ancestral polymorphism in life cycle length followed by collateral genetic evolution16 is considered the most parsimonious explanation for the parallel divergence and the formation of synchronous broods among three species groups, because multiple independent acquisitions of identical life cycles are unlikely6. In addition, a hypothesis of life cycle switching via introgressive hybridisation of the putative 13-year allele from 13- to 17-year cicadas has been proposed10,11,18. This hypothesis was used to explain the existence of two 13-year species in the Decim group18, proposing that introgressive hybridisation from the preexisting 13-year species M. tredecim to the 17-year M. septendecim produced the new 13-year species M. neotredecim. However, the hybrid origin hypothesis of M. neotredecim was rejected based on population genetic studies5,19,20. The hybrid origin hypothesis of 13-year species is unlikely to be applicable to the Cassini and Decula groups, which have no early diverged 13-year species (unless hybridisation between species groups drove life-cycle switching from 17- to 13-year cycles).
To understand the process and genetic basis of the parallel life cycle divergence observed in periodical cicadas, we inferred the demographic histories of broods of three Magicicada species groups. We used reduced representation sequences from transcriptomes (mRNA sequences) because Magicicada genomes are likely large in size (>6 Gbp) as in other cicadas21 and the whole genomes have not yet been sequenced. We focused on three pairs of 13- and 17-year species (excluding M. tredecim, which diverged earlier). In addition, we surveyed the genes responsible for life cycle control by comparing divergent loci of 13- and 17-year species pairs in the three species groups, which evolved in parallel. In general, comparisons of populations with parallel character divergence can be an effective means for discovering diverged portions of the genome and genes responsible for the character divergence22,23.
Our study reveals the historical process of the parallel life-cycle divergence in the three species groups. First we confirm the relationships of four major lineages (Cassini and Decula groups, and two lineages within Decim) and the absence of introgressive hybridisation among these four lineages. Then we estimate demographic histories in the three species groups and find that, in each group, 13-year broods are monophyletic, sister to or derived from 17-year broods. Interestingly, we find evidence of gene flow between the 13- and 17-year species in each species group. Finally we search for single-nucleotide polymorphisms (SNPs) or loci showing elevated divergence between life cycles, but do not find any divergent SNPs or loci shared among all 13- and 17-year species pairs, nor any evidence for parallel genomic divergence across all pairs. Thus, the genetic background of the life cycle divergence in periodical cicadas remains unclear.
Results
Assembly of transcriptome sequences and orthologous loci
We sequenced mRNA from head tissues of 28 individuals (Supplementary Data 1) from two representative 17-year broods (eastern and western broods, II and III, respectively) and the two major 13-year broods, XIX and XXIII (Fig. 2a). (Note that 12 broods with 17-year cycles and three broods with 13-year cycles currently exist). De novo assemblies of the RNAseq reads were generated separately for four distinct groups: Decim (M. septendecim, M. neotredecim); M. tredecim; Cassini (M. cassini, M. tredecassini); and Decula (M. septendecula, M. tredecula). For each group, we obtained 76,519–90,287 contigs (length: 300–26,405 bp) with an average N50 length of 1476 bp (Table 1). Using these contigs, 7511 clusters orthologous to contigs of the outgroup Okanagana villosa transcriptome sequence from Genbank were identified, of which 5270 were shared by all four Magicicada groups (Fig. 2b). Among the 5270 clusters, we identified 2636 clusters (orthologous loci) that contained data from O. villosa and at least 27 Magicicada samples for phylogenetic and demographic analyses. Of the 2636 loci, 99% had BLAST hits with e-values <1×10–5 in the RefSeq protein database (Supplementary Data 2). The average alignment length of the loci was 1627 bp, and the average nucleotide diversity (π) of the loci for all Magicicada sequences (n = 28) was 0.0019 (range: 0–0.0238). The nucleotide diversity of the loci within the seven species (n = 4 for each species) was generally low, with a mean of 0.00045–0.00071 and a median of 0–0.00018 (Fig. 2c).

Sampling sites and the number and nucleotide diversity of orthologous loci. a Ranges of Magicicada broods used in this study (enclosed with broken lines) and locations of sampling sites (coloured dots). b Venn diagram for 7511 clusters (gene loci) orthologous to those of Okanaga villosa (outgroup). *A total of 2636 loci were used in the following analyses. c Nucleotide diversity values (π) of loci in seven species are plotted as correlation plots between 13- and 17-year species in each species groups. Note that M. septendecim (17-year) appears twice because there are two 13-year species in the Decim group. Median, mean and maximum values of π for each species: M. tredecim, median = 0.7×10−4, mean = 6.5×10−4, max = 0.2165; M. septendecim, median = 1.8×10−4, mean = 7.1×10−4, max = 0.02876; M. neotredecim, median = 1.3×10−4, mean = 6.9×10−4, max = 0.02984; M. tredecassini, median = 1.5×10−4, mean = 5.6×10−4, max = 0.02165; M. cassini, median = 1.2×10−4, mean = 5.6×10−4, max = 0.03015; M. tredecula, median = 0.0×10−4, mean = 5.3×10−4, max = 0.1865; M. septendecula, median = 0.0×10−4, mean = 4.5×10−4, max = 0.02567
Molecular phylogeny of periodical cicadas
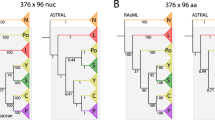
To characterise the historical relationship of species groups and broods, we first reconstructed phylogenetic trees using concatenated sequence data from the orthologous loci. The concatenated alignment was ca. 4.3 Mb in length, with 18% missing sites, and it contained 18,243 informative sites. The maximum-likelihood tree reliably recovered the monophyly of three species groups and the two lineages within the Decim group (M. tredecim and the lineage containing M. neotredecim and M. septendecim), but the relationships among broods within the Decim group (excluding M. tredecim), the Cassini group, and the Decula group were unresolved (Fig. 3). We also applied a species-tree method (SVDquartets24) to resolve the relationships among allochronically-separated broods, but it again poorly resolved the relationships among 13- and 17-year broods within each species group (Fig. 4a). In this tree, monophyly of the two 13-year broods was weakly supported in the Decim and Decula groups, whereas they were not monophyletic in the Cassini group.

Maximum-likelihood (ML) tree resulting from the concatenated sequences for individual cicadas. The tree was made ultrametric with the time of most recent common ancestor (tMRCA) of all Magicicada set to 3.89 million years ago6 (the 95% confidence interval of this node is shown by the broken line). Numerals above the branches are the bootstrap percentages resulting from the ML analysis. Estimated ages and 95% confidence intervals based on two calibrations using the upper and lower confidence limits of Magicicada tMRCA are shown for five major nodes (grey bars)

Relationship and demographic history of periodical cicada broods. a Phylogenetic relationship of broods reconstructed using SVDquartets. Numerals show bootstrap percentages of node recovery. b Demographic history of broods in each species group reconstructed using fastsimcoal2. Divergence of broods is shown with divergence times and effective population sizes (Ne; width of column). Vertical bars for divergence times show 95% confidence intervals. Demographic parameters were scaled with the effective population size of one brood (brood XIX for Decim; brood II for Cassini and Decula). c Estimated gene flow (NeM) between broods (open circles). Solid lines show 95% confidence intervals
Lack of hybridisation between four major lineages
To reconstruct the process of life cycle divergence, we first tested whether introgressive hybridisation among the four lineages (i.e. M. tredecim and three paired 13-year and 17-year species) was involved in life-cycle divergence events using the ABBA-BABA test with the D-statistic25,26 for SNPs. In particular, we tested the possibility that the earliest-diverged 13-year species, M. tredecim, introduced the 13-year life cycle into another lineage of Decim, or the Cassini and Decular groups through hybridisation, but we found no evidence for introgressive hybridisation (Table 2). We also tested for hybridisation between M. neotredecim and the Cassini or Decula group, and between the Cassini and Decula groups, but found no positive evidence (Table 2). Thus, we excluded the possibility that introgressive hybridisation between species groups or between the distinct Decim lineages was involved in the life-cycle divergence process.
Demographic histories within species groups
To further investigate the historical process of life-cycle divergence, we inferred the demographic histories of broods within species groups using the program fastsimcoal227, which analyses the joint site frequency spectra of synonymous SNPs. We used only high-quality SNPs from loci for which we could reliably infer reading frames. We considered three alternative scenarios of the relationships among broods (scenarios S1–S3), which reflected the possible diversification of the broods (Supplementary Fig. 1). In addition, we included three alternative models with gene flow between broods under each scenario, because recent divergence alone may not explain the low nodal support on the brood phylogenies. The three models were no gene flow, all possible recent and past gene flow (between ancestral populations and between current populations), and possible recent gene flow (between current populations). Thus, a total of nine models were compared in each of three species groups (Supplementary Fig. 1). For the Decim group, we included only samples of M. septendecim and M. neotredecim because M. tredecim had clearly diverged from the two species and gene flow between M. tredecim and parapatric M. neotredecim is virtually absent as was shown in our previous study20 and the ABBA-BABA test in the previous section.
We selected the best models of brood diversification based on model comparison using Akaike information criterion (AIC) weights and bootstrap proportions (Table 3). In all species groups, models with recent gene flow exhibited better fit than did models with no gene flow and those with both past and recent gene flow (Table 3). The best-fit scenarios were monophyly of both life cycles in the Decim and Cassini groups and monophyly of the 13-year species in the Decula group (Table 3, Fig. 4b). Note that the likelihood difference between recent gene flow models and past/recent gene flow models was marginal; the former models were favoured in AIC-based model comparisons because they had fewer parameters.
The estimated divergence times of 13- and 17-year life cycles in the three groups were 197, 121 and 95 ky ago (kya) in the Decim, Cassini and Decula groups, respectively (Fig. 4b, Supplementary Table 1). These three divergences occurred between the Illinoian glacial period and the last glacial period. These divergence times are comparable to the times of the most recent common ancestor (tMRCA) for 13- and 17-year species pairs estimated in the maximum-likelihood tree, 213, 131 and 111 kya for the Decim, Cassini and Decula groups, respectively (Fig. 4b). The most recent common ancestor for 13-year cicada broods occurred 74, 64 and 17 kya in the Decim, Cassini and Decula groups, respectively (Supplementary Table 1). Thus, the split of the two major 13-year broods likely occurred during the last glacial period (Fig. 4b).
The estimated effective population size (Ne) was consistent with the known biology of Magicicada (Fig. 4b, Supplementary Table 1). In the Cassini and Decula groups, Ne was larger in 13-year broods than in 17-year broods, which generally reflects the widespread range of 13-year species in these groups. By contrast, in the Decim group containing M. septendecim and M. neotredecim, Ne of the 17-year broods (M. septendecim) was larger than that of the 13-year broods (M. neotredecim), which suggests a recent origin for the narrowly distributed 13-year species M. neotredecim4. The current population sizes of 13-year and 17-year cicadas in each species group were larger than ancestral population sizes (except 17-year broods in the Decula group), which suggests recent population expansion associated with divergence of broods.
Estimated gene flow (NeM) between broods with the best models ranged from 0.01 to 23.8 migrants (individuals) per generation (Fig. 4c, Supplementary Table 1). For brood pairs in 13- or 17-year cicadas, gene flow was small between 17-year broods II and III, which are geographically separated, but 13-year brood pairs XIX and XXIII, which share lengthy boundaries, showed substantial gene flow (>1.0)28 in all species groups. For brood pairs between the 13- and 17-year cicadas, a substantial amount of gene flow (>1.0) was estimated to have occurred between all pairs in the Decim and Decula groups and between two of the four pairs in the Cassini group. Although the NeM confidence intervals were wide in each instance, the lower confidence limits of the gene flow between adjacent broods III and XIX were higher than 1.0 in the Decim and Decula groups, as well as between broods III and XXIII in the Decim group. In the Decula group, the NeM between broods II and XIX was greater than 1.0 despite the geographic separation of the samples, which indicates gene flow between eastern and western populations of brood XIX. In the Cassini group, the lower confidence limits of the gene flow between 13- and 17-year broods were low but non-zero (>0.003; Supplementary Table 1).
Genomic divergence between 13-year and 17-year cicadas
We measured genomic divergence between four 13- and 17-year species pairs of three groups (the three pairs detailed above and the M. tredecim/M. septendecim pair) using the fixation index Fst for individual SNPs and loci (Fig. 5). In general, Fst did not indicate divergence between 17- and 13-year species except in the anciently diverged pair, M. tredecim and M. septendecim (Fig. 5). At the locus level, Tajima’s D values were generally negative, and only 16–22% of loci exhibited positive values (Fig. 5), which indicates that the loci were mainly under purifying selection, although population size expansion (Fig. 4b) may have also affected Tajima’s D. We also calculated d xy between 13- and 17-year species as an absolute measure of nucleotide divergence, but the values of d xy were strongly correlated with π (Supplementary Fig. 2) and did not capture the divergence between species. Therefore, we used only Fst for the analyses of shared outliers.

Genomic sequence divergence between 13- and 17-year species within species groups. Frequency distributions of Fst of individual SNPs (left), Fst of loci (middle) and Tajima’s D (right) are shown for M. tredecim vs. M. septendecim (a), M. neotredecim vs. M. septendecim (b), M. tredecassini vs. M. cassini (c), and M. tredecula vs. M. septendecula (d). Median and mean values for SNP Fst: a median = 0.000, mean = 0.135; b median = 0.000, mean = −0.007; c median = 0.000, mean = −0.002; d median = 0.000, mean = −0.007; for locus Fst: a median = 0.1111, mean = 0.2146; b median = 0.0000, mean = −0.0012; c median = 0.0000, mean = 0.0055; d median = 0.0000, mean = −0.0049; for Tajima’s D: a median = −0.7995, mean = −0.6886; b median = −0.8015, mean = −0.7065; c median = −0.8015, mean = −0.7046; d median = −0.7806, mean = −0.5952
One possible mechanism of parallel life-cycle divergence is parallel divergence at the same nucleotide sites or loci, which may be accompanied by divergence of linked genomic portions. Therefore, we searched for divergent SNPs or loci shared between the four pairs of 13- and 17-year species (six comparisons) using Fst values for each 13- and 17-year species pair (Fig. 6). We defined elevated Fst by simulating SNPs under the best demographic models inferred in the previous section and taking the 95% quantiles of simulated Fst. Among 23,524 SNPs examined, 30 SNPs (0.1%) with elevated Fst (hereafter divergent SNPs) were shared by two species pairs (Fig. 6a, Supplementary Data 3), of which 27 SNPs were found in the within-Decim group comparison (i.e., M. tredecim/M. septendecim vs. M. neotredecim/M. septendecim). The other five comparisons yielded few or no shared divergent SNPs (Fig. 6a). The proportion of non-synonymous changes in the shared divergent SNPs was 0.47 (14/30) and did not significantly differ from genome wide proportion, 0.32 (P = 0.11, binomial test). At the locus level, we found 21 ‘divergent loci’ (0.7% of 2636 loci) with elevated Fst (Weir−Cockerham weighted Fst)29, which were shared by two or more species pairs (Fig. 6b, Supplementary Data 3). Further, we selected the maximum SNP Fst for each locus as an alternative measure of locus-level divergence. We discovered 15 divergent loci (0.6%) with elevated maximum Fst shared by two or three pairs (Fig. 6c, Supplementary Data 3). In the only divergent locus shared by three pairs (the exception being the Decula pair), the SNPs with maximum Fst were located in different positions among the three pairs.

Pairwise comparisons of sequence divergence between two pairs of 13- and 17-year species to identify shared divergent SNPs and loci between groups. Comparisons of Fst of SNPs (a), Weir−Cockerham weighted Fst of loci (b), and maximum Fst among SNPs within loci (c). Dashed lines indicate 95% quantile of simulated neutral distribution of Fst values, which we defined as outliers. Closed red dots are outliers shared between species groups. Open black and grey dots show non-shared outliers and non-outliers, respectively. The observed number of shared outliers (Nshared) and the probability (P) that observed numbers of shared outliers are obtained randomly for each of (a–c) are as follows (from left to right, and top to bottom). a Nshared = 25, P = 0.003; Nshared = 2, P = 0.81; Nshared = 0, P = 1; (row 2) Nshared = 1, P = 0.99; Nshared = 0, P = 0.99; (row 3) Nshared = 2, P = 0.81; b (row 1) Nshared = 9, P = 0.18; Nshared = 3, P = 0.99; Nshared = 3, P = 0.90; (row 2) Nshared = 3, P = 0.34; Nshared = 2, P = 0.31; (row 3) Nshared = 2, P = 0.43; c (row 1) Nshared = 6, P = 0.10; Nshared = 5, P = 0.31; Nshared = 1, P = 0.84; (row 2) Nshared = 1, P = 0.11; Nshared = 2, P = 0.004; (row 3) Nshared = 2, P = 0.01
The above three analyses suggest that parallel genomic divergence associated with life-cycle divergence is uncommon. To clarify this, we conducted a permutation test to estimate the probability that the number of shared divergent SNPs or loci observed in each comparison were obtained by chance alone22. We found non-random occurrence of outliers of that number only for the within-Decim comparison in the SNP Fst, none in the locus Fst, and only two comparisons involving the Decula or Cassini groups and M. neotredecim/M. septendecim in the locus maximum Fst (the number of shared divergent SNPs or loci, Nshared, and the permutational P values are given in the legend of Fig. 6a–c).
In the above outlier analyses, we obtained a total of 45 loci that exhibited elevated Fst at SNP or locus level between pairs of 13- and 17-year species (Supplementary Data 4). The functional annotation of these genes did not indicate enrichment of any kind of gene function. The 2636 loci studied included 21 genes involved in pathways potentially related to life cycle control (circadian clock, insulin signalling, insect hormone biosynthesis, MAPK signalling, and phototransduction30,31,32,33; Supplementary Data 5). However, the genes involved in these pathways were not found in the shared divergent loci.
Discussion
Our phylogenetic analysis using mRNA sequences clearly resolved the branching pattern of the four major periodical cicada clades, consistent with those of previous studies that used mitochondrial and genome-wide (restriction-site-associated DNA; RAD) markers6,20. However, neither mRNA and RAD sequence data resolved the relationships among broods within species groups despite the vast amounts of data, whereas the mitochondrial gene tree partly resolved phylogeographic (eastern, middle and western) patterns within species groups6. However, phylogenetic and population genetic inferences with mitochondrial gene sequences can be distorted by introgressive hybridisation and incomplete lineage sorting of ancestral polymorphism34. Therefore, it was necessary to revisit the results of our previous study based mainly on mitochondrial data, especially to confirm the divergence process of broods with different life cycles.
Our demographic inference using a site frequency-based method provides new insights into the parallel divergence process of 13- and 17-year life cycles, revealing that the three species groups have nearly parallel demographic histories, with 13-year broods monophyletically diverged from 17-year broods in each species group. The Decim and Cassini groups share a common diversification pattern in which 13- and 17-year groups diversified first, whereas the Decula group had a slightly different history, as the 13-year group was derived from brood III (representing the western 17-year brood). However, the divergence time of the 13-year group from brood III is close to that of broods II and III in the Decula group; thus the differences in divergence patterns among the species groups may not be substantial. The present results differ from those found through mitochondrial phylogenetics6, in which 13-year broods in both the Decim and Cassini groups were found to have been derived from the western haplotype group, including brood III, whereas the origin of 13-year broods appears to have been polyphyletic in the Decula group. Our demographic inference also suggests population expansions following brood splits in each species group. This finding is consistent with the previous results using mitochondrial data that population expansions occurred after the last glacial period in the Decim and Cassini groups6 although the present study does not restrict the timing of population expansion to the post LGM except for Decula 13-year broods.
We estimated that life cycle divergence (i.e., the split between 13- and 17-year species) in the Decim group (excluding M. tredecim) occurred at the beginning of the Illinoian glacial period (200–130 kya), and those of the Cassini and Decula groups during the Sangamon interglacial period (130–115 kya) or early in the last (Wisconsin) glacial period (115–12 kya). Although the confidence intervals for these estimated times are wide (between 247 and 19 ky overall), the present estimates are much older than the divergence times estimated using mitochondrial gene sequences in our previous study6, suggesting divergence within the last 23 kya (i.e. after the last glacial maximum). Because estimated tMRCA for the species groups are similar between this study and our previous study (see Fig. 3 and Supplementary Table 2), the short divergence times revealed by mitochondrial data may reflect recent mitochondrial introgression between geographically adjacent broods. If our new inferences are correct, it follows that both life cycles have persisted in all species groups since at least the beginning of the last glacial period.
Notably, our demographic inference showed that gene flow has occurred between the 13- and 17-year species in each species group, particularly between species with geographically adjacent 13- and 17-year broods. Although it is difficult to discriminate ‘diverged populations with gene flow’ from ‘recently diverged populations with no gene flow after the divergence,’ our model comparison showed that models with no gene flow had the lowest likelihoods compared to other models with gene flow (Table 2). In addition, the SVDquartets tree (Fig. 4a) showed low resolution for brood relationships; this lack of resolution makes sense because SVDquartets is designed to accommodate cases where gene flow is absent and incomplete lineage sorting is the source of gene tree incongruence35.
Gene flow between neighbouring populations of 13- and 17-year broods (species) may have occurred in the year of their co-emergence, every 221 (13×17) years or during occasional off-schedule emergences of smaller number of individuals (called ‘stragglers’36). The 13- and 17-year cicadas within species groups do not show clear morphological or behavioural differentiation4,5; hence they could potentially hybridise7,37. The finding of gene flow between sister 13- and 17-year species may be odd, because historical records indicate stability of the boundary between 13- and 17-year broods38. It is possible that the synchronised life cycle among individuals of each brood has been strongly selected, and thus is stable in the face of occasional gene flow9.
We initially hypothesised that the difference between the two life cycles was controlled by a locus that regulates juvenile development and an ancestral polymorphism at the locus may have caused the parallel life cycle divergence through collateral genetic evolution. Our comparison of orthologous gene sequences between 13- and 17-year species, however, has not provided any substantial clues resolving the genetic basis of life cycle divergence. We searched for shared SNPs or diverged loci among the four pairs of 13- and 17-year species (i.e., including M. tredecim), which may be related to the regulation of life cycles. Such shared SNPs/loci would show elevated Fst and deep divergence if ancestral polymorphisms were responsible for cycle shifts; alternatively, shared SNPs/loci with shallow divergence would be detected if independent mutations were responsible. However, we found no divergent SNPs or loci that were shared by all pairs. Even if life cycle loci exist, they may be undetectable in reduced representation sequences such as the RNAseq used in this study, likely because the responsible regions are small regardless of whether they are ancestral polymorphisms or independent mutations. We also found that non-random parallel genomic divergence (in terms of Fst) has not occurred among the four pairs of 13- and 17-year species, which may be expected in the parallel evolution of alternative phenotypes in different lineages22,23. If life cycle is controlled at multiple genetic levels rather than by a single mutation or a single diverged locus, any mutation in a group of genes within the same pathway could trigger a life cycle shift17. However, the results of functional annotation for the divergent loci between 13- and 17-year species showed no evidence of enrichment for a particular pathway or gene function. Thus, we have no conclusive information on the genetic control of life cycles at present.
Considering that we did not observe definitive genomic differences between the two life cycles, a non-genetic explanation for life cycle differences based on life cycle plasticity may not be ruled out completely. In a non-genetic scenario, different life cycles may be maintained by a threshold response of nymphs to clinal climatic factors such as the cumulative temperature during growing seasons. In fact, the geographic life cycle boundary (Fig. 1b) is predictable by local temperature data39. However, such an environmentally cued life cycle control may be unstable under fluctuating climatic conditions. In either case (i.e., genetic or non-genetic control of life cycle regulation), it would be necessary to conduct a thorough comparison of the whole genomic sequences between closely related 13- and 17-year species to fully explore the nature of life-cycle divergence in periodical cicadas.
Methods
RNA preparation and sequencing
We sampled 28 individuals from the seven known species of Magicicada (Supplementary Data 1). Four 13-year species were sampled from brood XIX (2011) and XXIII (2015), and three 17-year species from brood II (2013) and III (2014) during their emergences (Fig. 2a, Supplementary Data 1). Total RNA was extracted from head tissues using QIAGEN RNeasy. Libraries for sequencing were constructed and sequenced using the Illumina Hiseq2000 platform. Quality-filtered raw reads were deposited at the DNA Data Bank of Japan (DDBJ), in the DDBJ Read Archive (DRA).
De novo assembly and SNP calling
The quality-filtered sequence reads were de novo assembled using the Trinity assembler version r2014071740 with the default parameter settings. Samples from the species groups (Decim, Cassini and Decula) were pooled, and consensus contigs of species groups were assembled. Within the Decim group, M. tredecim samples were separately assembled because M. tredecim is clearly diverged from the monophyletic group that includes M. septendecim and M. neotredecim6,20. Thus, we obtained consensus assemblies for M. tredecim and the remaining Decim (M. septendecim/M. neotredecim), Cassini (M. cassini/M. tredecassni), and Decula (M. septendecula/M. tredecula) species.
SNPs for each sample were called as follows. Reads of samples were mapped to the consensus contigs using bowtie2 version 4.1.241, and variants were called with the ‘mpileup’ command in SAMtools version 1.2.0 and the ‘call’ command in BCFtools version 1.2.042, which implements the likelihood method for multi-sample SNP calling. Only SNPs supported with coverage of ≥3 and a quality score ≥20 were retained. These SNPs were inserted into the contigs using the BCFtools ‘consensus’ command, with heterozygous sites retained using IUPAC-style ambiguity coding. Bases with coverage <3 were masked with N, and terminal Ns were removed. Contigs shorter than 300 bp were filtered out, and the longest isoform for each trinity sequence cluster was selected for downstream orthology clustering.
Okanagana villosa was selected as the outgroup species for clustering; this is the closest species available in the NCBI database. Contigs of the O. villosa transcriptome43 were downloaded from the Transcriptome Shotgun Assembly database (Accession: GAWQ02000001–GAWQ02051314) and filtered with the same criteria as used for the Magicicada trinity contigs; only contigs longer than 300 bp and the longest isoforms were retained for the following clustering.
Orthology clustering
The consensus contigs of the samples were clustered into putative orthologous groups (loci) following the approach of Yang and Smith44. In brief, all-by-all BLASTN45 searches were conducted on all pairs of coding sequences of contigs, and then sequences with high similarity scores (evalue <1×10–5 and sequence identity >50%) were then clustered using MCL46. Then these homologous sequence clusters were aligned using MAFFT version 7.12347, and initial homologous trees were built using RAxML version 8.2.448. Orthologous clusters were obtained following the ‘monophyletic outgroup’ criterion44, i.e. keeping the largest subtree that consisted exclusively of ingroup samples without duplication and monophyletic outgroup samples. Clustering was conducted using the phylogenomic dataset construction scripts available at https://bitbucket.org/yangya/phylogenomic_dataset_construction. To obtain the final alignments, consensus contigs were replaced by contigs with SNPs, and the sequences were realigned using PRANK version 1400349 using the default parameters. We retained orthologous clusters containing ≥27 Magicicada samples (>95% of samples) as a final data set. Clusters with overall genetic variation greater than 0.05 were removed as putative erroneous clusters. The longest cluster sequences were used for BLAST searches in the RefSeq protein database (see Supplementary Data 2 for annotated clusters).
Phylogenetic inference
The maximum-likelihood (ML) phylogeny of individual samples was estimated using RAxML version 8.2.448 with the concatenated alignment. RAxML was run using the ‘rapid bootstrap analysis and search for best-scoring ML tree’ algorithm with a GTR-Γ model and 100 bootstrap replicates. To estimate divergence time, the ML tree was converted to an ultrametric tree using LSD version 0.3beta50, with a calibration time of 3.89 mya at the node of the most recent common ancestor of all Magicicada6. Confidence intervals of node ages were obtained by 1000 bootstrap analysis. To account for the uncertainty for the time of the Magicicada MRCA, we also estimated divergence times with the calibration times of 3.08 and 4.69 mya, which were the lower and upper values of the 95% highest probability density interval. For each node, the confidence interval was determined as the oldest and youngest ages of 95% confidence intervals obtained from 1000 bootstrap replicates. A brood-level population tree was constructed using SVDquartets24 implemented in PAUP* version 4.0a14751. All clusters were concatenated, and SVDquartets was run using the ‘species tree’ option with 100 bootstrap replicates.
ABBA-BABA test
We used the ABBA-BABA test with the D-statistic25,26 to test whether introgressive hybridisation has occurred between different 13-year species from different species groups or between distinct lineages of the same species group (i.e., M. tredecim vs. M. neotredecim in the Decim group). Under the assumption that population P1 and P2 are derived from population P3 and outgroup O, the ABBA-BABA test searches for evidence of hybridisation between P3 and P1 or P2 by comparing the frequencies of the site patterns ABBA and BABA. We set 17- and 13-year broods in the same species group as P1 and P2, respectively, and set one of 13-year broods from different species group as P3. An outgroup (O) was chosen from the closest available outgroup taxa. We tested the hybridisation of seven pairs of 13-year species with all four combinations of broods, totalling 28 comparisons (Table 1). D-statistics were calculated by a modified version of PyRAD version 3.0.6652, which accepts a fasta alignment as an input. The standard deviation of the D-statistic was obtained by a bootstrap resampling with 1000 replications.
Demographic inference and model selection
We conducted demographic inference and model comparison using a method based on the site frequency spectrum (SFS) implemented in fastsimcoal2 version 2.5.2.2127. Synonymous SNPs were selected from the alignments of clusters, and folded joint SFSs of four populations representing 17-year broods II and III and 13-year broods XIX and XXIII were obtained with minimum site frequencies (5%) using Arlequin version 3.553. Then the likelihoods of demographic scenarios were calculated using fastsimcoal2. Monomorphic sites were excluded from the likelihood calculations with the ‘removeZeroSFS’ option because we could not estimate the accurate number of monomorphic sites for synonymous SNPs. According to this option, the effective population size (Ne) of one population (brood XIX for Decim, brood II for Decula and Cassini) was fixed to the value calculated from the average genetic variation (π) of the population and the relationship, π = 4Neμ, where μ is the Magicicada-specific mutation rate estimated as below.
We estimated the mean mutation rate from the present mRNA sequence data using the previously estimated age of several major nodes in the Magicicada phylogenetic tree6 and the node heights of the ML tree as estimated from the present mRNA sequence data as described above, and assuming a time-dependent substitution rate54. We also assumed a generation time of 15 years, the average of 13 and 17 years. Based on the ML tree resulting from concatenated mRNA sequences, node heights for seven clades are obtained (Supplementary Table 2). Using the corresponding node ages and a generation time of 15 years, the substitution rate per site per generation at each node was calculated. The substitution rate decayed over time towards an asymptote, as predicted54. Then, using the R package ‘nls’, the substitution rate and node age data were fitted to a non-linear model with the time-dependent evolutionary rate equation54:
where µ is the instantaneous mutation rate, and λ is inversely proportional to the half-life of the rate decay, and k is a finite asymptotic evolutionary rate. As a result, we obtained estimates of these variables as µ = 0.008494, λ = 2.9185 and k = 0.006849 (per site per million generations). At t = 0, the rate µ + k equals 0.0153 per million generations (=1.53×10−8 per generation). This value was used as the mutation rate in the demographic analysis.
We included the following three alternative scenarios in the model comparison, which are based on known phylogeographic trees and the two life cycles:
Scenario S1: 13- and 17-year broods form monophyletic groups ((II, III), (XIX, XXIII));
Scenario S2: geographically adjacent sampled broods form clades irrespective of their life cycles (II, (XXIII, (III, XIX)));
Scenario S3: 13-year species are monophyletic, and adjacent 17-year broods are closer to these (II, (III, (XIX, XXIII))).
To assess the effects of gene flow, we included three models of gene flow between broods under the three population divergence scenarios listed above. The three models were ‘no gene flow’; ‘past and recent gene flow’, where gene flow exists between all current and ancestral populations; and ‘recent gene flow only’, where gene flow only exists between current populations. In total, nine models were used in the model comparison (Supplementary Fig. 1).
We chose the best model using AIC values and AIC weights55 calculated from composite likelihoods of the models, as recommended by Excoffier et al.27. In addition to model comparison with maximum likelihood inference, we performed bootstrap resampling of 100 replicates with Poisson approximation56 and recorded the bootstrap proportions, i.e., the proportions of replicates for which a given model was repeatedly chosen as the best model57.
Population genomic measures
To characterise the within- and between-species genetic profiles of 17- and 13-year Magicicada species, population genetic measures were calculated for each orthologous cluster (locus). Genetic variation (π) and the number of segregating sites (S) within seven species were calculated. Tajima’s D58 was calculated to detect purifying or balancing selection in each species group. As a measure of net divergence between 13- and 17-year species, Fst29 was calculated for each SNP as an SNP-level measure of divergence and for each cluster as a locus-level measure of divergence using the R version 3.3.359 package ‘pegas’60. We used a weighted average of Fst values in a locus as a locus-level estimator per the method of Weir and Cockerham29. Maximum Fst values within a locus were collected as an alternative measure of locus-level divergence. We also calculated the average number of pairwise differences, d xy , for each locus between 13- and 17-year species because this index is recommended as an absolute measure of population divergence61.
Due to the sparse number of SNPs within loci and small sample sizes within populations, we were not able to reliably phase the genotypes. Therefore, we employed the repeated random haplotype sampling (RRHS) strategy62 when phase information was required. RRHS randomly assigns one of two possible genotypes at heterozygous sites. Thus, π, d xy and Tajima’s D were repeatedly calculated with 100 RRHS replicates, and their averages were used as estimates.
Outlier analysis for diverged genomic portions
To detect diverged genomic portions associated with the divergence of 13- and 17-year species, we conducted outlier analyses of Fst for each SNP, Fst for each locus, and maximum Fst among all SNPs within each locus. Fst is an inappropriate measure of population differentiation when it is highly negatively correlated with nucleotide diversity63. However, in our case, Fst was not correlated with mean nucleotide diversity at the locus level except for a weak negative correlation in the M. tredecim/M. septendecim pair (Supplementary Fig. 2). Note that Fst and mean nucleotide diversity are expected to be uncorrelated with each other when demographic factors (e.g., gene flow, genetic drift) outweigh the effect of mutations, whereas a negative correlation is expected between these measures in the opposite situation63. Meanwhile, the nucleotide divergence d xy , which is considered a more appropriate measure of population differentiation61, was strongly positively correlated with nucleotide diversity and hence may lead to false discovery of elevated d xy at regions with high nucleotide diversity64 (Supplementary Fig. 2). Thus, the use of Fst, rather than d xy , was considered appropriate in the present case.
We defined the SNPs/loci with elevated Fst values as ‘divergent SNPs/loci’. To determine thresholds to define elevated Fst, we simulated up to 10,000 unlinked SNPs for the best-fitting demographic models selected above using fastsimcoal2 and calculated Fst between 13- and 17-year broods. The 95% quantile of the simulated statistics was chosen as the threshold to define elevated Fst. The divergent SNPs/loci shared by two or more comparisons between two pairs of 13- and 17-year species were considered as ‘shared divergent SNPs/loci’, which are the candidate SNPs/loci responsible for the parallel life cycle divergence. The threshold value for the maximum Fst for a locus was determined by repeatedly taking a maximum of five Fst values of simulated SNPs to generate a distribution of maximum of Fst and obtaining the 95% quantile of this distribution. To determine the threshold to define elevated locus-level Fst, we simulated linked sites of 2500 bp long for 5000 times (replicates) under the same demographic model. The weighted average of Fst values for SNPs in the linked sites was calculated each time, and the 95% quantile of the 5000 average Fst values were chosen as the threshold.
The number of divergent SNPs or loci with elevated Fst shared by two or more comparisons (i.e., ‘shared divergent SNPs/loci’) was considered an indicator of parallel divergence. The statistical significance of the numbers of shared divergent SNPs or loci was tested with permutation tests with 1000 replicates, which estimated the probability that the number of shared divergent SNPs or loci observed in each comparison were obtained by chance alone. For the shared divergent loci, functional annotations were made using DAVID Bioinformatics Resources 6.865,66.
Data availability
The raw sequence reads used in the present study are available from the DDBJ Read Archive (DRA) of the DNA Data Bank of Japan (DDBJ) (BioProject, PRJDB4567; BioSample, SAMD00047121–SAMD0004712147148). Other relevant data and input files used in the fastsimcoal2 runs are available via Figshare at https://doi.org/10.6084/m9.figshare.c.401152067.
References
Roff, D. A. Life History Evolution (Sinauer, Sunderland, MA, 2002).
Williams, K. S. & Simon, C. The ecology, behavior, and evolution of periodical cicadas. Annu. Rev. Entomol. 40, 269–295 (1995).
Alexander, R. D. & Moore, T. E. The evolutionary relationships of 17-year and 13-year cicadas, and three new species (Homoptera, Cicadidae, Magicicada). Misc. Publ. Mus. Zool. Univ. Mich. 121, 1–59 (1962).
Marshall, D. C. & Cooley, J. R. Reproductive character displacement and speciation in periodical cicadas, with description of a new species, 13-year Magicicada neotredecim. Evolution 54, 1313 (2000).
Cooley, J. R., Simon, C., Marshall, D. C., Slon, K. & Ehrhardt, C. Allochronic speciation, secondary contact, and reproductive character displacement in periodical cicadas (Hemiptera: Magicicada spp.): genetic, morphological, and behavioural evidence. Mol. Ecol. 10, 661–671 (2001).
Sota, T. et al. Independent divergence of 13-and 17-y life cycles among three periodical cicada lineages. Proc. Natl. Acad. Sci. USA 110, 6919–6924 (2013).
Lloyd, M. & Dybas, H. S. The periodical cicada problem II. Evolution. Evolution 20, 466–505 (1966).
Cox, R. T. & Carlton, C. E. Paleoclimatic influences in the evolution of periodical cicadas (Insecta: Homoptera: Cicadidae: Magicicada spp.). Am. Midl. Nat. 120, 183–193 (1988).
Yoshimura, J. The evolutionary origins of periodical cicadas during ice ages. Am. Nat. 149, 112–124 (1997).
Lloyd, M., Kritsky, G. & Simon, C. A simple Mendelian model for 13- and 17-year life cycles of periodical cicadas, with historical evidence of hybridization between them. Evolution 37, 1162–1180 (1983).
Cox, R. T. & Carlton, C. E. Evidence of genetic dominance of the 13-year life cycle in periodical cicadas (Homoptera: Cicadidae: Magicicada spp.). Am. Midl. Nat. 125, 63–74 (1991).
White, J. A. & Lloyd, M. Growth rates of 17- and 13-year periodical cicadas. Am. Midl. Nat. 94, 127–143 (1975).
Koyama, T. et al. Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptation. J. Evol. Biol. 28, 1270–1277 (2015).
Marshall, D. C., Cooley, J. R. & Hill, K. B. R. Developmental plasticity of life-cycle length in thirteen-year periodical cicadas (Hemiptera: Cicadidae). Ann. Entomol. Soc. Am. 104, 443–450 (2011).
West-Eberhard, M. J. Developmental Plasticity and Evolution (Oxford University Press, New York, 2003).
Stern, D. L. The genetic causes of convergent evolution. Nat. Rev. Genet. 14, 751–764 (2013).
Elmer, K. R. & Meyer, A. Adaptation in the age of ecological genomics: insights from parallelism and convergence. Trends Ecol. Evol. 26, 298–306 (2011).
Cox, R. T. & Carlton, C. E. A comment on gene introgression versus en masse cycle switching in the evolution of 13-year and 17-year life cycles in periodical cicadas. Evolution 57, 428–432 (2003).
Simon, C. et al. Genetic evidence for assortative mating between 13-year cicadas and sympatric ‘17-year cicadas with 13-year life cycles’ provides support for allochronic speciation. Evolution 54, 1326–1336 (2000).
Koyama, T. et al. Genomic divergence and lack of introgressive hybridization between two 13-year periodical cicadas support life cycle switching in the face of climate change. Mol. Ecol. 25, 5543–5556 (2016).
Hanrahan, S. J. & Johnston, J. S. New genome size estimates of 134 species of arthropods. Chromosom. Res. 19, 809–823 (2011).
Soria-Carrasco, V. et al. Stick insect genomes reveal natural selection’s role in parallel speciation. Science 344, 738–742 (2014).
Westram, A. M. et al. Do the same genes underlie parallel phenotypic divergence in different Littorina saxatilis populations? Mol. Ecol. 23, 4603–4616 (2014).
Chifman, J. & Kubatko, L. Quartet inference from SNP data under the coalescent model. Bioinformatics 30, 3317–3324 (2014).
Green, R. E. et al. A draft sequence of the neandertal genome. Science 328, 710–722 (2010).
Durand, E. Y., Patterson, N., Reich, D. & Slatkin, M. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28, 2239–2252 (2011).
Excoffier, L., Dupanloup, I., Huerta-Sanchez, E., Sousa, V. C. & Foll, M. Robust demographic inference from genomic and SNP data. PLoS Genet. 9, e1003905 (2013).
Zhang, C., Zhang, D. X., Zhu, T. & Yang, Z. Evaluation of a bayesian coalescent method of species delimitation. Syst. Biol. 60, 747–761 (2011).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984).
Young, M. & Kay, S. A. Time zones: a comparative genetics of circadian clocks. Nat. Rev. Genet. 2, 702–715 (2001).
Koštál, V. Insect photoperiodic calendar and circadian clock: independence, cooperation, or unity? J. Insect Physiol. 57, 538–556 (2011).
Yamanaka, N., Rewitz, K. F. & O’Connor, M. B. Ecdysone control of developmental transitions: lessons from Drosophila research. Annu. Rev. Entomol. 58, 497–516 (2013).
Nijhout, H. F. et al. The developmental control of size in insects. Wiley Interdiscip. Rev. Dev. Biol. 3, 113–134 (2014).
Ballard, J. W. O. & Whitlock, M. C. The incomplete natural history of mitochondria. Mol. Ecol. 13, 729–744 (2004).
Chou, J. et al. A comparative study of SVDquartets and other coalescent-based species tree estimation methods. Bmc Genom. 16, S2 (2015).
Marlatt, C. L. A consideration of the validity of the old records bearing on the distribution of the broods of the periodical cicada, with particular reference to the occurrence of broods VI and XXIII in 1898. Bull. U.S. Bur. Entomol. 18, 59–78 (1898).
Cooley, J. R., Marshall, D. C., Hill, K. B. R. & Simon, C. Reconstructing asymmetrical reproductive character displacement in a periodical cicada contact zone. J. Evol. Biol. 19, 855–868 (2006).
Marshall, D. C. Periodical cicada (Homoptera: Cicadidae) life-cycle variations, the historical emergence record, and the geographic stability of brood distributions. Ann. Entomol. Soc. Am. 94, 386–399 (2001).
Cooley, J. R., Marshall, D. C., Simon, C., Neckermann, M. L. & Bunker, G. At the limits: habitat suitability modelling of northern 17-year periodical cicada extinctions (Hemiptera: Magicicada spp.). Glob. Ecol. Biogeogr. 22, 410–421 (2013).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data. Nat. Biotechnol. 29, 644–652 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Misof, B. et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 346, 763–767 (2014).
Yang, Y. & Smith, S. A. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Mol. Biol. Evol. 31, 3081–3092 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
van Dongen, S. Graph Clustering by Flow Simulation. http://dspace.library.uu.nl/handle/1874/848 (2000).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Löytynoja, A. & Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320, 1632–1635 (2008).
To, T., Jung, M., Lycett, S. & Gascuel, O. Fast dating using least-squares criteria and algorithms. Syst. Biol. 65, 82–97 (2015).
Swofford, D. L. PAUP*. Phylogenetic Analysis Using Parsimony (* and Other Methods) (Sinauer Associates, Sunderland, MA, 2002).
Eaton, D. A. R. PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30, 1844–1849 (2014).
Excoffier, L. & Lischer, H. E. Arlequin suite ver. 3.5. A new series of program to perform population genetics analyses under Linux and Winsows. Mol. Ecol. Resour. 10, 564–567 (2010).
Ho, S. Y. W., Phillips, M. J., Cooper, A. & Drummond, A. J. Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 22, 1561–1568 (2005).
Burnham, K. P. & Anderson, D. R. Model Selection and Multimodel Inference: A Practical Information-Theoritic Approach2nd edn (Springer Science+Business Media, Inc, New York, 2002).
Hanley, J. A. & MacGibbon, B. Creating non-parametric bootstrap samples using Poisson frequencies. Comput. Methods Prog. Biomed. 83, 57–62 (2006).
Buckland, S. T., Burnham, K. P. & Augustin, N. H. Model selection: an integral part of inference. Bioinformatics 53, 603–618 (1997).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
R Core Team. A Language and Environment for Statistical Computing (The R Foundation for Statistical Computing, Vienna, Austria, 2016).
Paradis, E. pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–420 (2010).
Cruickshank, T. E. & Hahn, M. W. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23, 3133–3157 (2014).
Lischer, H. E. L., Excoffier, L. & Heckel, G. Ignoring heterozygous sites biases phylogenomic estimates of divergence times: implications for the evolutionary history of Microtus voles. Mol. Biol. Evol. 31, 817–831 (2014).
Wang, J. Does G ST underestimate genetic differentiation from marker data? Mol. Ecol. 24, 3546–3558 (2015).
Riesch, R. et al. Transitions between phases of genomic differentiation during stick-insect speciation. Nat. Ecol. Evol. 1, 82 (2017).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Fujisawa, T. et al. Data from: Triplicate parallel life cycle divergence despite gene flow in periodical cicadas. https://doi.org/10.6084/m9.figshare.c.4011520 (2018).
Acknowledgements
This work was supported by the following funding: JSPS KAKENHI (26257405, 22255004 to J.Y.; 25128707, 23128507 to T.S.; JP26840126, JP13J03600 and JP17K15182 to S.K.); SPIRITS at Kyoto University (to T.S.); the Asahi Glass Foundation (to S.K.). C.S. and J.R.C. acknowledge support from NSF DEB 0955849 and DEB 1655891.
Author information
Authors and Affiliations
Contributions
T.S. and T.F. conceived this study. T.K., S.K., J.Y., J.R.C., C.S., and T.S. conducted field work. T.K. and T.S. conducted laboratory works. T.F. designed and conducted the analyses. T.F. and T.S. drafted the manuscript. All authors read, revised and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fujisawa, T., Koyama, T., Kakishima, S. et al. Triplicate parallel life cycle divergence despite gene flow in periodical cicadas. Commun Biol 1, 26 (2018). https://doi.org/10.1038/s42003-018-0025-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-018-0025-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
