
Abstract
Sunlight-driven H2 generation is a central technology to tackle our impending carbon-based energy collapse. Colloidal photocatalysts consisting of plasmonic and catalytic nanoparticles are promising for H2 production at solar irradiances, but their performance is hindered by absorption and multiscattering events. Here we present a two-dimensional bimetallic catalyst by incorporating platinum nanoparticles into a well-defined supercrystal of gold nanoparticles. The bimetallic supercrystal exhibited an H2 generation rate of \(139\,{\mathrm{mmol}}\,{\mathrm{g}}_{\mathrm{cat}}^{-1}\,{\mathrm{h}}^{-1}\) via formic acid dehydrogenation under visible light illumination and solar irradiance. This configuration makes it possible to study the interaction between the two metallic materials and the influence of this in catalysis. We observe a correlation between the intensity of the electric field in the hotspots and the boosted catalytic activity of platinum nanoparticles, while identifying a minor role of heat and gold-to-platinum charge transfer in the enhancement. Our results demonstrate the benefits of two-dimensional configurations with optimized architecture for liquid-phase photocatalysis.

Similar content being viewed by others

Main
Molecular hydrogen (H2) will inevitably be a key player in the quest for clean and sustainable energy. Its high exothermic combustion makes it a suitable replacement for carbon-based fuels, thereby reducing the carbon footprint1. Similar to what occurs in nature, capturing the energy from sunlight in H2 bonds is a promising strategy to store and manipulate solar energy in further processes, and materials that facilitate this energy transduction are of great interest2,3,4. In this regard, subwavelength metallic nanoparticles (NPs) offer unique possibilities for photocatalysis driven by localized surface plasmon resonances. Plasmonic metals such as gold, aluminium, silver, magnesium and copper display these resonances in the visible range of the electromagnetic spectrum, enabling them to efficiently absorb light in the spectral region of maximum solar irradiance5. This light–matter interaction results in intensified electric fields at the NP surface, highly energetic holes and electrons (hot carriers), and local heat. Despite these outstanding optical properties, the plasmonic metals used so far exhibit comparatively low catalytic activities6,7. Therefore, to take advantage of these remarkable optical properties in chemical transformations, plasmonic NPs were integrated into multicomponent nanostructures to produce photocatalysts with better performance with respect to the individual constituents8,9. In particular, bimetallic structures consisting of catalytic and plasmonic metals combine strong absorbance of visible light and high reactivity10,11,12. The term ‘antenna–reactor’ was coined for configurations in which the plasmonic and catalytic NP were placed in close proximity, but was also extended to arrangements in which the active materials either form islands or are dispersed as single atoms at the surface of the plasmonic antenna. This configuration presents the highest efficiency in converting light into reaction products compared with other configurations13,14,15,16,17.
Despite the enhanced activity of these bimetallic photocatalysts, their performance in liquid environments faces several experimental challenges. For instance, concentrated solutions suffer from multiple scattering and absorption events, impeding the simultaneous activation of all nanostructures present by reducing the light penetration into the reactor down to hundreds of micrometres (∼500 μm)18. This restricts the working range for catalyst concentration in solution and increases the challenge of manufacturing suitably engineered reactors. In addition, ligands providing stability to colloidal NPs can undergo photochemical desorption19,20, resulting in uncontrolled aggregation of nanostructures leading to altered optical properties, sedimentation of the aggregates and less available surface for catalytic activity. Moving from colloidal suspensions to a film configuration is an appealing strategy to circumvent the limitations due to light penetration and aggregation. This approach allows for the collective excitation of every individual NP in a relatively fixed position, thus presenting a solution to the experimental limitations described above21,22,23. In this regard, plasmonic two-dimensional (2D) supercrystals24,25,26, devices in which the photoresponse is governed by the periodic organization of individual components in a plane27,28, promote the transition from 3D to quasi-2D materials, aiming for better exploitation of the benefits of bimetallic photocatalysts. In this context, 2D expresses the expansion of the crystals in two dimensions. It is not limited to monolayers, but rather implies a defined number of layers in a given domain29.
The proximity of the individual NPs in the 2D periodic array results in the formation of plasmonic hotspots—spatial regions characterized by enhanced electric fields—at the interparticle gaps30,31. The intense electric fields sustained in these hotspots boost the otherwise weak absorption of the catalytic centre in the visible range32,33,34, and stand out as an important factor to be modulated in the elusive interplay between the thermally driven and electric-field-driven photoactivity of plasmonic bimetallic catalysts15,35,36,37,38,39. Thus, creating a large number of hotspots in densely packed supercrystals, which is unlikely to be achieved in colloidal solution or disordered catalysts, is expected to maximize the use of active metals for catalytic applications.
We achieved plasmonic structures by assembling individual gold nanoparticles (AuNPs) and platinum nanoparticles (PtNPs), obtaining highly ordered bimetallic supercrystals by a colloidal approach24,25,30. This fabrication methodology yielded 2D bimetallic photocatalysts in which AuNPs were assembled into a periodic hexagonal array and PtNPs were placed in the interparticle gaps of the AuNPs, scaling the preferred antenna–reactor configuration up to the mm2 regime. Here we show that, when operated under white-light illumination and sunlight irradiance, the rationally designed bimetallic structures generate higher H2 production than existing top performers for formic acid dehydrogenation. The remarkable activity was achieved with very low platinum loadings. The well-defined structure of the photocatalyst made it possible to spectrally resolve the plasmonic effects. Wavelength-dependent experiments and electrodynamic simulations revealed that the electric field enhancement in hotspots dictates the photoactivation of the PtNPs, which is also supported by transient absorption spectroscopy. Charge injection from gold to platinum, and photothermal contributions, were confirmed to play a minor role in photocatalytic enhancement. The results show that bimetallic 2D supercrystals are promising candidates for photocatalytic applications.
Results
Synthesis and characterization of 2D supercrystals
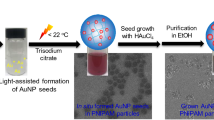
Figure 1 presents the microscopic characterization of the synthesized plasmonic supercrystals. The monometallic 2D supercrystal (Au supercrystal) that serves as reference/control sample was fabricated according to a previously reported method in which individual AuNPs are used as building blocks25. The precise control of the synthesis of AuNPs, their surface chemistry and the self-assembly step resulted in a well-packed hexagonal array of 22 nm AuNPs with an edge-to-edge distance of ∼2 nm (Fig. 1a). No crystalline arrangement other than hexagonal close packing of the large AuNPs was found in the sample when investigating different areas by transmission electron microscopy (TEM). The homogeneity of the structure over a large area is shown in the TEM image in Fig. 1b. These layers can extend up to a few square millimetres (Fig. 1c). In addition to monolayers, areas with multilayers also emerge during the crystallization process (Fig. 1c). The multilayers maintain the same crystalline phase25, but result in different optical properties. With optical microscopy in transmission mode, we were able to identify the different layer numbers due to contrast difference, indicating the different sample thicknesses24,40. One example of the Au supercrystal in transmittance is depicted in Fig. 1c. The lighter the blue, the thinner the supercrystal. The unambiguous colour contrast was utilized to measure the areas of the different layer numbers.

a,b, TEM images of Au supercrystal made of 22 nm AuNPs at two different magnifications. The interparticle gap of the NPs is ∼2 nm. c, Representative transmission microscopy image of the supercrystal. The different blue hues indicate different numbers of particle layers. 1L, monolayer; 2L, bilayer; 3L, trilayer; 3L+, multilayer. d,e, TEM images of a bimetallic 2D AuPt supercrystal at two different magnifications. PtNPs (∼3 nm) are hosted at the interparticle gap (∼3.5 nm) between the 22 nm AuNPs. No interface is created between the different metals.
The colloidal approach to supercrystal fabrication was extended to bimetallic structures comprised of AuNPs and PtNPs. Through the same preparation methodology as used for Au supercrystals, we managed to assemble both 22 nm AuNPs and ∼3 nm PtNPs simultaneously, yielding a bimetallic supercrystal (AuPt supercrystal). The integration of platinum, a catalytically highly active metal, realized a supercrystal with combined optical and catalytic properties. In this composition the AuNPs dominate the optical behaviour, whereas the PtNPs act as the active site for chemical reactions. The homogeneity and periodicity of the supercrystal was not affected by the presence of PtNPs (Fig. 1e). A higher-magnification TEM image revealed that during the assembly step the PtNPs were placed at the interparticle gaps between the AuNPs, which increased the gap size to 3.5 nm (Fig. 1d and Supplementary Figure 1). At this point, it is important to emphasize that the small PtNPs were located in the interstices (gaps) of the 2D hexagonal array of AuNPs and no direct contact exists between the two components of the bimetallic AuPt supercrystal. This structure is acknowledged to be an antenna–reactor configuration. The crystallinity of both AuNPs and PtNPs and the material distribution were confirmed by high-resolution TEM and energy-dispersive X-ray spectroscopy (Supplementary Figure 2).
The tunability of physical parameters, such as AuNP size, PtNP loading, interparticle gap size and number of layers, was confirmed by assembling larger particles with longer thiolated polystyrene (PSSH) ligands, which yielded similar results (Supplementary Fig 3). The assembly methodology was also proven to work for different catalytic metals, as shown by replacing PtNPs with palladium NPs (Supplementary Figure 4). Hence, the developed approach enables the creation of bimetallic supercrystals with a desired antenna–reactor combination, spanning several square millimetres consistently, while providing a broad range of tunable physical parameters. For a detailed explanation of the manufacturing process, see Methods.
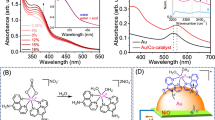
To investigate the optical properties of the indium tin oxide (ITO)-supported Au and AuPt supercrystals, microtransmission and reflection measurements were performed on both supercrystals within the spectral range of 450–850 nm. The spectra were taken in both cases on monolayers, bilayers and trilayers (1L, 2L, 3L). The absorbance (A) was calculated afterwards as the difference between the transmitted (T) and reflected (R) light (Methods). The resulting spectra for the pure gold supercrystals are shown in Fig. 2a and the spectra for the bimetallic sample in Fig. 2b. The obtained experimental spectra are in good agreement with the finite-difference time-domain (FDTD) simulations of both supercrystal structures (Supplementary Figure 5). For the Au and AuPt supercrystals, the monolayer spectrum is dominated by interband transitions of the gold31. For bilayers and trilayers, the characteristic polaritonic modes that occur due to deep strong light–matter coupling start to appear between 650 and 750 nm (ref. 24). The energetic position of these polaritonic resonances depends on the film thickness, the diameter of the particles, the gap size and the dielectric environment24,30. Therefore, the small redshift of the bimetallic spectra with respect to the resonances presented by the monometallic supercrystal can be explained by the larger gap size, and the changes in the refractive index of the background are due to the presence of PtNPs. These interesting optical properties presented by the supercrystals are intricately related to the photocatalytic activity of the bimetallic structures.

a,b, Experimental layer-dependent reflectance, transmittance and absorbance spectra of the pure Au supercrystal (a) and the bimetallic AuPt supercrystal (b). c,d, Weighted reflectance, transmittance and absorbance spectra of the pure Au supercrystal (c) and the bimetallic AuPt supercrystal (d) from analysis of the composition of the different layer numbers.
As mentioned above, the colour contrast of the supercrystal layers imaged with an optical microscope in transmission mode made it possible to estimate the size of the domains. Thus, it was possible to calculate the overall optical response of the samples by weighting the experimental response of the individual layers by the different areas. It is important to know the ratios of the various layers because during the catalysis experiments a large area of the sample will be illuminated containing different numbers of layers. The details of the sample composition are given in Table 1. The study revealed that monolayers with ∼80% coverage for the control sample and ∼60% for the AuPt supercrystal make up the main part of the samples. The bimetallic sample also has a notable amount of trilayers and multilayers (∼30%), whereas the monometallic sample only consists of ∼3% multilayers.
The weighted optical behaviour of the control and AuPt sample are depicted in Fig. 2c,d. As expected, due to the large contribution of monolayers the overall response of the ITO-supported supercrystals was dominated by the interband transitions of the gold. In contrast to the pure Au supercrystal, the optical response of the AuPt sample shows an influence of the multilayers. Two shoulders are clearly visible in the absorbance spectrum at 671 and 732 nm due to the presence of the polaritonic modes in the bilayers and trilayers. Once the optical response of the samples was determined, they were tested as catalysts for formic acid dehydrogenation.
Photocatalysis
The photocatalytic power of the resulting supercrystals was investigated by testing their performance on formic acid decomposition without any further additives. The probe reaction was chosen based on the lower activity of gold versus platinum in this context, and also because it is a carbon-neutral H2 carrier1,41,42. The experiments were conducted in the absence (dark) and presence of light and repeated at least three times each to ensure reproducibility. It has been shown that the probe redox reaction can follow two different pathways, namely, dehydrogenation or dehydration. The former produces H2 and CO2 and is favoured thermodynamically (ΔG = −48.4 kJ mol−1), whereas the latter produces H2O and CO (ΔG = −28.5 kJ mol−1)1. CO was not detected throughout our experiments, excluding dehydration. For these studies a broadband lamp, equipped with an ultraviolet long-pass filter (λ > 400 nm) and an infrared filter, was used as a source of light, to work exclusively in the visible range of the spectrum and rule out heat generated from solvent absorption. An external cryostat set at 25 °C was used to control the temperature during the reaction. The experiment was set up for the reactor to be illuminated with ∼110 mW cm−2, which corresponds to sunlight irradiance. The stirring conditions helped to reduce any temperature gradient43. Further details on the experimental set-up and conditions are given in Methods.
Figure 3a shows the performance of ITO-supported gold and AuPt supercrystals when tested for formic acid decomposition. The mass of the catalysts was obtained by inductively coupled plasma mass spectrometry (ICP-MS), while the preservation of the optical properties was monitored by optical spectroscopy (Supplementary Figs. 8 and 9). Details of the normalization are provided in Supplementary Fig. 6 and Supplementary Table 5. The Au supercrystal presented an activity of \(36\,{\mathrm{mmol}}\,{\mathrm{g}}_{\mathrm{cat}}^{-1}\,{\mathrm{h}}^{-1}\) and a negligible enhancement upon illumination. Conversely, the inclusion of small amounts of PtNPs in the supercrystal increased the performance of the binary AuPt supercrystal up to \(67\,{\mathrm{mmol}}\,{\mathrm{g}}_{\mathrm{cat}}^{-1}\,{\mathrm{h}}^{-1}\). When illuminated with white light, the performance was increased by approximately a factor of two, reaching \(139\,{\mathrm{mmol}}\,{\mathrm{g}}_{\mathrm{cat}}^{-1}\,{\mathrm{h}}^{-1}\). The improved performance of platinum under illumination in the experiment suggests that the interaction of the incoming light with the gold array results in energized platinum, useful for catalytic purposes. Indeed, the AuPt supercrystal appears to be the best plasmonic performer when formic acid is used as the H2 carrier8. Additionally, to assess the boost in PtNP performance, the activity of the AuPt supercrystal was only normalized by the platinum mass and compared with a platinum control (Supplementary Fig. 7), evidencing its better use when incorporated into the 2D supercrystal (Supplementary Fig. 10).

a, H2 generation rate normalized by the total mass of catalyst in both conditions, dark and light, for Au and AuPt supercrystals. b, Arrhenius plot for AuPt supercrystal. Ea was reduced from 33.4 kJ mol−1 to 29.8 kJ mol−1 upon white-light illumination. Irradiance used in all experiments, ∼110 mW cm−2, T = 25 °C. Data are presented as the mean values of at least three independent measurements. Error bars represent the s.d.
Temperature-dependent experiments were conducted to evaluate the activation barrier (Ea)16. We first investigated the temperature-dependent activity of the Au supercrystal versus the AuPt supercrystal over the 10–40 °C temperature range, without illumination. As expected, the bimetallic sample always presented a larger activity (Supplementary Fig. 11a). While the activation barrier for the Au supercrystal was 40.7 kJ mol−1 (Supplementary Fig. 11b), the one for the AuPt supercrystal was 33.4 kJ mol−1 (blue curve in Fig. 3b), evidencing the superior activity due to the platinum. The same temperature-dependent study was carried out again, but now comparing the activity of AuPt supercrystal in dark and light over the same temperature range. Interestingly, we found that the H2 production was always enhanced across the range upon light assistance, although the enhancement factor was not constant. Despite this, the use of white light for photocatalysis resulted in a 10% decrease in the activation barrier for the Pt–FA interface (Fig. 3b and Supplementary Fig. 12). All these experimental results highlight the synergy between the optical properties of the hexagonal array of 22 nm AuNPs and the interstitial PtNPs for sunlight-driven H2 production. A similar trend was observed for the same system with 39 nm AuNPs (Supplementary Fig. 13). In the next section, we intended to disentangle the plasmonic contributions to the improved performance of AuPt with respect to H2 generation.
Mechanistic insight
The superior photocatalytic activity of the AuPt clearly suggests that PtNPs benefit from the optical properties of the supercrystal. Several possible contributions are discussed for plasmonic photocatalysis with mono- or bimetallic systems: enhanced near fields, thermalized hot carriers, non-thermalized hot carriers and local heating44,45,46,47. Hence, unravelling the dominant energy pathway improving PtNP performance at the interparticle gap of the gold array is crucial to fully exploit the advantages of such structures. A set of experiments was conducted to gain insight into the PtNP activation due to the interaction between the incoming light and the supercrystal. Figure 4 summarizes our efforts to elucidate the plasmonic contributions to the increased reaction rates.

a, Wavelength-dependent H2 production (blue) plotted simultaneously with the electric (E) field strength (black) in the hotspots within the visible range and the weighted absorbance (grey). The photoactivity of the films peaks at 650 nm. Data are presented as the mean values of at least three independent measurements. Error bars represent the s.d. b, Weighted electric field intensity in between two AuNPs with a PtNP located in the centre of the gap. The asymmetry of the electric field to the left and right side of the PtNP is due to numerical inaccuracy. c, Weighted electric field intensity map of a bimetallic supercrystal. d, Transient absorption (A) spectra of Au and AuPt supercrystals when pumped at λ = 650 nm. Orange and red dashed lines, gold probing wavelengths; purple dashed line, platinum probing wavelength. e, Kinetics in AuNPs on Au and AuPt supercrystals at the probing wavelengths. λpump = 650 nm. No differences are observed in plasmon decay, suggesting no modification in hot electron dynamics upon platinum inclusion. f, Kinetics at λprobe = 420 nm. λpump = 650 nm. Additional contrast is presented in the bimetallic supercrystal, indicating platinum activation.
First, wavelength-dependent catalytic experiments were performed on the bimetallic supercrystal, which made it possible to discriminate the photocatalyst response in different spectral regions48. For these tests, the broadband lamp was equipped with bandpass filters centred at 450, 550, 650, 750 and 800 nm, with a bandwidth of 20 nm. The power (5 mW) was corrected for each wavelength to conduct the experiments with a constant irradiance and to match the power used previously with unfiltered light. The resulting wavelength-dependent H2 generation rates are shown in Fig. 4a, plotted together with the simulation of the enhanced electric field intensity at the hotspot for the topmost gold layer within the same spectral range, and the weighted absorbance. The relevance of hotspots in bimetallic plasmonic structures for photocatalysis has been proven for different systems15,35. Only the top layer of each domain was considered in the simulations, as this is where the catalysis mainly occurs, as we will discuss in the following paragraph. The average electric field enhancement at the hotspots was calculated by FDTD simulations (Methods). The simulations were evaluated in the centre of an interparticle gap between two gold particles because this is the location where we expect the PtNPs to exhibit the highest photocatalytic activity due to the strongest hotspots existing there.
As can be observed, the photoactivity of the AuPt supercrystal correlates with the electric field enhancement: the reaction rate peaks at the enhancement maximum. Based on this result, it can be inferred that at around 650 nm, the plasmonic components are funnelling more energy to the PtNPs, enhancing their absorption, and thus increasing the generation of excited carriers at the active metal which contribute to the redox reactions9,15,35. This manifests as an improved performance in catalysis at that wavelength. Concomitantly, the maximized reaction rate does not coincide with the maximum absorbance (Fig. 2). This is an indication that the experimentally observed enhancement upon illumination is not primarily influenced by heat generated upon absorption by the supercrystals, but rather that the electric field enhancement facilitates the reaction. If heat had caused the rate increase, a larger enhancement would have been expected at shorter wavelengths, where the absorbance is higher (Fig. 4a)49. The sample fabrication process yields 2D supercrystals with monolayer, bilayer and multilayer domains (Table 1). Therefore, it is worth investigating how the electric field enhancement depends on the number of layers. We simulated the spatial distribution of the electric field enhancement in the third dimension for different numbers of layers illuminated at 600 nm excitation wavelength (Supplementary Figure 14), the spectral range where we saw the highest catalytic activity. Consistent with Mueller et al.30, it is almost exclusively the topmost layer that is plasmonically activated, giving a strong electric field that boosts the catalytic process. This is due to the plasmonic stop band, which unlike the polaritonic modes, does not allow the light to enter the supercrystal24,30, maximizing the field enhancement at the topmost layer.
Additional optical simulations were performed to screen the electric field enhancement across the gap (in the x,y plane) when exciting the AuPt supercrystal in and out of the field enhancement maximum at 550, 650 and 800 nm (Supplementary Figure 15), covering the wavelength range used in photocatalytic experiments. While the presence of PtNPs led to an electric field intensity enhancement of 103 times, a superior confinement is achieved at 650 nm (Fig. 4b). The intensity of the weighted electric field at 650 nm was almost three times larger than the intensity at the other wavelengths (Fig. 4a and Supplementary Figure 15). Figure 4c depicts the lattice unit cell of the FDTD simulations, showing the PtNPs in the strong hotspots of the AuNPs where they are responsible for the enhancement and therefore for the high photocatalytic activity. It is important to emphasize that Fig. 4a was simulated without PtNPs in the gap, while Fig. 4b,c was calculated with PtNPs in the gaps between the AuNPs, leading to different electric field enhancements. However, both kinds of simulation show the strong enhancement at 650 nm and the same enhancement ratio between different wavelengths. The electric field enhancement simulations are in line with the experimental data. Interestingly, the H2 generation rate at 800 nm was 1.6 times larger than at 550 nm, even though the absorbance at lower energy was negligible compared to the one at 550 nm (Fig. 2). This further supports our interpretation that heat is not the main plasmonic contribution to the enhanced performance of PtNPs.
Studying the plasmon relaxation of the excited sample makes it possible to gain an insight into the interaction between the constituents of the samples, and to precisely evaluate tunnelling from the plasmonic to the catalytic component driven by the enhanced electric field50,51. To this end, the electronic dynamics of AuPt and Au supercrystals, and the platinum control, were investigated by transient absorption spectroscopy. These results complemented the bulk wavelength-dependent experiments and optical simulations.
The resulting normalized transient absorption spectra of both Au and AuPt supercrystals, when pumped at the largest photoactivity (λ = 650 nm), are shown in Fig. 4d. The red (λ = 610 nm) and orange (λ = 640 nm) dashed lines correspond to the probing wavelengths for the kinetics in the Au and AuPt supercrystals, respectively. These wavelengths were selected as they correspond to the center wavelength of the plasmon-related resonance (bleach) in each system, and thereby give access to information about the relaxation of excited carriers in gold52. It is worth mentioning that the emergence of polaritonic modes does not affect the electron dynamics in each individual AuNP52,53. The dynamics at the plasmon bleach are displayed in Fig. 4e. Essentially no changes in the dynamics were observed, suggesting the absence of meaningful gold-to-platinum charge transfer when exciting the optically active layers in the wavelength regime under investigation54. The similarities in the kinetics also make it possible to neglect the charge transfer through the conductive ITO substrate. The immediate onset of additional contrast in the platinum spectral region of the hybrid material points towards a direct excitation of platinum when the system is excited at 650 nm (Fig. 4f). Plain PtNPs show no contrast under these excitation conditions (Supplementary Figs. 16 and 17). The fact that this larger contrast is only observed for the AuPt supercrystal at wavelengths where stronger fields act on the PtNPs is an indication of enhanced absorption processes due to the strongly confined electric field. This probably corresponds to increased d-to-s transition rates within platinum32,33. Even when pumping the 2D supercrystals at 400 and 500 nm, the spectral region at which interband transitions dominate (Supplementary Fig. 18), no notable differences between the materials and no strong indication of platinum excitation is observed. This further supports the hypothesis that the electric fields in the hotspots drive the reaction.
The evidence presented in this section sheds light on the mechanisms for harnessing light to boost the performance of plasmonic-catalytic nanoreactors. Our findings indicate that by incorporating PtNPs into the interparticle gaps of hexagonally arrayed 22 nm AuNPs, it is possible to turn a catalyst with weak visible light absorption into an active sunlight-type photocatalyst via enhanced excitation. From our experiments, we have effectively determined that the improved photoactivity is a consequence of the interaction of strongly confined electromagnetic fields (hotspots) with interstitial PtNPs, whereas plasmonically generated heat, and charge injection from gold to platinum, appear in comparison to have minimal effect on the reaction rate enhancement. As such, and according to our pump–probe results, the main contribution of the plasmon in this hybrid structure is the activation of the catalytic sites (platinum) via the plasmonic (gold) enhanced near-fields. The increased performance of PtNPs upon illumination can be attributed to the generation of excited carriers (holes and electrons) participating in the redox cycle of formic acid dehydrogenation. The light-enhanced reaction rate is primarily due to the cleavage of the C–H bond, which represents the rate-limiting step of the reaction11,15,46,55. These results highlight that accurately positioning catalytic metals in optical hotspots is a crucial parameter to modulate in these types of bimetallic photocalatysts. Various parameters, ranging from interparticle distance, constituent size or even the number of layers, can be modified to achieve greater electric fields at the hotspots, which in turn will impact the performance of these optocatalytic structures.
With these established design rules, efforts will be directed towards inexpensive and sustainable devices, which can be achieved with both Earth-abundant plasmonic (that is, copper, aluminium, magnesium) and catalytic (that is, iron) metals17,56, and the development of supports preserving the achieved supercrystal structure (that is, mesoporous silica). In addition to formic acid dehydrogenation, we anticipate that the underlying physical principles described in our work will be exploited towards other important chemical transformations, such as CO2 reduction or carbon–carbon coupling57,58.
Conclusions
In this study, we introduce a plasmonic bimetallic supercrystal in which we achieved the desired antenna–reactor configuration from colloidal suspension. The size of the supercrystal can be extended up to several square millimetres while maintaining the configuration, and monolayer, bilayer and multilayer domains can be achieved. We prove the synergistic effect of plasmonic-catalytic components in supercrystals by observing a two-fold increase in the rate of generation of H2 in the formic acid dehydrogenation reaction at solar irradiances. By using well-defined supercrystals, we were able to distinguish the plasmonic contributions to the catalytic enhancement. Our findings reveal that the performance of the PtNPs is primarily dictated by the intensity of the electromagnetic fields at the hotspots, whereas thermal contributions and charge injection from AuNP into the catalytic centre have a lower impact on the reaction enhancement.
Methods
Materials
Tetrachloroauric(III) acid (≥99.9% trace metals basis), hexadecyltrimethylammonium bromide (≥98%) and chloride (≥98%), l-ascorbic acid (reagent grade), chloroplatinic acid solution (8 wt% in H2O), sodium citrate trihydrate (≥99%), sodium borohydride (≥98%) and ITO-coated glass slides (surface resistivity, 8–12 Ω cm−2) were from Sigma-Aldrich. Toluene (≥99.5%), tetrahydrofuran (≥99.5%) and ethanol (denatured, >96%) were from VWR. Diethylene glycol (DEG, reagent grade) was from Merck. PSSHs (PSSH2k: MN = 2,000 g mol−1, MW = 2,300 g mol−1; PSSH5k: MN = 5,300 g mol−1, MW = 5,800 g mol−1; PSSH10k: MN = 11,500 g mol−1, MW = 12,400 g mol−1) were from Polymer Source. All reagents were used without further treatment. H2 standards (100, 500 and 1,000 ppm) were purchased from Linde Gas. Ultrapure water with a resistivity of 18 MΩ·cm was used in all experiments.
Synthesis of AuNPs
AuNPs were synthesized based on the seeded growth protocol from Zheng et al.59 and functionalized with PSSH as described previously25. Several batches were prepared: AuNPs with a core diameter of <D> = 22.0 ± 1.0 nm and a particle concentration of c = 9.5 nM were functionalized with PSSH2k; AuNPs with <D> = 39.0 ± 1.0 nm and c = 2.4 nM and AuNPs with <D> = 38.7 ± 0.9 nm and c = 2.0 nM were functionalized with PSSH5k; and AuNPs with <D> = 35.5 ± 1.0 nm and c = 2.6 nM were functionalized with PSSH5k and with PSSH2k.
Synthesis of PtNPs
PtNPs with diameters of 2.7 ± 0.6 nm were synthesized as follows: 500 μl of H2PtCl6 0.05 M were added to 8.6 ml of ultrapure water (stirred at 500 r.p.m.), succeeded by the addition of 500 μl of sodium citrate 0.1 M, which served as the stabilizing agent. The platinum precursor was reduced upon rapid addition of 500 μl of NaBH4 0.015 M, evidenced by the dark brown colour of the final solution. Finally, the solution was left undisturbed for 10 min60.
The as-synthesized PtNPs were functionalized with PSSH2k by phase transfer as described in a previous report61. In detail, the aqueous PtNP dispersion (1,000 μl) was mixed with PSSH2k (1,100 μl, 0.18 mM) in toluene and ethanol (1,000 μl). After vigorous shaking, phase separation was facilitated by addition of a few milligrammes of sodium chloride. The toluene phase with the PtNPs was then carefully removed and used for self-assembly experiments without further purification. The PtNPs had a diameter of 2.7 ± 0.6 nm and the platinum concentration in the final dispersion in toluene was 9.6 mg l−1, corresponding to 65 nM particle concentration.
Self-assembly
Self-assembly of pure and mixed samples of polystyrene-functionalized NPs took place on DEG as a liquid subphase as described previously25,62. Different volume ratios of AuNP@PSSH and PtNP@PSSH2k were pipetted onto the DEG and the sample cell was covered with a glass cover-slip to slow down the evaporation of the toluene. After 16–24 h crystalline films had formed, which were carefully skimmed of with TEM grids or transferred to ITO substrates for catalysis experiments. Crystalline films with 2D hexagonal symmetry and incorporated PtNPs formed under all tested conditions (Supplementary Figs. 1 and 3). The samples differed in platinum loading and the resulting interparticle spacing. For the controls, just AuNP@PSSH or just PtNP@PSSH2k were used accordingly.
TEM
The TEM measurements for standard characterization were performed using a JEOL JEM-1011 instrument operating at 100 kV. Samples of self-assembled films were carefully skimmed off with a carbon-coated copper grid held by a tweezer. The grid was then dried in vacuum for at least 1 h. Quantitative analyses of size distributions and nearest-neighbour distances based on TEM measurements were performed with ImageJ v.1.50i. The indicated mean values are obtained from analyses of at least three images from different spots on the samples.
Layer identification and area calculation
The identification of the layer number and the size calculation of the different layers was done with an optical microscope (Nikon Eclipse LV 100) equipped with a microscope camera (Nikon DS-Ri2). The different contrasts in the transmission images enabled identification of the layers. Subsequently, the layer sizes where measured using Nikon NIS-Elements software.
Optical spectroscopy
A home-built microabsorbance set-up was used to measure the optical spectra of both Au supercrystals and AuPt supercrystals. A supercontinuum laser (NKT-FIU15) with a broad spectral range was used as the light source. The light was guided to an inverse microscope (Olympus IX71) equipped with a 100× objective (numerical aperture, 0.9) that focuses the light onto the sample, and a second 100× objective (numerical aperture, 0.8) to collect the transmitted light (T). Via an optical fibre the collected light was guided to the spectrometer (Avantes AvaSpec 3648). For the reflectance measurements the reflected light (R) was guided through two beam-splitters to a collector lens that was connected via an optical fibre with the Avantes spectrometer. Afterwards, the absorbance (A) was calculated by A = 100% − R − T.
FDTD simulations
For the FDTD simulations the commercial Lumerical FDTD Solutions software package was used as described in refs. 24,31,40. Spherical AuNPs with a diameter of 22 nm and interparticle gaps of 2 nm were packed into a hexagonal lattice. To simulate the different crystal layers one to three layers were stacked in the ABC sequence of an fcc crystal. The cell size of the mesh was set to 0.2 nm to accurately model the geometry of the supercrystal. The dielectric function of gold was determined by fitting the experimental data from Olmon et al.63. For the ligand molecules (polystyrene), a dielectric medium with refractive index n = 1.4 was assumed to fill the space between the gold particles. The lattice unit cell of the crystal was chosen as the FDTD simulation region with periodic boundary conditions. A broadband plane-wave source along the z direction was used as excitation source. The reflectance (R) was recorded with a monitor behind the source while the transmittance (T) was measured with a monitor behind the supercrystal. The absorbance (A) was subsequently calculated by A = 1 − R − T. For the AuPt simulations, 3 nm platinum particles where added in the gaps of the AuNPs and the gap size was changed to 3.5 nm. The dielectric function of the platinum was determined by fitting the experimental data from Palik64. All the other parameters were kept the same. To simulate unpolarized light, every simulation was performed twice, once with with x-polarized light and once with y-polarized light. Subsequently, the mean value was taken to represent unpolarized light. For the simulations of the electric field enhancement, the simulated spectra of the topmost layer for 1L, 2L and 3L were summed up and weighted by the percentage of the area (Table 1) to simulate the case of the photocatalysis experiments.
Catalysis
The catalytic experiments were performed in a clean 15 ml reactor containing a flat quartz window (diameter, 1.22 cm) and a rubber septum for sampling. The samples (supercrystals on ITO) were glued to a sample holder facing the quartz window, ensuring a complete and homogeneous illumination. The reactor is fitted with a water jacket where water was pumped in and out, allowing accurate control of the temperature for the duration of the experiments using an external cryostat (±0.5 °C). The reaction mixture was stirred at 550 r.p.m. using a magnetic bar. The catalysis time was set to 1 h and a renewed degassed reaction mixture (N2 for 30 min) was used in all the experiments. The reactor was thoroughly rinsed for every new experiment. For illumination, a commercial broadband arc lamp (Newport, 66921) was used; the ultraviolet and infrared contributions were filtered out by a long-pass filter (λ > 400 nm) and with an infrared liquid filter (6123NS), respectively. The power was determined using a commercial thermal detector (Thorlabs, S401C). For gas detection, we used a gas chromatograph (Perkin Elmer, Clarus 590 GC) equipped with both a flame-ionization detector to detect organic traces and a thermal conductivity detector for H2 detection. In addition, the flame-ionization detector was coupled to an H2 generator (Perkin Elmer, NM plus H2 100).
ICP-MS
Elemental mass concentrations were measured with an Agilent 7700 ICP-MS set-up. The calibration curves were prepared with corresponding elemental standards (Carl Roth). Film samples on ITO were dissolved overnight in 200 μl freshly prepared aqua regia and then diluted to 4,000 μl with 2% hydrochloric acid. To determine the PtNP concentration, 50 μl of the dispersion was dried, dissolved in 240 μl aqua regia overnight and then diluted to 2,500 μl with 2% hydrochloric acid. The elemental concentrations in these solutions were determined and used to calculate the according masses.
Transient absorption measurements
Transient absorption data were obtained using an Ultrafast Systems Helios transient absorption spectrometer. The system was seeded with 800 nm, 35 fs laser pulses generated by a mode-locked titanium:sapphire oscillator (SpectraPhysics, MaiTai) and amplified by chirped pulse amplification (SpectraPhysics, Spitfire Ace) operating at 1 kHz. The output of the amplifier was split in two arms: one arm was used to generate pump pulses of varying wavelengths, either using an optical parametric amplifier (Light Conversion, TOPAS Prime) or by simple frequency doubling in a beta barium borate crystal. Every second one of these pump pulses was blocked by a chopper operating at 500 Hz before they were focused onto the sample, which was fixed in space using a 3D translation stage. The spot diameter of the pump pulses was 500 μm (I = Imax × 1/e2). Using a neutral density filter, the fluence of the pump was adjusted to 150 μJ cm−2. The second arm was led through a delay stage and then used for white-light generation in a CaF crystal. The generated white light was then focused onto the sample with a spot diameter of 250 μm (1/e2). Spectra were recorded for varying delay times in transmission with an average time of 1 s. Differential spectra were obtained by subtracting the pumped and unpumped spectra for each delay time. Afterwards, the differential spectra were corrected for chirp and pump scattering and normalized to the bleach signal at the time of maximum contrast. Single dA versus delay time kinetics were extracted for fixed probe wavelengths.
Data availability
All data in the manuscript and Supplementary Information are stored in Zenodo: https://doi.org/10.5281/zenodo.8354800.svg
References
Eppinger, J. & Huang, K. W. Formic acid as a hydrogen energy carrier. ACS Energy Lett. 2, 188–195 (2017).
Aslam, U., Rao, V. G., Chavez, S. & Linic, S. Catalytic conversion of solar to chemical energy on plasmonic metal nanostructures. Nat. Catal. 1, 656–665 (2018).
Rodrigues, M. Pd. S. et al. Gold–rhodium nanoflowers for the plasmon-enhanced hydrogen evolution reaction under visible light. ACS Catal. 11, 13543–13555 (2021).
Holmes-Gentle, I., Tembhurne, S. & Suter, C. Kilowatt-scale solar hydrogen production system using a concentrated integrated photoelectrochemical device. Nat. Energy 8, 586–596 (2023).
Linic, S., Aslam, U., Boerigter, C. & Morabito, M. Photochemical transformations on plasmonic metal nanoparticles. Nat. Mater. 14, 567–576 (2015).
Hammer, B. & Norskov, J. K. Why gold is the noblest of all the metals. Nature 376, 238–240 (1995).
Esterhuizen, J. A., Goldsmith, B. R. & Linic, S. Theory-guided machine learning finds geometric structure–property relationships for chemisorption on subsurface alloys. Chem 6, 3100–3117 (2020).
Ezendam, S. et al. Hybrid plasmonic nanomaterials for hydrogen generation and carbon dioxide reduction. ACS Energy Lett. 7, 778–815 (2022).
Linic, S., Chavez, S. & Elias, R. Flow and extraction of energy and charge carriers in hybrid plasmonic nanostructures. Nat. Mater. 20, 916–924 (2021).
Zhao, S. et al. Introducing visible-light sensitivity into photocatalytic CeO2 nanoparticles by hybrid particle preparation exploiting plasmonic properties of gold: enhanced photoelectrocatalysis exemplified for hydrogen peroxide sensing. Nanoscale 13, 980–990 (2021).
Zhao, S. et al. Tailoring of the photocatalytic activity of CeO2 nanoparticles by the presence of plasmonic ag nanoparticles. Nanoscale 14, 12048–12059 (2022).
Khalid, W. et al. Immobilization of quantum dots via conjugated self-assembled monolayers and their application as a light-controlled sensor for the detection of hydrogen peroxide. ACS Nano 5, 9870–9876 (2011).
Xie, W., Walkenfort, B. & Schlücker, S. Label-free SERS monitoring of chemical reactions catalyzed by small gold nanoparticles using 3D plasmonic superstructures. J. Am. Chem. Soc. 135, 1657–1660 (2013).
Swearer, D. F. et al. Heterometallic antenna–reactor complexes for photocatalysis. Proc. Natl Acad. Sci. USA 113, 8916–8920 (2016).
Herran, M. et al. Tailoring plasmonic bimetallic nanocatalysts toward sunlight-driven H2 production. Adv. Funct. Mater. 32, 2203418 (2022).
Zhou, L. et al. Quantifying hot carrier and thermal contributions in plasmonic photocatalysis. Science 362, 69–72 (2018).
Yuan, Y. et al. Earth-abundant photocatalyst for H2 generation from NH3 with light-emitting diode illumination. Science 378, 889–893 (2022).
Hogan, N. J. et al. Nanoparticles heat through light localization. Nano Lett. 14, 4640–4645 (2014).
Kim, Y., Smith, J. G. & Jain, P. K. Harvesting multiple electron–hole pairs generated through plasmonic excitation of Au nanoparticles. Nat. Chem. 10, 763–769 (2018).
Jain, P. K., Qian, W. & El-Sayed, M. A. Ultrafast cooling of photoexcited electrons in gold nanoparticle-thiolated DNA conjugates involves the dissociation of the gold–thiol bond. J. Am. Chem. Soc. 128, 2426–2433 (2006).
Boong, S. K. et al. Superlattice-based plasmonic catalysis: concentrating light at the nanoscale to drive efficient nitrogen-to-ammonia fixation at ambient conditions. Angew. Chem. Int. Ed. 62, e202216562 (2023).
Yalavarthi, R., Henrotte, O., Kment, Š. & Naldoni, A. Determining the role of Pd catalyst morphology and deposition criteria over large area plasmonic metasurfaces during light-enhanced electrochemical oxidation of formic acid. J. Chem. Phys. 157, 114706 (2022).
Yalavarthi, R. et al. Multimetallic metasurfaces for enhanced electrocatalytic oxidations in direct alcohol fuel cells. Laser Photonics Rev. 16, 2200137 (2022).
Mueller, N. S. et al. Deep strong light–matter coupling in plasmonic nanoparticle crystals. Nature 583, 780–784 (2020).
Schulz, F. et al. Structural order in plasmonic superlattices. Nat. Commun. 11, 1–9 (2020).
Mueller, N. S. et al. Direct optical excitation of dark plasmons for hot electron generation. Faraday Discuss. 214, 159–173 (2019).
Cortés, E. et al. Optical metasurfaces for energy conversion. Chem. Rev. 122, 15082–15176 (2022).
Schulz, F. & Lange, H. Optimizing interparticle gaps in large-scale gold nanoparticle supercrystals for flexible light–matter coupling. Adv. Opt. Mater. 10, 2202064 (2022).
Hamon, C. et al. Collective plasmonic properties in few-layer gold nanorod supercrystals. ACS Photonics 2, 1482–1488 (2015).
Mueller, N. S. et al. Surface-enhanced Raman scattering and surface-enhanced infrared absorption by plasmon polaritons in three-dimensional nanoparticle supercrystals. ACS Nano 15, 5523–5533 (2021).
G. M. Vieira, B., Mueller, N. S., Barros, E. B. & Reich, S. Plasmonic properties of close-packed metallic nanoparticle mono- and bilayers. J. Phys. Chem. C 123, 17951–17960 (2019).
Aslam, U., Chavez, S. & Linic, S. Controlling energy flow in multimetallic nanostructures for plasmonic catalysis. Nat. Nanotechnol. 12, 1000–1005 (2017).
Chavez, S., Aslam, U. & Linic, S. Design principles for directing energy and energetic charge flow in multicomponent plasmonic nanostructures. ACS Energy Lett. 3, 1590–1596 (2018).
Zhang, C. et al. Al–Pd nanodisk heterodimers as antenna–reactor photocatalysts. Nano Lett. 16, 6677–6682 (2016).
Lee, S. et al. Core–shell bimetallic nanoparticle trimers for efficient light-to-chemical energy conversion. ACS Energy Lett. 5, 3881–3890 (2020).
Elias, R. C. & Linic, S. Elucidating the roles of local and nonlocal rate enhancement mechanisms in plasmonic catalysis. J. Am. Chem. Soc. 144, 19990–19998 (2022).
Baffou, G. et al. Photoinduced heating of nanoparticle arrays. ACS Nano 7, 6478–6488 (2013).
Besteiro, L. V., Kong, X.-T., Wang, Z., Hartland, G. & Govorov, A. O. Understanding hot-electron generation and plasmon relaxation in metal nanocrystals: quantum and classical mechanisms. ACS Photonics 4, 2759–2781 (2017).
Sivan, Y., Baraban, J., Un, I. W. & Dubi, Y. Comment on “quantifying hot carrier and thermal contributions in plasmonic photocatalysis”. Science 364, eaaw9367 (2019).
Mueller, N. S. et al. Dark interlayer plasmons in colloidal gold nanoparticle bi- and few-layers. ACS Photonics 5, 3962–3969 (2018).
Tedsree, K. et al. Hydrogen production from formic acid decomposition at room temperature using a Ag–Pd core–shell nanocatalyst. Nat. Nanotechnol. 6, 302–307 (2011).
Czaun, M. et al. Iridium-catalyzed continuous hydrogen generation from formic acid and its subsequent utilization in a fuel cell: toward a carbon neutral chemical energy storage. ACS Catal. 6, 7475–7484 (2016).
Un, I.-W. & Sivan, Y. The role of heat generation and fluid flow in plasmon-enhanced reduction–oxidation reactions. ACS Photonics 8, 1183–1190 (2021).
Rodio, M. et al. Experimental evidence for nonthermal contributions to plasmon-enhanced electrochemical oxidation reactions. ACS Catal. 10, 2345–2353 (2020).
Zheng, Z., Tachikawa, T. & Majima, T. Plasmon-enhanced formic acid dehydrogenation using anisotropic Pd–Au nanorods studied at the single-particle level. J. Am. Chem. Soc. 137, 948–957 (2015).
Wu, B. et al. Plasmon-mediated photocatalytic decomposition of formic acid on palladium nanostructures. Adv. Opt. Mater. 4, 1041–1046 (2016).
Gargiulo, J. et al. Impact of bimetallic interface design on heat generation in plasmonic Au/Pd nanostructures studied by single-particle thermometry. Nat. Commun. 14, 1–11 (2023).
Baffou, G., Bordacchini, I., Baldi, A. & Quidant, R. Simple experimental procedures to distinguish photothermal from hot-carrier processes in plasmonics. Light Sci. Appl. 9, 1–16 (2020).
Li, K. et al. Balancing near-field enhancement, absorption, and scattering for effective antenna–reactor plasmonic photocatalysis. Nano Lett. 17, 3710–3717 (2017).
Link, S. & El-Sayed, M. A. Shape and size dependence of radiative, non-radiative and photothermal properties of gold nanocrystals. Int. Rev. Phys. Chem. 19, 409–453 (2000).
Savage, K. J. et al. Revealing the quantum regime in tunnelling plasmonics. Nature 491, 574–577 (2012).
Hoeing, D., Schulz, F., Mueller, N. S., Reich, S. & Lange, H. Dark plasmon modes for efficient hot electron generation in multilayers of gold nanoparticles. J. Chem. Phys. 152, 064710 (2020).
Staechelin, Y. U., Hoeing, D., Schulz, F. & Lange, H. Size-dependent electron–phonon coupling in monocrystalline gold nanoparticles. ACS Photonics 8, 752–757 (2021).
Engelbrekt, C., Crampton, K. T., Fishman, D. A., Law, M. & Apkarian, V. A. Efficient plasmon-mediated energy funneling to the surface of Au@Pt core–shell nanocrystals. ACS Nano 14, 5061–5074 (2020).
Han, L. et al. Anchoring Pt single atoms on Te nanowires for plasmon-enhanced dehydrogenation of formic acid at room temperature. Adv. Sci. 6, 1900006 (2019).
Ringe, E. Shapes, plasmonic properties, and reactivity of magnesium nanoparticles. J. Phys. Chem. C 124, 15665–15679 (2020).
Yu, S., Wilson, A. J., Heo, J. & Jain, P. K. Plasmonic control of multi-electron transfer and C–C coupling in visible-light-driven CO2 reduction on Au nanoparticles. Nano Lett. 18, 2189–2194 (2018).
Li, Z. & Kurouski, D. Probing the plasmon-driven Suzuki–Miyaura coupling reactions with cargo-TERS towards tailored catalysis. Nanoscale 13, 11793–11799 (2021).
Zheng, Y., Zhong, X., Li, Z. & Xia, Y. Successive, seed-mediated growth for the synthesis of single-crystal gold nanospheres with uniform diameters controlled in the range of 5–150 nm. Part. Part. Syst. Charact. 31, 266–273 (2014).
Sanles-Sobrido, M. et al. Highly catalytic single-crystal dendritic Pt nanostructures supported on carbon nanotubes. Chem. Mater. 21, 1531–1535 (2009).
Schulz, F., Tober, S. & Lange, H. Size-dependent phase transfer functionalization of gold nanoparticles to promote well-ordered self-assembly. Langmuir 33, 14437–14444 (2017).
Dong, A., Chen, J., Vora, P. M., Kikkawa, J. M. & Murray, C. B. Binary nanocrystal superlattice membranes self-assembled at the liquid–air interface. Nature 466, 474–477 (2010).
Olmon, R. L. et al. Optical dielectric function of gold. Phys. Rev. B 86, 235147 (2012).
Palik, E. D. (ed.) Handbook of Optical Constants of Solids (Academic Press, 1985).
Acknowledgements
We acknowledge funding and support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (EXC 2089/1, 390776260) e-conversion Cluster of Excellence, the Bavarian programme Solar Technologies Go Hybrid (SolTech), the Center for NanoScience (CeNS) and the European Commission through the ERC Starting Grant CATALIGHT (802989) and the Consolidator Grant DarkSERS (772108). A.S.-C. acknowledges Xunta de Galicia, Spain, for her postdoctoral fellowship. F.S. acknowledges support by PIER Hamburg (PIF-2021-02). H.L., S.R. and F.S. acknowledge funding by the DFG (504656879). The work is also supported by the Cluster of Excellence ‘Advanced Imaging of Matter’ of the DFG (EXC 2056, 390715994) and the SupraFAB research building at Freie Universität Berlin. We thank J. Barrios and S. Jonak Jutta for help with palladium nanoparticle synthesis.
Funding
Open access funding provided by Universität Hamburg.
Author information
Authors and Affiliations
Contributions
Idea and coordination: F.S. and E.C. Colloidal synthesis: M.H., A.S.-C. and F.S. Colloidal assembly: F.S. Photocatalytic experiments: M.H. Electron microscopy analysis: F.S. and A.K. Optical spectroscopy and optical microscopy characterization: S.J., M.K. and S.R. Transient absorption spectroscopy: D.H. and H.L. FDTD simulations: S.J. Data analysis and discussion: all authors. Supervision and funding: E.C., S.R., W.J.P., H.L. and F.S. Figures: M.H. and S.J. Writing: M.H. and S.J. Reviewing and editing: all authors.
Corresponding authors
Ethics declarations
Competing interests
M.H., F.S. and E.C. are listed as co-inventors of a European patent application, ‘Embedding of catalytically active nanoparticles into superstructures of plasmonic nanoparticles to enhance the photocatalytic activity’ (EP22186109.9). The patent describes the fabrication of bimetallic supercrystals and the photocatalytic performance presented in this manuscript. The remaining authors declare no competing interests
Peer review
Peer review information
Nature Catalysis thanks Sanchari Chowdhury, Zong-Yan Zhao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–18 and Tables 1–7.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Herran, M., Juergensen, S., Kessens, M. et al. Plasmonic bimetallic two-dimensional supercrystals for H2 generation. Nat Catal 6, 1205–1214 (2023). https://doi.org/10.1038/s41929-023-01053-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41929-023-01053-9
This article is cited by
-
Catalysis in the hot spots
Nature Catalysis (2023)

{kind=link}