
Abstract
RNA editosomes selectively deaminate cytidines to uridines in plant organellar transcripts—mostly to restore protein functionality and consequently facilitate mitochondrial and chloroplast function. The RNA editosomal pentatricopeptide repeat proteins serve target RNA recognition, whereas the intensively studied DYW domain elicits catalysis. Here we present structures and functional data of a DYW domain in an inactive ground state and activated. DYW domains harbour a cytidine deaminase fold and a C-terminal DYW motif, with catalytic and structural zinc atoms, respectively. A conserved gating domain within the deaminase fold regulates the active site sterically and mechanistically in a process that we termed gated zinc shutter. Based on the structures, an autoinhibited ground state and its activation are cross-validated by RNA editing assays and differential scanning fluorimetry. We anticipate that, in vivo, the framework of an active plant RNA editosome triggers the release of DYW autoinhibition to ensure a controlled and coordinated cytidine deamination playing a key role in mitochondrial and chloroplast homeostasis.

Similar content being viewed by others
Main
Plant RNA editing specifically converts several hundreds of cytidines to uridines in mitochondrial and chloroplast transcripts1,2,3,4. The RNA editing activity has to be stringently suppressed in the cytosol and stimulated only in the organelles at the target site, as no C-to-U RNA editing has been described in nuclear transcripts in plants5,6. Nuclear-encoded pentatricopeptide repeat (PPR) proteins with a C-terminal DYW domain have been characterized as site-specific factors for C-to-U RNA editing in plant mitochondria and plastids7,8. RNA substrate recognition is conferred by the PPR tract, whereas the exact role of the DYW domain, which can be also recruited to an editing site in trans, has not been clarified9,10,11,12,13,14,15,16,17. The DYW domain, which was named by the highly conserved last three amino acids, aspartate, tyrosine and tryptophan, has been proposed as the best candidate to elicit deamination employing a HxE(x)nCxxC zinc ion binding signature18,19. Indeed, DYW domains from DYW1 and ELI1 demonstrated zinc ion binding capacity, and recent orthogonal E.coli as well as in vitro experiments with a single DYW containing PPR protein strongly support their function as catalytic entities within the RNA editosome20,21,22,23. Apart from PPR proteins and DYW domains, several other factors (for example MORF or ORRM proteins) were shown to be part of RNA editosomes6,10,24,25. Until now, only MORF proteins and PPR repeats bound to the target RNA are structurally characterized13,26,27,28. As DYW domains share only low sequence conservation with known deaminase structures (from 5 to 19% residue identities), modelling attempts have been conducted, albeit with a limited reliability18,20,21,29. Finally, missing structural information has left the exact mechanistic function, regulation and catalytic properties of DYW domains within the RNA editosome open.
Herein we describe structures of a DYW domain and find that, apart from a cytidine deaminase fold, DYW domains contain a characteristic DYW motif, stabilized by a zinc atom, as well as a gating domain that controls zinc-mediated catalysis sterically and catalytically. The catalytic regulation hallmarks an unusual protein regulation principle where, upon activation, a major movement of the gating domain alters the coordination around the catalytic zinc atom while in the inactive state, the zinc is inhibited by its coordination setting. We employed in vivo RNA editing assays to map the potential RNA path on the DYW domain and identify key residues required for regulation and catalysis to occur. Finally, RNA in vitro editing and thermal shift assays consolidate the structural data and confirm a tetrahydrouridine or nucleotide triphosphate-triggered activation mirroring the two different conformational states. Beyond the identification of an unusual principle in metalloenzyme regulation, our results reveal key mechanisms in plant organellar RNA editing catalysis, its autoinhibition and have far-reaching implications for mitochondrial and chloroplast homeostasis.
Results
Crystal structure of the Arabidopsis thaliana OTP86DYW
Here we report crystal structures of the DYW domain of an Arabidopsis thaliana (A. thaliana) plastid RNA editing factor, OTP86, as the outcome of a solubility and crystallization screening of over 100 different DYW domain constructs from 30 PPR proteins. OTP86 was characterized as a site-specific factor for an editing site in rps14 transcripts30. The protein consists of 20 N-terminal PPR repeats, E1 and E2 motifs, which are predicted to have a PPR- or tetratricopeptide-repeat-like (TPR-like) fold and a C-terminal DYW domain31.
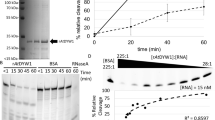
To initially assess whether the OTP86 DYW domain (OTP86DYW) is an active editing factor, we conducted in vivo orthogonal RNA editing assays in Escherichia coli (E. coli) and in vitro assays with purified proteins. Both methods verified the cytidine deaminase activity of the OTP86 DYW domain when fused with the PPR tract of the moss Physcomitrium PPR56 protein (Supplementary Fig. 1a–c)22,23. When the catalytically important E894 of OTP86DYW was replaced by an alanine, editing was abolished.
We then set out to pioneer the structural characterization of DYW domains exemplified by OTP86DYW. Several years of crystallization attempts were severely hampered by the very limited amounts of soluble OTP86DYW (residues G826 to W960), which migrates at a molecular weight of about 15 kDa in size-exclusion chromatography, indicating a monomeric state (Supplementary Fig. 2a). Finally, we obtained crystals of OTP86DYW belonging to space group C2 and diffracting to a resolution of 2.5 Å (Supplementary Fig. 3a and Supplementary Table 1). The structure was solved by single-wavelength anomalous dispersion (SAD) phasing harnessing four zinc atoms (see Methods, Supplementary Table 1 and Supplementary Fig. 3b,c for details).
The fold of OTP86DYW is highly similar to cytidine deaminases but has prominent additional features (Fig. 1a–e). A comparison of OTP86DYW with E. coli cytidine deaminase (PDB ID: 1CTU; ref. 32) reveals an overall similarity (r.m.s.d. = 2.4 Å for 72 of 132 residues superimposed) to the typical core deaminase fold comprising five β-strands flanked by two α-helices32. The region previously termed PG box covers the first two β-strands of the deaminase domain21,29,33. Remarkably, the deaminase fold of OTP86DYW is interrupted by an insertion of about 55 residues that bridge β-strand 2 and α-helix 2 (Figs. 1b and 2). The insertion is composed of an amphipathic α-helix that runs across one face of the entire structure contacting both α-helices of the deaminase fold with conserved hydrophobic residues (Supplementary Fig. 3d and Fig. 2) and re-enters the deaminase fold via a highly conserved β-finger at α-helix 2, which in turn harbours the HxE(x)nCxxC motif, crucial to catalysis and substrate binding29 (Figs. 1b,c and 2). This motif has a high similarity to the cytidine deaminase signature HxE(x)nPCxxC and contains a catalytically important glutamate residue (E894 in OTP86), only the proline is not conserved in DYW domains (Fig. 2)18. Contrasting the large inserted domain of OTP86DYW, E. coli cytidine deaminase only contains a smaller loop which instead points away from the active site permitting nucleotide entry (Fig. 1a). We conclude that the OTP86DYW active site seems to have limited accessibility for substrate cytidines, which is conferred by an insertion (H837-G891), and we thus term this insertion gating domain.

a, Superimposition of OTP86DYW (marine) with E. coli cytidine deaminase (EcCDZ, cyan) bound to the inhibitor zebularine (not shown) (PDB-ID: 1CTU; ref. 32). The consensus deaminase zinc ions are shown as green (OTP86DYW, Zn1) and light-green (EcCDZ, Zn) spheres, a zinc ion partially coordinated by the DYW motif is shown in yellow (Zn2). b, The OTP86DYW structure defines a paradigmatic organization for DYW domains. The cytidine deaminase domain (slate) coordinates a zinc ion (Zn1, green) three-fold with H892, C920 and C923, the fourth position is occupied by a water molecule (W, white sphere). The deaminase domain is interrupted by a gating domain (orange) and terminates with a DYW motif (red), partially coordinating a second zinc ion (Zn2, yellow). c, A close-up view on the cytidine deaminase active site, with catalytically relevant residues shown as sticks. d, A close-up view of the DYW motif and the flanking β-strand 7 as well as α-helix 3. e, Electrostatic surface potentials as indicated by the colour scale bar (bottom), obtained by APBS version 1.5 and plotted on the surface of OTP86DYW. Residues involved in zinc coordination are shown as sticks; zinc atoms are as in b. Rotation symbols indicate the views relative to b. Interacting residues are shown as sticks and coloured by atom type. Blue, nitrogen; red, oxygen; yellow, sulfur; carbons take the colour of the respective molecule. Dashed lines represent hydrogen bonds, whereas thick grey dashed lines indicate zinc coordination. Dashed lines in the ribbon plots represent residues 842–844 not clearly defined by electron density.

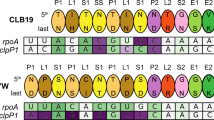
The alignment compares OTP86DYW with plant organellar DYW domains, hornwort putative U-to-C aminases53,72, C-to-U deaminases from E. coli (EcCD, PDB-ID: 1AF2; ref. 43), and Mus musculus (MmCD, PDB-ID: 2FR6; ref. 44). The alignment was prepared by Chimera employing Clustal Omega and shaded with ALSCRIPT73,74. Proteins are identified on the left of the aligned sequences with residues numbered (see Supplementary Table 2). Higher conservation is indicated by a darker background. Full conservation within DYW domains is indicated by dark blue background shading. Single-residue Cα r.m.s.d. values (calculated with CCP4i) between OTP86DYW and OTP86DYW* (or structural changes upon activation) are displayed on the top of each alignment block as a colour gradient from white to pink (prepared with Python Matplotlib) and are quantified in the top-right box. The overall r.m.s.d. between the two structures was 1.99 Å, the gating domain residues 870–891 (β-finger) amounted to 2.98 Å. The numbering on top of each block refers to A. thaliana OTP86. Below the alignment blocks, secondary structure elements (α–α-helix, β–β-sheet) of OTP86DYW are shown in slate for the deaminase domain, orange for the gating domain and red for the DYW motif. A dashed line or no line indicates residues missing from the structure or the expression construct, respectively. Green shading indicates residues coordinating the catalytic Zn1 ion; yellow shading indicates residues coordinating the Zn2 atom within the DYW motif and strand β7/helix α3; and purple shading indicates residues relevant for catalysis according to past studies. At, Arabidopsis thaliana; Pp, Physcomitrium patens; Aa, Anthoceros agrestis.
The gating domain is shared by DYW domains of all land plant clades (Figs. 1b and 2, and Supplementary Fig. 4), suggesting a conserved C-to-U RNA editing mechanism. DYW1 and DYW2 of Arabidopsis thaliana, however, show a less conserved N-terminus of the gating domain (Fig. 2)12,15,17, but interaction with E+ type PPR proteins that carry C-terminally truncated DYW domains may restore the functionality of these deviant gating domains again.
The arrangement of the active-site zinc ion coordination corroborates past in vivo studies in which mutants of the HxE(x)nCxxC zinc ion binding signature showed no editing activity (Fig. 1c)20,23,34. The highly conserved OTP86DYW E894, which was previously hypothesized to transfer a proton from the substrate water molecule to ammonia during catalysis (see Supplementary Note 1), was shown to be essential for in vivo editing20,23,35,36,37. Notably, R895 hydrogen bonds to C920 and compensates the negative charge of the active site together with the dipole moment of helix α3 in a similar fashion as observed for Bacillus subtilis cytidine deaminase38. As a third hallmark of the DYW domain structure, the nine C-terminal residues form a structural element that we termed a DYW motif, which is represented by an additional β-strand and a short loop. In OTP86DYW, the motif terminates with the DSW sequence and provides two ligands (C954, C956) for a second zinc ion (Zn2); two more ligands are part of the deaminase domain (H924 and H947), indicating that Zn2 only has a structural role within DYW domains (Figs. 1d and 2). We employed X-ray fluorescence spectroscopy on OTP86DYW crystals to assess whether divalent metals other than zinc were present in our structure. A comparison of the spectrum taken from solvent area in the sample loop with an OTP86DYW crystal confirmed zinc as the only relevant signal detected between calcium and copper (Supplementary Fig. 2c,d). Other ions do not fit in the observed coordination geometries and electron densities. Any alteration of the residues involved in the coordination of Zn2 abolished RNA editing in vivo, which is probably due to destabilization of the entire motif34. The tryptophan at position 960 in OTP86DYW flanks Zn2, is highly conserved in DYW domains and was shown to be essential for deaminase function in vivo for DYW1 and PpPPR6520,23. Notably, the surface charge distribution of OTP86DYW reveals a region of positively charged residues spanning across the active site and passing in between the base of the gating domain’s β-finger and the DYW motif. As RNA bases around the editing site are not conserved, this probably represents the path of the negatively charged RNA backbone, which is placed for catalysis by the PPR tract after or concomitant to activation of the DYW domain (Fig. 1e).
Crystal structure of an activated A. thaliana OTP86DYW
As crystal soaking experiments with substrate, product or different short RNA trinucleotides were unsuccessful, we attempted co-crystallization of OTP86DYW with the well-characterized deaminase inhibitor tetrahydrouridine (THU)30. Along this approach, we observed several new crystallization conditions that indicated a different crystallization behaviour due to the presence of THU. Finally, we obtained crystals of OTP86DYW with space group P21212, which diffracted to a resolution of 1.65 Å (Supplementary Table 1). When employing the coordinates of OTP86DYW, structure solution by molecular replacement failed; however, four copies of a truncated model missing the gating domain were successfully placed in the asymmetric unit (see Methods, Supplementary Table 1 and Supplementary Fig. 5). Explaining the failed molecular replacement, OTP86DYW had clearly changed its conformation substantially in the presence of THU towards an activated (OTP86DYW*) state (Fig. 3a, Supplementary Fig. 6 and Supplementary Video 1). The conformational change mainly involved the β-hairpin of the gating domain (which now adopts an extended β-strand conformation), and its connection to α-helix 1 (gating domain) and α-helix 2 (deaminase domain). It is widely accepted that conformational switches of β-fingers may take part in the regulation of macromolecular complexes as observed for the RNAse H domain in the spliceosomal Prp8 protein39,40. The conformational change has a marked effect on the active site architecture, in particular zinc coordination. The inactive structure zinc coordination is maintained by coordinating H892, C920, C923 and a more distant water molecule, whereas the catalytically important E894 is ionically bonded to K915 (Fig. 3b); K915 also hydrogen bonds to S828 and S893. In this configuration, the ion pair will reduce the basic character of E894 and hinder the required deprotonation of the deaminating water, which is not productively coordinated by Zn1 and also not contacted by E894. We reason that beyond the steric inhibition through the gating domain, K915 has to be released before efficient catalysis can occur. This notion is corroborated by the activated OTP86DYW* (Fig. 3d), in which K915 points away from E894. Although E894 is conserved in all deaminases, K915 is restricted to DYW domains (Figs. 1c and 2)38. The conformational changes upon activation involve several larger backbone torsion angle movements of the gating domain’s β-hairpin.

a, Superimposition of inactive OTP86DYW (colouring and dashed lines are as in Fig. 1b) and activated OTP86DYW* (deaminase domain, cyan; gating domain, ochre; DYW motif, dark red). Zn1 (green) and Zn2 (yellow) of OTP86DYW are shown. b, A close-up view of the OTP86DYW active site in the inhibited state. c, A close-up view of the activated OTP86DYW* active site in a catalytically competent conformation. d, A close-up view of the Zn1-coordination environment of the OTP86DYW active site in its catalytically inhibited state. e, A close-up view of the Zn1-coordination environment of the OTP86DYW active site in its catalytically active state. Distances within the vicinity of Zn1 are given in Å. Rotation symbols indicate the views relative to a. Interacting residues are shown as sticks and coloured by atom type. Water molecules (W) shown as white spheres. Carbon — as for the respective molecule; nitrogen, blue; oxygen, red; sulfur, yellow. Dashed lines represent hydrogen bonds, thick grey dashed lines indicate zinc coordination.
The direct effects of the gating domain’s conformational change on OTP86DYW catalytic activation via Zn1 coordination are evident from the detailed structural comparison of the zinc coordination and E894 (Fig. 3d,e, and Supplementary Videos 1 and 2). Remarkably, the conformational change of H892 from the main chain dihedral angles angles ϕ/ψ = 58°/42° (inactive) to ϕ/ψ = −74°/152° (active) and a concomitant repositioning of its side chain elicits a pervasive impact on the Zn1 coordination geometry (Fig. 3b,c). When superimposing residues C920–C923 of both structures, activation moves the coordinating nitrogen of H892 by 2 Å, with a concomitant rotation around the Zn1 coordination sphere by about 35° that harnesses the zinc ligands C920 and C923 as a rotation axis; C920, C923 and Zn1 remain largely unaffected during activation. The restructuring of the active site reduces the zinc–water/water–E894 distances from 3.07 Å/3.93 Å (inactive conformation) to 2.15 Å/2.53 Å, thereby activating the mechanistically important water molecule (Supplementary Video 2). The altered H892 positioning permits the remotely located water molecule to be attracted to Zn1 as a fourth coordination ligand poised for the deamination reaction, and the water molecule is now situated in close vicinity to E894 as well as the C920 amide (Fig. 3d,e and Supplementary Figs. 5 and 6). In OTP86DYW*, distances and angles of the zinc ligands are in agreement with a catalytically competent reaction centre41,42. Furthermore, the strand length of the β-finger is extended upon activation, still maintaining the original backbone hydrogen bonding residue pairs of the inactive OTP86DYW. The side chain of H890, which shields the active site as a counterpart relative to the zinc coordination sphere (formed by H892, C920 and C923) in the inactive OTP86DYW, is repositioned far away from the active site by about 13 Å in OTP86DYW* (compare Fig. 3b,c, Supplementary Video 1). Unexpectedly, THU could not be located in the electron density, which implies a crucial role in triggering activation but not as tightly bound inhibitor. We are not aware of a comparable mechanism and thus coin this catalytic activation mechanism of DYW domains, and probably other metalloenzymes, gated zinc shutter.
Structural comparison of the OTP86 DYW domain to other cytidine deaminases
A comparison of OTP86DYW with known ligand-bound deaminase domains confirms the presence of a complete active site for catalysis and fortifies the notion of a steric autoregulatory mechanism for DYW domains (Fig. 4a–i). When comparing OTP86DYW with cytidine deaminase from mouse bound to cytidine (MmCD), or human APOBEC3A in complex with a short DNA (HsAPOBEC3A), nearly all of the residues required for nucleotide binding are present in OTP86DYW and located at corresponding positions (Fig. 3b–d,g)43,44. For example, all atoms of residues coordinating Zn1 and E894, as well as the backbone R918 carbonyl, C920 and S893 amides (contacting the respective base in MmCD and HsAPOBEC3A) of OTP86, superimpose to their mouse and human equivalents with r.m.s.d. values of 1.0 and 0.9 Å, respectively. The backbone carbonyl oxygen of R918 or the backbone amide of C920 are within hydrogen bonding distance to the amine of the base or the activated water molecule, respectively (compare Fig. 4b,c with Fig. 4d,g). Likewise, the backbone carbonyl oxygen of S893 (OTP86) may contact the keto group of the bound cytidine as for A66 in MmCD or A71 in HsAPOBEC3A. L917 of OTP86DYW (Fig. 4b,c) has equivalent residues (I87 in MmCD or W98 in human APOBEC3A) that stack on the edited base (Fig. 4b–d,g). H70 in HsAPOBEC3A adopts a similar side chain conformation as H892, however, only in OTP86DYW*, implying a role in base stacking upon activation (compare Fig. 4b,c,g). The OTP86-equivalent residue for MmCD N54 or HsAPOBEC3A N57, both of which contact the sugar 3′ oxygen, could not be identified. This residue may also be part of a region preceding the OTP86DYW deaminase fold (or PG box), which is missing in our structure. Hence we conclude that the OTP86DYW active site and the positioning of the base targeted for deamination is nearly identical to other cytidine deaminases. The absence of the region preceding the PG box from our crystallization constructs may have impeded our attempts to obtain structures of OTP86DYW bound to substrate-related molecules.

a, Superimposition of inactive OTP86DYW and activated OTP86DYW* (as in Fig. 3a). b, A close-up-view of the OTP86DYW active site in the inhibited state, depicting catalytic and potential RNA binding residues. c, A close-up-view of the OTP86DYW* active site. d, M. musculus cytidine deaminase (MmCD; light orange) in complex with cytidine (yellow), coordinated zinc (green sphere) and an activated water molecule (white sphere) (PDB-ID: 2FR6; ref. 44) e,f, A surface display of the active site cavity of OTP86DYW (e) and OTP86DYW* (f) with the superimposed cytidine from d. g, Human APOBEC3A (dark ochre) with bound DNA (orange, only the active site cytidine is shown for clarity), coordinated Cl (light pink) (PDB-ID: 5KEG; ref. 75). h,i, A surface display of the active site cavity of OTP86DYW (h) and of OTP86DYW* (i) with the superimposed DNA from g. The cytidine deaminase structure of M. musculus and human APOBEC3A were superimposed employing only the zinc-coordinating residues and the equivalents of OTP86DYW E894. The colouring and dashes are as in Fig. 3. Rotation symbols indicate the views relative to a.
A superimposition of bound nucleotides of known deaminase structures has further implications for the OTP86 activation mechanism. To investigate the nucleotide binding mode of OTP86DYW, we superimposed active site residues of MmCD and HsAPOBEC3A onto OTP86DYW in its activated and inactive states and compared the substrate positions (Fig. 4e,f,h,i). For example, the cytidine bound to MmCD causes steric clashes with the β-finger of the OTP86DYW gating domain when positioned in the active site of OTP86DYW. Due to the conformational change of the gating domain upon OTP86DYW activation, this inhibition is released (compare Fig. 4e,f). When comparing OTP86 structures with DNA-bound human APOBEC3A, the +1 nucleotide (3′ of the active site) causes steric clashes with the gating domain only in the inactive OTP86DYW conformation, but not in OTP86DYW*. In conclusion, several superimposed substrate nucleotides suggest a steric inhibition by the OTP86 gating domain and probably other DYW deaminases as key residues (1) for nucleotide positioning and (2) participating in the conformational changes show a high degree of conservation (Fig. 2). These observations consolidate the notion of an autoinhibited ground state of OTP86DYW, which is paradigmatic for all PPR proteins with a DYW domain.
In vivo RNA editing assays with OTP86DYW and variants
To cross-validate the structural data and also probe the DYW domain surface, we conducted orthogonal in vivo RNA editing assays in E. coli employing PPR56PPRE1E2–OTP86DYW mutants (Fig. 5)23. The solubility of the mutants was assessed by a western blot employing the soluble fraction of the respective cell lysates (Supplementary Fig. 7). To this end, the reduced activities of for example, L856, R912, T914, D922 can be explained by the very limited solubility of the respective fusion proteins. By contrast, K555A (PPR56 numbering, corresponding to position K823 in OTP86) is soluble and the mutant has a dramatically reduced editing activity. In OTP86, the equivalent lysine is located directly before the PG box at position 823 and may contact the sugar of the edited nucleotide or the acidic phosphate backbone, for example as N54 in MmCD (see Fig. 4d). L889 directly precedes the active site as part of the β-finger, changes its position upon activation and may contact the RNA substrate remotely from the edited base, probably explaining its reduced in vivo RNA editing activity (Fig. 3b,c). H892 is a key regulatory residue as it alters its zinc coordination position upon activation, which poises the active site for the reaction. An alanine at this position is inactive as it is not suitable as a zinc coordination ligand. A cysteine may coordinate the zinc; however, H892C is also inactive. We reason that either the cysteine side chain does not provide the necessary flexibility to undergo a dramatic repositioning as that of histidine does. Alternatively, cysteine is a strong coordination ligand of zinc compared with histidine and may thus reduce zinc reactivity. S828 and S893 apparently play an important inhibitory role when contacting K915 and tethering it to the catalytically important E894 (Fig. 3b and Supplementary Note 3).

a, The C-to-U editing activities at the nad4eU272SL site in E. coli expressed with the PPR56PPRE1E2–OTP86DYW fusion protein (OTP86DYW), or its mutants, are plotted. The activities of mutants are relative to that of PPR56PPRE1E2–OTP86DYW (82.4 ± 2.1% edited). The bars represent the mean values, with each mutant protein ±s.d. based on three independent experiments (shown as yellow diamonds). The soluble protein expression of each mutated construct in E. coli was verified by western blot analysis (Supplementary Fig. 7). b, The activities of OTP86DYW mutants shown in a plotted on the surface of the inactive OTP86DYW and the activated OTP86DYW* structure as a heatmap (activity is scaled in the bar on the bottom), with untested residues shown in grey.
Mutants of the catalytic residue E894 retain their solubility upon mutation to alanine or the structurally analogous uncharged glutamine; however, both mutants are inactive, which consolidates their important role in deamination catalysis22,23,35. Albeit soluble to a low degree (compare with D922), the R895A mutant is probably inactive due to a structural destabilization of the active site. Alternatively, R895 may be crucial to catalysis according to the previously described zinc charge compensation38. Interestingly, the hydrogen bond donor of R895 changes from the terminal Nη to the weaker bridging Nε during activation. In the active conformation, R895 hydrogen bonds to D872 (Fig. 3b,c). This stabilizing effect is missing in the R895A mutant, which could possibly be an explanation for high conservation and R895A inactivity. Conversely, D922 stabilizes the inactive state of OTP86DYW; however, an alanine mutant has reduced activity. We can explain this effect as a result of the inactive ground state (destabilized through D922A) very likely being required for repetitive reactions elicited by a single DYW domain. Mutations of R945, D958 and W960 to alanine show reduced activity, which can be structurally explained by destabilizing effects on OTP86DYW. W960 is tightly embedded in the DYW motif and stabilizes it as it stacks on top of zinc-coordinating H924 beneath highly conserved R918 and maintains a hydrogen bond to the backbone oxygen of V919.
Likewise, D958 consolidates the DYW motif by formation of a hydrogen bond to highly conserved K928, which explains the impaired function of a respective aspartate mutant in in vivo editing assays with DYW120 and finally our catalytically impaired D958A mutant. S959 in OTP86 (or tyrosine in most DYW domains) points into the solvent, thus, mutation of the corresponding tyrosine to alanine has no effect on DYW1 in vivo activity20 and the reverse mutation has no effect on OTP86 activity in this work (Fig. 5a); however, an phenylalanine to alanine mutation at this position in Physcomitrium PPR65 showed a severe negative impact on editing23. Our structure may help to interpret these past in vivo mutagenesis studies in several ways. Most likely, an impaired stability of the DYW motif as pictured above triggers a destabilization of the active-site Zn1 as they are directly linked via helix α3, which provides residues coordinating Zn1 and Zn2 (Figs. 1b and 2, and Supplementary Fig. 3b,c). In this context, the DYW motif may also play a role in regulation, for example, the release of the gating domain, repositioning of K915 or binding nucleotides adjacent to the editing site. Finally, we replaced the gating domain’s β-finger residues 875–890 with three glycine residues (Δ875–890GGG). The removal of the entire β-finger markedly reduces the editing activity, which implies its important functional role—probably during activation, dimerization or RNA binding—conferred by this region in DYW domains. Although size-exclusion chromatography of isolated OTP86DYW and in the presence of activators (Supplementary Fig. 2a,b) did not indicate dimer formation, prominent protein–protein contacts within the crystal lattice may be physiologically relevant (Supplementary Fig. 8 and Supplementary Note 3)45.
Validation of the OTP86DYW activation mechanism in vitro
We next set out to cross-validate these distinct structural changes of isolated OTP86DYW in solution. The very low amounts of available OTP86DYW led us to conduct differential scanning fluorimetry (DSF). In a typical DSF experiment, an increase of the protein’s melting point (Tm) upon ligand binding is observed46. The substrate (CMP), product (UMP) and a K915A mutation do not have an effect on the overall high Tm of OTP86DYW, which is about 71–72 °C in each case (Fig. 6a)47. These results imply a limited accessibility of the active site due to steric inhibition, consistent with the structures and corroborated by mutants. The well-characterized transition-state analogue THU lowers the Tm of OTP86DYW to 60 °C, corresponding to the structural changes we observed following THU co-crystallization. We reason that THU, a potent cytidine deaminase inhibitor, outcompetes the gating domain from the active site, for example, by releasing H890, opening the protein up for substrate access and thereby destabilizing OTP86DYW markedly47,48. Interestingly, this effect is less severe with the K915A mutation, implying a functional role of K915A during activation but not catalysis. Next we asked whether the effect of THU is reversible. A THU-pre-treated OTP86DYW sample was therefore subjected to size-exclusion chromatography to remove THU. Indeed, repurified OTP86DYW resembled the inactive state with a Tm of 72 °C despite THU exposure beforehand. The active state could be restored by the addition of 2 mM THU, resulting in a Tm of 61 °C. We conclude that DYW domains have an inhibited ground state that is restored after the activation and an editing event.

a, Melting points of OTP86DYW and selected variants in the presence of substrate, product and activators. Thermal shift assays were performed in triplicates. Curves were subjected to sigmoidal fitting; error bars indicate the s.d. of the three measurements (shown as yellow diamonds); regular samples are shown as blue bars, whereas SEC-related experiments are shown as orange bars; ‘pre’ indicates a sample pre-incubated with the respective activator and purified by SEC. b, C-to-U conversion for in vitro reactions with recombinant PPR56 or PPR56PPRE1E2–OTP86DYW with the addition of THU, ATP or GTP are displayed in a bar plot. In vitro editing with recombinant PPR56PPRE1E2–OTP86DYW-E894A mutant protein showed no editing activity. Bars indicate the mean value ±s.d. based on three independent experiments (shown as yellow diamonds). A grey dashed line indicates a Tm of 71 °C for wild type protein (WT in a) or the activity of the untreated proteins (in b).
ATP was reported to activate in vitro RNA editing reactions with a recombinant Physcomitrium PPR65 protein as well as with plant organellar lysates22,49,50; we thus also tested ATP and a concentration of 2 mM was required to drop the Tm to 65 °C. A very similar picture was obtained by addition of GTP, where 2 mM GTP reduced the Tm to 65.3 °C. Like THU, the activators ATP and GTP also do not stably bind to OTP86DYW. When ATP- or GTP-treated OTP86DYW is subjected to size-exclusion chromatography, the higher Tm of the untreated protein (inactive state) is restored in the eluate fractions; however, addition of ATP or GTP to the eluted samples leads again to a decrease of the Tm, indicating a reversible structural change and a stable ground state in the absence of activators (Fig. 6a). In the size-exclusion chromatograms of isolated OTP86DYW and OTP86DYW pre-treated with 2 mM ATP, the A260/280 ratios of the respective eluted OTP86DYW peaks are identical (0.54), which further supports the dissociation of the activators from the DYW domain (Supplementary Fig. 2c,d). Contrasting ATP as efficient activator, the addition of 2 mM AMP has a very mild effect. The three phosphates of ATP seem particularly important for activation as the non-hydrolysable analogue AMPPCP had only a mild effect on activation (comparable to AMP); that is, lowering the Tm to 68 °C. In summary, the activation of OTP86 seems to be either triggered by THU or triphosphate nucleotides.
To gain more insight into whether the H892C mutation effect (activity loss) is of a catalytic or structural nature, we assessed the Tm of H892C in the presence of the activators. The H892C mutant closely resembles the wild-type protein regarding activation, albeit less pronounced. The detrimental effect of H892C on activity in the in vivo assays therefore relies on the stronger zinc ligand properties of cysteine rather than an impaired structural rearrangement of the catalytic site due to activation (see Fig. 5a).
The L917A mutation showed a prominent decrease in Tm when THU was added; however, a milder effect with ATP comparable to wild type was observed. This may indicate that the activation via THU and ATP relies on different mechanisms. Finally, the R918A mutant showed a weaker decrease in Tm in the presence of activator compared with the wild-type. This may be a result of the impaired dimerization capability or an indirect destabilization of the active site via α-helix 3 and thus reduced activation (see Supplementary Fig. 8).
To consolidate and cross-validate our structural data, in vivo activities and DSF, and to gain more control about the reaction conditions, we conducted in vitro RNA editing assays with purified PPR56 and PPR56PPRE1E2–OTP86DYW (Fig. 6b). Contrasting an earlier report, and consistent with the proposed DYW domain activation mechanism, the cytidine deaminase inhibitor THU increases deaminase activity markedly for both proteins in a concentration-dependent manner22. Within this study we were not able to structurally explain this effect due to the absence of THU in the electron density. In agreement with past in vitro editing assays, ATP activates PPR56 and PPR56PPRE1E2–OTP86DYW in a concentration-dependent fashion22. We observe that higher ATP concentrations inhibit deaminase activity and thus confirm a highly sensitive regulation of DYW domains by ATP which may be of an allosteric type. Other trinucleotides such as GTP also activate both PPR proteins in a concentration-dependent manner, albeit with a higher sensitivity, confirming the DSF measurements. All assays cross-validate our structural data of the OTP86 DYW domain in its inactive and active states along with a complex regulation mechanism, which suggests an intricate activation of the plant organellar RNA editosome in vivo.
Discussion
Our results draw a uniform picture of an unexpected autoinhibition mechanism elicited by DYW domains, which is released in the context of a plant RNA editosome at the site of editing. The data presented here is consistent with past in vivo mutagenesis studies and underlines the cytidine deaminase function of DYW domains in RNA editing8,11,20,23,35,51. Typically, cytidine deaminases are highly active enzymes29. With regulated DYW domains, which only exert catalysis specifically and in the context of the RNA editosome, unspecific side reactions that result in an overarching distortion of the organellar transcriptome—and lastly proteome—would be avoided. Likewise, a strict autoinhibition of the deaminase activity protects the cytosol as all RNA editosome proteins originate from nuclear transcripts and are imported into organelles9.
The higher target specificity of DYW type RNA editing factors in plant organelles compared with animal RNA editing deaminase enzymes suggests that the specific binding of RNA by the PPR tract can be a trigger of the DYW activation (Supplementary Note 4). It is also possible that other co-factors in the plant RNA editosome, for example MORF proteins, support moving the gating domain either directly or through changing the conformation of PPR, E1 and E2 domains.
When we compare OTP86DYW to other ligand-bound cytidine deaminases we can extrapolate that the −1 and −2 nucleotide positions relative to the editing site fall into the region of the DYW motif, indicating a head-to-tail arrangement of PPR tract and DYW domain with respect to the direction of the protein sequence of the respective proteins32,44 (Fig. 7). Our observations are in line with a past study in which the 0 to −3 nucleotides bind to the DYW domain, whereas the E1 and E2 motifs do not contribute to binding the target RNA52. In this scenario, the DYW motif bridges the PPR tract and the deaminase/gating domain, which may be the reason for its important structural role within the plant organellar RNA editosome. Furthermore, our structural data suggest a potential multimerization of DYW domains (Supplementary Note 5).

Activation of OTP86DYW is triggered by a conformational change of its gating domain, which may be elicited by an activator such as ATP, PPR–RNA complex formation, via the E1/E2 domains or different factors such as MORF proteins. The direction and path of the RNA can be extrapolated from comparisons to known cytidine deaminase co-structures such as human APOBEC3A (see Fig. 4h,i; PDB-ID: 5KEG75), our comprehensive mutations of the domain surface (see Fig. 5) and the charged surface of OTP86DYW (see Fig. 1e). Colours and labels are as in Fig. 3a.
Reverse U-to-C RNA editing is observed only in hornwort, most lycophytes and ferns and might be elicited by PPR DYW:KP proteins53,54,55,56,57,58. Our work has several implications that this process may not depend on a strong autoinhibition (Supplementary Note 6). We searched for gating domain-like sequences in proteins of all kingdoms using a phmmer search (HmmerWeb version 2.41.1)59. Only PPR proteins that included a conserved gating domain sequence were detected. Finally, a comparison of members of the deaminase superfamily identified the gating domain as exclusive insertion in DYW-type PPR proteins29.
On the basis of our observations, we propose a regulation mechanism of RNA editing by ATP or other triphosphate nucleotides via the DYW cytidine deaminase activity. RNA editing is directly coupled to the organellar nucleotide metabolism downstream as ATP production is dependent on RNA editing. Conversely, nucleotide levels seem to regulate RNA editing, thus creating a feedback loop. In this scenario, organellar ATP synthesis and RNA editing are mutually regulated to achieve homeostasis. In the light of our artificial in vitro system with isolated proteins, we anticipate a high sensitivity of this feedback loop in vivo possibly owing to the generally low abundance of editing factors observed in mitochondria60.
We have further identified a very unusual regulation mechanism involving zinc coordination. In this protein regulation principle, a major domain movement alters the coordination around a zinc atom. In the inactive state, the zinc is inhibited by its coordination setting, which restricts the access of a water molecule as fourth zinc ligand required for catalysis. Upon activation, the DYW gating domain changes its conformation, which triggers the repositioning of a histidine involved in zinc coordination. The altered zinc coordination permits a water molecule to be recruited as a fourth ligand between zinc and the catalytic residue E894 to attack the base for deamination. This regulation principle may also apply to other metalloenzymes beyond DYW deaminases and we are not aware of any similar mechanism described in the current literature.
Our observations explain three decades of previously failed attempts to establish an in vitro RNA editing assay and impaired nucleotide binding of DYW domains6,11,51. We anticipate our results to be a valuable basis for follow-up experiments for example, a ligand-bound DYW domain structure or cryo electron microsocropy studies of a complete editosome. Based on our structure, further in vitro activity assays with structure-guided DYW domain mutants become conceivable where ligand binding, substrate binding or dimerization dependent activation is enhanced or reduced upon mutagenesis.
Methods
Cloning, expression and protein purification
When we set out to determine the structure of a DYW domain, we first screened 18 different A. thaliana (CRR22, CRR28, OTP81, OTP82, OTP84, OTP85, OTP86, OTP90, LPA66, YS1, RARE1, MEF1, MEF8, MEF10, MEF11, MEF14, MEF22 and MEF29) and three different Physcomitrella patens (PpPPR_65, PpPPR_71 and PpPPR_79) DYW proteins with four different N-terminal starting points (PPR-E1E2-DYW, E1E2-DYW, DYW (according to Cheng et al.31) and DYW (according to Lurin et al.61)) and various DYW-containing constructs of nine additional DYW proteins, totalling 113 tested expression constructs. Of these, only one MEF22 and one OTP86 construct yielded small amounts of soluble protein. Only OTP86 (amino acid residues 826–960) crystallized.
A DNA fragment encoding the A. thaliana OTP86 DYW domain (amino acid residues 826–960) was cloned into pET28a to yield a protein (OTP86DYW) with a tobacco-etch-virus-cleavable (TEV-cleavable) N-terminal Strep-tag. After TEV cleavage, the protein retains the N-terminal tripeptide GAM from the tag. For protein production, E. coli Rosetta2 (DE3) cells were transformed with the respective plasmid, grown in terrific broth to an OD600 of 0.6 at 37 °C, cooled to 20 °C, induced with 0.5 mM isopropyl-β-d-thiogalactoside (IPTG) and cultivated at 16 °C overnight. Cells were harvested by centrifugation and stored at −80 °C. Cell pellets from expression cultures were resuspended in lysis buffer (20 mM Tris-HCl, pH 7.5, 200 mM NaCl), supplemented with 0,01% (w/v) CHAPS in the presence of a protease inhibitor cocktail (Roche)). Cells were lysed using a Sonoplus sonifier (Bandelin) and cell debris were removed by centrifugation. For purification of OTP86DYW, the soluble fraction was passed over a StrepTactin gravity flow column, pre-equilibrated with lysis buffer. The beads were washed with lysis buffer and fusion proteins were eluted with lysis buffer supplemented with 10 mM desthiobiotin. The eluate was treated with a 1:40 protein mass ratio of TEV protease (in lysis buffer) overnight to remove the N-terminal Strep-tag. Cleaved proteins were further purified via Superdex 75 gel filtration chromatography (GE Healthcare, Unicorn Software 5.20) in 20 mM Tris, pH 7.5 and 150 mM NaCl. Peak fractions of the monomers were pooled, passed over an equilibrated StrepTactin gravity flow column, concentrated to 8–15 mg ml−1, flash frozen in liquid nitrogen and stored at −80 °C. Any alteration to the expression construct described above (for example, variations of the N-terminus length) abolished protein solubility.
Crystallographic analyses
OTP86DYW, supplemented with 2 mM UMP, crystallized by sitting drop vapour diffusion (100 nl protein plus 100 nl reservoir and 30 nl 0.1 M 50% v/v Jeffamine M-600 pH 7.0 as an additive) at 4 °C with a reservoir containing 0.1 M glycine, pH 10.5, 1.2 M NaH2PO4, 0.8 M K2HPO4 and 0.2 M Li2SO4 (space group C2). Crystals were cryoprotected with reservoir solution supplemented with 15% (v/v) ethylene glycol. Diffraction data to 2.5 Å resolution were collected at 100 K at beamline 14.1 of the BESSY II storage ring62. All diffraction data were processed with XDS63. Activated OTP86DYW*—supplemented with 2 mM CMP and 2 mM THU—crystallized by sitting drop vapour diffusion (1 µl protein plus 1 µl reservoir) in 100 mM sodium acetate (pH 4.6) and 2 M sodium formate (space group P21212), with a pronounced degree of translational non-crystallographic symmetry. Crystals were cryoprotected with reservoir solution supplemented with 2 mM CTP, 2 mM THU, and adjusted to a concentration of 3 M sodium formate as a cryoprotectant. Diffraction data to 1.65 Å resolution were collected at 100 K at beamline 14.1 of the BESSY II storage ring62. All diffraction data were processed with XDS63.
The structure of OTP86DYW was solved by single-wavelength anomalous dispersion with four zinc sites in space group C2 and two molecules per asymmetric unit employing PHENIX.AUTOSOL64. The initial density modified map was iteratively improved by manual model building with Coot65 and refined with PHENIX.REFINE (including experimental phases in the initial stages); automated model building was performed with PHENIX.AUTOBUILD64,65,66. The structure of OTP86DYW* was solved by molecular replacement with PHASER67 employing the structural coordinates of a truncated OTP86DYW, encompassing the deaminase domain and DYW motif. Despite the translational non-crystallographic symmetry, structure solution and refinement were successful, albeit with slightly increased R-factors (see Supplementary Table 1). The remaining model parts were built manually with COOT65 and with PHENIX.AUTOBUILD64,65,66 in an iterative fashion to improve the model until completion. Structure figures were rendered with open source Pymol v.1.8, structural movies were made with Pymol 2.2.3 (Schrödinger) under an academic license. Electrostatic surface potential was obtained by APBS employing a Pymol addon68,69,70. C-alpha r.m.s.d. values for structural comparison were calculated with CCP4i71.
DSF
The DSF experiments were performed in a 96-well plate in a plate reader combined with a thermocycler (Stratagene Mx3005P). Purified OTP86DYW or mutants were diluted to 0.2 mg ml–1 in buffer A (20 mM Tris, pH 7.5; 150 mM NaCl) supplemented with 10× SYPRO orange (1:500 dilution of the stock) in a total volume of 10 μl and pipetted into a 96-well plate. Either 10 µl of buffer A or 10 µl of buffer A supplemented with the respective ligand were added to the SYPRO orange/protein mixture. The temperature was increased from 25 °C to 95 °C and the fluorescence emission was monitored in steps of 1 °C per min with hold steps of 30 s between reads. The fluorescence intensity was then plotted as a function of temperature. The sigmoidal curve from each condition was normalized and corrected for the background signal of the fluorophore in the buffer. The inflection points of the curves, representing the thermal melting temperature of the protein in the respective conditions, were compared. Each experiment was done in triplicate, averaged and a standard deviation of the respective melting temperatures was calculated.
Size-exclusion chromatography
OTP86DYW was analysed by analytical size-exclusion chromatography on a Superdex 75 PC3.2 column (GE Healthcare, Unicorn Software 5.20) in size-exclusion buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl) at a flow rate of 50–70 µl min–1. Eluted fractions were analysed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) or subjected to DSF. Calibration chromatograms for the column were obtained from GE healthcare online support.
Cloning of MBP-PPR56PPRE1E2–OTP86DYW and its OTP86DYW mutants
Plasmids containing wild-type Physcomitrium PPR56 (pETG41K::PPR56) were previously described in ref. 23. For pETG41K::PPR56PPRE1E2–OTP86DYW, DNA fragments for PPR domain, E1 and E2 domains of PPR56 (amino acid residues 213–556) and DYW domain of OTP86 (825–960) were separately amplified by polymerase chain reaction (PCR) and cloned into the BsrGI digested pETG41K::PPR56 with NEBuilder (New England Biolabs). For cloning mutants, the C-terminal part OTP86 DYW was amplified by PCR with a mutation introduced primer set, and the remainder of PPR56PPRE1E2–OTP86DYW was separately amplified to create a 15 bp overlap to the mutated PCR fragments. The two PCR fragments were simultaneously cloned into the BsrGI digested pETG41K::PPR56 by NEBuilder (New England Biolabs). The mutated OTP86DYW in pETG41K was amplified by PCR and cloned into pET28a for the OTP86DYW mutant protein expression constructs.
In E. coli RNA editing assay
Expression of recombinant PPR proteins was performed as previously described in ref. 23. Plasmids were transformed into E. coli BL21(DE3) (TAKARA), and 5 ml E. coli starter cultures (lysogeny broth medium with 50 μM kanamycin) were grown overnight; 40 µl of the pre-culture was transferred to 4 ml of the same media supplemented with 0.4 mM ZnSO4 in a 15 ml cell culture tube (IWAKI, http://www.atgc.co.jp). Cultures were grown at 37 °C until an OD600 of 0.4–0.7 was reached. Cultures were cooled on ice for 5 min before adding 0.4 mM IPTG for induction of construct expression. Cells were incubated at 180 rpm at 16 °C for 20 h. After stopping induction, 2 ml of the sample was transferred to a sample tube for SDS–PAGE, with another 1 ml to a tube for RNA editing analysis. The respective samples were harvested and the pellets were frozen in liquid nitrogen and stored at −80 °C until further use. For the RNA editing assay, the total RNA was extracted from the E. coli cells after adding 100 µl of the lysozyme buffer (10 mM Tris-HCl (pH 8.0), 0.1 mM EDTA, 10 mg ml–1 lysozyme) using a Maxwell RSC Plant RNA Kit system (Promega, www.promega.com). Isolated RNA was used for PCR with reverse transcription (RT–PCR). Data were analysed with Microsoft Excel and plotted with Python/Matplotlib.
Validation of the amount of mutated recombinant proteins in E.coli
Escherichia coli cells from 2 ml culture were resuspended in 1 ml of chilled lysis buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 10% glycerol, 5 mM imidazole, 0.07% mercaptoethanol, 0.1% Triton-X-100, 1× complete EDTA-free (Roche) and 1 mM PMSF) and the soluble fraction was isolated after sonication and centrifugation; 7.5 µl of the soluble protein lysate was loaded on SDS–PAGE gels for silver staining (Source Data for Supplementary Fig. 7). For the western blot analysis, 150 µl of the soluble protein lysate was precipitated with 400 µl acetone. After centrifugation at 4 °C for 30 min at 15,000 rpm, the pellet was extracted with 15 µl of 1× loading buffer (50 mM Tris-HCl (pH6.8), 2% SDS, 0.2% bromophenol blue, 100 mM DTT, 10% glycerol) and loaded onto an SDS–PAGE gel. Expression of recombinant proteins was assayed by western blot analysis with an anti His-Tag antibody (PGI proteintech Group; AB_11232599) at 1:20,000 dilution followed by incubation with Anti-Mouse IgG, HRP-Linked Whole Ab (GE Healthcare; AB_772209) at 1:50,000 dillution. Signals were detected with ECL Prime Western Blotting Detection Reagent (GE Healthcare) and visualized with an ImageQuant LAS4000 (GE Healthcare). The signal intensities of the western blot analysis were analysed using ImageQuant TL v.8.1 (GE healthcare).
Expression of PPR proteins in E.coli for in vitro assays
Lysogeny broth medium (50 ml) with 50 µM kanamycin was inoculated with 500 µl of overnight cultures and incubated at 37 °C for 2 h to an OD600 of around 0.4–0.6. The cultures were cooled on ice for at least 5 min and ZnSO4 was added to a final concentration of 0.4 mM and IPTG to 0.4 mM to induce expression. Cells were incubated at 16 °C and 180 rpm for 20 h. The cells were centrifuged at 4 °C, 5,000 rpm for 10 min and cell pellets were suspend in 5 ml lysis buffer (50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 0.07% mercaptoethanol and 1 mM phenylmethylsulfonyl fluoride). The E. coli cells were sonicated with six sets of 10 × 2 s pulses with 1 min breaks while on ice. After centrifugation at 4 °C for 10 min at 15,000 rpm, the supernatant from the 5 ml samples was mixed with 30 µl of amylose resin (New England Biolabs) equilibrated in lysis buffer and mixed with the rotary machine for 1 h at 4 °C. The amylose resin was washed three times using 1 ml of lysis buffer. Proteins were eluted with 30 µl of elution buffer (lysis buffer with 10 mM maltose).
Preparation of RNA editing substrates
Polymerase-chain-reaction fragments were amplified using the pETG41K::PPR56 as a template, and primers nad4FEcoRV:GGCCTCTTGCGGGATATCTCAAACATCAATTTTTATATAGGTATAGACGGTATCT and nad4RBamH: CCGGCGTAGAGGATCCAAAATGAAGAGATACCGTCTATACCTATA. This fragment was cloned into pACYC184 digested with EcoRV and BamHI by NEBuilder. Furthermore, using this clone (pACYC184-Ppnad4) as a template, a PCR amplicon was synthesized with primers T7KS_pACY184EF:
GTAATACGACTCACTATAGGGCTCGAGGTCGACGGTATCAATCTAACAATGCGCTCATC
and SKR-pACYC184_EB_R:
CGCTCTAGAACTAGTGGATCCAGCGACGGAATCTTACTTA produced amplicons with a 5′ T7 promoter sequence. The amplicon was purified with a PCR purification kit (QIAGEN). RNA was synthesized with T7 polymerase (TAKARA) using the PCR amplicon as a template. RNA was diluted to 100 fmol µl–1 and used for the reaction with purified recombinant proteins.
In vitro RNA editing activity assay
Standard in vitro RNA editing reaction mixtures contained 100 mM Tris-HCl (pH 7.5), 10 mM maltose, 0.017% mercaptoethanol, 10U of RNaseOUT (Invitrogen), 1× proteinase inhibitor mixture complete EDTA-free (Roche), 100 fmol of mRNA substrate, and 2.5 µg of purified recombinant PPR56 proteins (PPR56PPRE1E2–OTP86DYW) or its mutated variants. The reaction mixtures were incubated at 16 °C for 2.5 h and purified RNAs were used for RT–PCR reactions.
Detection of C-to-U RNA editing
Complementary DNA was synthesized with a random hexamer with ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO) for both in E. coli and in vitro editing assays. A reverse primer upstream of the T7 terminator sequence and a forward primer binding the PPR56 coding region for in E. coli assay and KS and SK primers for the in vitro assay were used for RT–PCR amplification with GoTaq Master Mixes (Promega). After 5 min initial denaturation at 94 °C followed by 35 cycles each with 30 s denaturation at 94 °C, 30 s annealing at 55 °C, 1 min synthesis at 72 °C. For purification of PCR products, 2U ExoI (TAKARA) and 0.5U Shrimp Alkaline Phosphatase (TAKARA) were added and incubated at 37 °C for 1 h followed by 15 min at 80 °C and sequenced directly (Macrogen, www.macrogen-japan.co.jp or GENEWIZ, https://www.genewiz.com). Sequencing chromatograms were analysed with DNADynamo v.1.608 (www.bluetractorsoftware.co.uk). RNA editing was quantified as the ratio of the resulting thymidine peak to the sum of the thymidine and cytidine peak heights at the respective editing site. Editing values are given as the mean of at least three replicates with standard deviations. Data were analysed with Microsoft Excel and plotted with Python/Matplotlib.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Structure coordinates and diffraction data were deposited with the Protein Data Bank (http://www.pdb.org) under accession codes 7O4E (OTP86DYW) and 7O4F (OTP86DYW*). Source data are provided with this paper. The data that support the findings of this study are available from the corresponding authors on reasonable request.
References
Hiesel, R., Wissinger, B., Schuster, W. & Brennicke, A. RNA editing in plant mitochondria. Science 246, 1632–1634 (1989).
Covello, P. S. & Gray, M. W. RNA editing in plant mitochondria. Nature 341, 662–666 (1989).
Gualberto, J. M., Lamattina, L., Bonnard, G., Weil, J. H. & Grienenberger, J. M. RNA editing in wheat mitochondria results in the conservation of protein sequences. Nature 341, 660–662 (1989).
Hoch, B., Maier, R. M., Appel, K., Igloi, G. L. & Kössel, H. Editing of a chloroplast mRNA by creation of an initiation codon. Nature 353, 178–180 (1991).
Takenaka, M., Zehrmann, A., Verbitskiy, D., Härtel, B. & Brennicke, A. RNA editing in plants and its evolution. Annu. Rev. Genet. 47, 335–352 (2013).
Small, I. D., Schallenberg‐Rüdinger, M., Takenaka, M., Mireau, H. & Ostersetzer‐Biran, O. Plant organellar RNA editing: what 30 years of research has revealed. Plant J. 101, 1040–1056 (2020).
Kotera, E., Tasaka, M. & Shikanai, T. A pentatricopeptide repeat protein is essential for RNA editing in chloroplasts. Nature 433, 326–330 (2005).
Zehrmann, A., Verbitskiy, D., van der Merwe, J. A., Brennicke, A. & Takenaka, M. A DYW domain-containing pentatricopeptide repeat protein Is required for RNA editing at multiple sites in mitochondria of Arabidopsis thaliana. Plant Cell 21, 558–567 (2009).
Schmitz-Linneweber, C. & Small, I. Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci. 13, 663–670 (2008).
Takenaka, M. How complex are the editosomes in plant organelles? Mol. Plant 7, 582–585 (2014).
Nakamura, T. & Sugita, M. A conserved DYW domain of the pentatricopeptide repeat protein possesses a novel endoribonuclease activity. FEBS Lett. 582, 4163–4168 (2008).
Boussardon, C. et al. Two interacting proteins are necessary for the editing of the NdhD-1 site in Arabidopsis plastids. Plant Cell 24, 3684–3694 (2012).
Yin, P. et al. Structural basis for the modular recognition of single-stranded RNA by PPR proteins. Nature 504, 168–171 (2013).
Barkan, A. et al. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 8, e1002910 (2012).
Andrés-Colás, N. et al. Multiple PPR protein interactions are involved in the RNA editing system in Arabidopsis mitochondria and plastids. Proc. Natl Acad. Sci. USA 114, 201705815 (2017).
Barkan, A. & Small, I. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 65, 415–442 (2014).
Guillaumot, D. et al. Two interacting PPR proteins are major Arabidopsis editing factors in plastid and mitochondria. Proc. Natl Acad. Sci. USA 114, 201705780 (2017).
Salone, V. et al. A hypothesis on the identification of the editing enzyme in plant organelles. FEBS Lett. 581, 4132–4138 (2007).
Schallenberg-Rüdinger, M., Lenz, H., Polsakiewicz, M., Gott, J. M. & Knoop, V. A survey of PPR proteins identifies DYW domains like those of land plant RNA editing factors in diverse eukaryotes. RNA Biol. 10, 1549–1556 (2013).
Boussardon, C. et al. The cytidine deaminase signature HxE(x)nCxxC of DYW1 binds zinc and is necessary for RNA editing of ndhD-1. N. Phytol. 203, 1090–1095 (2014).
Hayes, M. L., Giang, K., Berhane, B. & Mulligan, R. M. Identification of two pentatricopeptide repeat genes required for RNA editing and zinc binding by C-terminal cytidine deaminase-like domains. J. Biol. Chem. 288, 36519–36529 (2013).
Hayes, M. L. & Santibanez, P. I. A plant pentatricopeptide repeat protein with a DYW-deaminase domain is sufficient for catalyzing C-to-U RNA editing in vitro. J. Biol. Chem. 295, 3497–3505 (2020).
Oldenkott, B., Yang, Y., Lesch, E., Knoop, V. & Schallenberg-Rüdinger, M. Plant-type pentatricopeptide repeat proteins with a DYW domain drive C-to-U RNA editing in Escherichia coli. Commun. Biol. 2, 1–8 (2019).
Takenaka, M. et al. Multiple organellar RNA editing factor (MORF) family proteins are required for RNA editing in mitochondria and plastids of plants. Proc. Natl Acad. Sci. USA 109, 5104–5109 (2012).
Sun, T. et al. An RNA recognition motif-containing protein is required for plastid RNA editing in Arabidopsis and maize. Proc. Natl Acad. Sci. USA 110, E1169–E1178 (2013).
Yang, J., Zhang, M. & Wang, X. Crystal structure of the chloroplast RNA editing factor MORF2. Biochem. Biophys. Res. Commun. 495, 2038–2043 (2018).
Haag, S. et al. Crystal structures of the Arabidopsis thaliana organellar RNA editing factors MORF1 and MORF9. Nucl. Acids Res. 45, 4915–4928 (2017).
Yan, J. et al. MORF9 increases the RNA-binding activity of PLS-type pentatricopeptide repeat protein in plastid RNA editing. Nat. Plants 3, 17037 (2017).
Iyer, L. M., Zhang, D., Rogozin, I. B. & Aravind, L. Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucl. Acids Res. 39, 9473–9497 (2011).
Hammani, K. et al. A study of new Arabidopsis chloroplast RNA editing mutants reveals general features of editing factors and their target sites. Plant Cell 21, 3686–3699 (2009).
Cheng, S. et al. Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. Plant J. 85, 532–547 (2016).
Xiang, S., Short, S. A., Wolfenden, R. & Carter, C. W. Cytidine deaminase complexed to 3-deazacytidine: a ‘valence buffer’ in zinc enzyme catalysis. Biochemistry 35, 1335–1341 (1996).
Okuda, K., Myouga, F., Motohashi, R., Shinozaki, K. & Shikanai, T. Conserved domain structure of pentatricopeptide repeat proteins involved in chloroplast RNA editing. Proc. Natl Acad. Sci. USA 104, 8178–8183 (2007).
Wagoner, J. A., Sun, T., Lin, L. & Hanson, M. R. Cytidine deaminase motifs within the DYW domain of two pentatricopeptide repeat-containing proteins are required for site-specific chloroplast RNA editing. J. Biol. Chem. 290, 2957–2968 (2015).
Hayes, M. L., Dang, K. N., Diaz, M. F. & Mulligan, R. M. A conserved glutamate residue in the C-terminal deaminase domain of pentatricopeptide repeat proteins is required for RNA editing activity. J. Biol. Chem. 290, 10136–10142 (2015).
Xiang, S., Wolfenden, R., Carter, C. W. & Short, S. A. Transition-state selectivity for a single hydroxyl group during catalysis by cytidine deaminase. Biochemistry 34, 4516–4523 (1995).
Diaz, M. F., Bentolila, S., Hayes, M. L., Hanson, M. R. & Mulligan, R. M. A protein with an unusually short PPR domain, MEF8, affects editing at over 60 Arabidopsis mitochondrial C targets of RNA editing. Plant J. 92, 638–649 (2017).
Johansson, E., Neuhard, J., Willemoës, M. & Larsen, S. Structural, kinetic, and mutational studies of the zinc ion environment in tetrameric cytidine deaminase. Biochemistry 43, 6020–6029 (2004).
Schellenberg, M. J. et al. A conformational switch in PRP8 mediates metal ion coordination that promotes pre-mRNA exon ligation. Nat. Struct. Mol. Biol. 20, 728–734 (2013).
Fica, S. M. & Nagai, K. Cryo-electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat. Struct. Mol. Biol. 24, 791–799 (2017).
Laitaoja, M., Valjakka, J. & Jänis, J. Zinc coordination spheres in protein structures. Inorg. Chem. 52, 10983–10991 (2013).
Maret, W. & Li, Y. Coordination dynamics of zinc in proteins. Chem. Rev. 109, 4682–4707 (2009).
Xiang, S., Short, S. A., Wolfenden, R. & Carter, C. W. The structure of the cytidine deaminase-product complex provides evidence for efficient proton transfer and ground-state destabilization. Biochemistry 36, 4768–4774 (1997).
Teh, A. H. et al. The 1.48 Å resolution crystal structure of the homotetrameric cytidine deaminase from mouse. Biochemistry 45, 7825–7833 (2006).
Krissinel, E. Crystal contacts as nature’s docking solutions. J. Comput. Chem. 31, 133–143 (2010).
Niesen, F. H., Berglund, H. & Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 (2007).
Costanzi, S. et al. Delineation of the molecular mechanisms of nucleoside recognition by cytidine deaminase through virtual screening. ChemMedChem 6, 1452–1458 (2011).
Cohen, R. M. & Wolfenden, R. Cytidine deaminase from Escherichia coli. Purification, properties and inhibition by the potential transition state analog 3,4,5,6-tetrahydrouridine. J. Biol. Chem. 246, 7561–7565 (1971).
Hegeman, C. E., Hayes, M. L. & Hanson, M. R. Substrate and cofactor requirements for RNA editing of chloroplast transcripts in Arabidopsis in vitro. Plant J. 42, 124–132 (2005).
Takenaka, M. & Brennicke, A. In vitro RNA editing in pea mitochondria requires NTP or dNTP, suggesting involvement of an RNA helicase. J. Biol. Chem. 278, 47526–47533 (2003).
Okuda, K. et al. Pentatricopeptide repeat proteins with the DYW motif have distinct molecular functions in RNA editing and RNA cleavage in Arabidopsis chloroplasts. Plant Cell 21, 146–156 (2009).
Okuda, K. et al. Quantitative analysis of motifs contributing to the interaction between PLS-subfamily members and their target RNA sequences in plastid RNA editing. Plant J. 80, 870–882 (2014).
Gerke, P. et al. Towards a plant model for enigmatic U‐to‐C RNA editing: the organelle genomes, transcriptomes, editomes and candidate RNA editing factors in the hornwort Anthoceros agrestis. N. Phytol. 225, 1974–1992 (2020).
Kugita, M., Yamamoto, Y., Fujikawa, T., Matsumoto, T. & Yoshinaga, K. RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucl. Acids Res. 31, 2417–2423 (2003).
Grewe, F. et al. A unique transcriptome: 1782 positions of RNA editing alter 1406 codon identities in mitochondrial mRNAs of the lycophyte Isoetes engelmannii. Nucl. Acids Res. 39, 2890–2902 (2011).
Knie, N., Grewe, F., Fischer, S. & Knoop, V. Reverse U-to-C editing exceeds C-to-U RNA editing in some ferns—a monilophyte-wide comparison of chloroplast and mitochondrial RNA editing suggests independent evolution of the two processes in both organelles. BMC Evol. Biol. 16, 134 (2016).
Oldenkott, B., Yamaguchi, K., Tsuji-Tsukinoki, S., Knie, N. & Knoop, V. Chloroplast RNA editing going extreme: more than 3400 events of C-to-U editing in the chloroplast transcriptome of the lycophyte Selaginella uncinata. RNA 20, 1499–1506 (2014).
Gutmann, B. et al. The expansion and diversification of pentatricopeptide repeat RNA-editing factors in plants. Mol. Plant 13, 215–230 (2020).
Potter, S. C. et al. HMMER web server: 2018 update. Nucl. Acids Res. 46, W200–W204 (2018).
Fuchs, P. et al. Single organelle function and organization as estimated from Arabidopsis mitochondrial proteomics. Plant J. 101, 420–441 (2020).
Lurin, C. et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16, 2089–2103 (2004).
Mueller, U. et al. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 19, 442–449 (2012).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
Zwart, P. H. et al. Automated structure solution with the PHENIX Suite. Methods Mol. Biol. 426, 419–435 (2008).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Terwilliger, T. C. et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D 64, 61–69 (2008).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA 98, 10037–10041 (2001).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 (2004).
Lerner, M. G. & Carlson, H. A. APBS plugin for PyMOL (University of Michigan, 2006).
Potterton, L. et al. CCP4i2: The new graphical user interface to the CCP4 program suite. Acta Crystallogr. D 74, 68–84 (2018).
Li, F. W. et al. Anthoceros genomes illuminate the origin of land plants and the unique biology of hornworts. Nat. Plants 6, 259–272 (2020).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Barton, G. J. Alscript: a tool to format multiple sequence alignments. Protein Eng. Des. Sel. 6, 37–40 (1993).
Kouno, T. et al. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 8, 1–8 (2017).
Acknowledgements
In remembrance of Axel Brennicke (1953–2017), the co-discoverer of RNA editing in plant organelles and who initiated this project in 2011. We thank B. Girbardt, D. Pruchner, A. Müller, Y. Yew, N. Holton, O. Ganichkin, K. Vester, A. Bergfort, A. Jörg, M. Burger, J. Wollenhaupt and A. Maeda for excellent technical assistance and experimental support. We are grateful to C. Schmitz-Linneweber, V. Knoop, M. Wahl and T. Shikanai for their valuable support and discussions. We acknowledge access to beamline P13, operated by EMBL Hamburg at the PETRA III storage ring (Hamburg, Germany). We would like to thank I. Bento, G. Bourenkov and T. Schneider for their assistance in using the beamline. We acknowledge access to beamlines BL14.1/2/3 of the BESSY II storage ring (Berlin, Germany) via the Joint Berlin MX-Laboratory sponsored by the Helmholtz-Zentrum Berlin für Materialien und Energie, the Freie Universität Berlin, the Humboldt-Universität zu Berlin, the Max-Delbrück Centrum, and the Leibniz-Institut für Molekulare Pharmakologie. This work was supported by DFG grant SCHA 1952/2-2 to M.S.R., JSPS Grants-in-Aid for Scientific Research no. 18H02462 and DFG grant no. TA624/10-1 to M.T, and start-up funding from the University of Greifswald to G.W.
Author information
Authors and Affiliations
Contributions
M.T. and G.W. coordinated and supervised the project. M.T., T.B., S.T., S.H and D.V. cloned OTP86 constructs and performed test expressions, M. S.-R. and B.O. designed initial bacterial expression constructs and developed the bacterial RNA editing assay. S.T. cloned mutants of PPR56PPRE1E2–OTP86DYW, performed the bacterial editing assays and western blot analysis. M.T. and S.T. analysed the data. B.F. expressed, purified and assayed in vitro activity of PPR56PPRE1E2–OTP86DYW. T.B. and G.W. conducted DSF experiments, expressed, purified and crystallized OTP86DYW. G.J.P., C.F. and G.W. collected diffraction data. C.F, M.S.W, G.J.P. and G.W. analysed the data. M.S.-R. performed evolutionary conservation analyses. M.T. and G.W. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Catalysis thanks Philippe Giegé, Mamoru Sugita and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–8, Tables 1 and 2, Notes 1–6 and references.
Supplementary Video 1
Catalytic activation of the A. thaliana OTP86 DYW domain. Colouring and labels as in Fig. 3.
Supplementary Video 2
Catalytic activation of the A. thaliana OTP86 DYW domain active site. Colouring and labels as in Fig. 3.
Source data
Source Data Fig. 2
Source data for r.m.s.d. calculations.
Source Data Fig. 5a
Source data for in vivo RNA editing assays.
Source Data Fig. 6a
Source data for DSF experiments, melting curves, fitting, Tm values (6a); Source data for in vitro RNA editing assays (6b).
Source Data Supplementary Fig. 1c
Source data for exemplary RNA editing assessment.
Source Data Supplementary Fig. 2
Source data for size exclusion chromatograms, Source data for X-ray fluorescence spectroscopy.
Source Data Supplementary Fig. 4
Source data for phylogenetic analyses.
Source Data Supplementary Fig. 7
Source data for the calculation of Western Blot intensities.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Takenaka, M., Takenaka, S., Barthel, T. et al. DYW domain structures imply an unusual regulation principle in plant organellar RNA editing catalysis. Nat Catal 4, 510–522 (2021). https://doi.org/10.1038/s41929-021-00633-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41929-021-00633-x
This article is cited by
-
Gene expression and physiological roles of post-transcriptional editing in plant organellar systems
Theoretical and Experimental Plant Physiology (2024)
-
The dicot homolog of maize PPR103 carries a C-terminal DYW domain and may have a role in C-to-U editing of some chloroplast RNA transcripts
Plant Molecular Biology (2024)
-
A ribonuclease activity linked to DYW1 in vitro is inhibited by RIP/MORF proteins
Scientific Reports (2023)
-
U-to-C RNA editing by synthetic PPR-DYW proteins in bacteria and human culture cells
Communications Biology (2022)
