
Abstract
We report on outstanding photo-responsivity, R > 103 A/W, fast response (~0.1 s), and broadband sensitivity ranging from the UV to the NIR in two terminal graphene/MoS2 photodetectors. Our devices are based on the deterministic transfer of MoS2 on top of directly grown graphene on sapphire, and their performance outperforms previous similar photodetectors using large-scale grown graphene. Here we devise a protocol for the direct growth of transparent (transmittance, Tr > 90%), highly conductive (sheet resistance, R□ < 1 kΩ) uniform and continuous graphene films on sapphire at 700 °C by using plasma-assisted chemical vapor deposition (CVD) with C2H2/H2 gas mixtures. Our study demonstrates the successful use of plasma-assisted low-temperature CVD techniques to directly grow graphene on insulators for optoelectronic applications.
Similar content being viewed by others

Introduction
Since the demonstration of scalable growth of graphene by catalytic CVD1,2,3,4, great efforts have been devoted5,6,7, and much progress has been made8 on the metal-assisted growth of large-area high-quality graphene. Nonetheless, big issues still exist, mainly related to the relative thermal expansion coefficients of graphene and catalyst9,10, suitable large-scale uniformity11, and reliable polymer and metal elimination during the transfer step, as metal residues in transferred graphene are above the standard specifications requested for on-fab integration12. Alternative routes have been explored as the direct pyrolytic growth of graphene on substrates of technological relevance13,14. This approach simplifies the processing of graphene as it avoids transfer and yields ultraclean layers. However, obtaining uniform and smooth graphene by these synthetic routes remains challenging, as the relative thermal expansion between graphene and substrate at high temperature produces wrinkles in graphene15,16. Another alternative method is the plasma-assisted CVD growth that enables the synthesis at low temperature (T) and renders wrinkle-free material17,18. Unfortunately, the as-grown graphene often exhibits high nucleation density and small grain size, due to the low surface mobility of species at low T19,20. Also, the poor coalescence of grains, along with the non-limiting character of the process, triggers the vertical growth of carbon layers or pillars21. However, the optimization of these protocols would provide graphene at a large scale with properties suitable for many applications22,23,24.
Here, we apply our previously developed two-step strategy25,26, to directly grow graphene on sapphire by plasma-assisted CVD with C2H2/H2 gas mixtures at 700 °C. In the first step, small graphene nuclei are grown on sapphire with strict control of the nuclei density. In the second step, the nuclei size is increased, growing from the nuclei edges until completing a continuous film. We find that, among the synthesis parameters, low gas pressure and flow are critical to decrease the nucleation density to extremely low values not observed previously19,20,25,26 in order to subsequently yield graphene films with large grain sizes and good coalescence27. Our graphene films exhibit high transmittance (Tr > 90%), and are highly conductive (R□< 1 kΩ), competing with state-of-the-art high T directly grown graphene15,28. Moreover, the morphology of the films is wrinkle-free and presents better continuity, uniformity, and smoothness, being essential features for material integration. In order to demonstrate the suitability of our graphene films for optoelectronic devices, van der Waals 2D heterostructures have been prepared by micromechanical exfoliation and deterministic transfer of MoS2 on top of our mechanically patterned graphene-sapphire substrates to fabricate two terminal photodetectors. Van der Waals 2D heterostructures circumvent the lattice-matching constraints encountered in heteroepitaxial synthesis of thin films for photonic and optoelectronic devices29,30,31, usually combining semiconducting 2D materials, as optically active layers, and graphene electrodes32,33,34. Previous work has been reported on graphene-MoS2 2D van der Waals heterostructures on optoelectronics devices using all micromechanically exfoliated materials35, catalytic CVD grown graphene, and transferred materials34 or high T graphene grown on insulators16,36,37 but not using low T direct growth of graphene on insulators. Prior to the MoS2 transfer, our graphene layers are mechanically patterned, defining a simple architecture of two graphene contacts separated by a continuous graphene channel on top of which the photosensitive MoS2 layer is placed. This simple device architecture eliminates the need for other metal contacts and related technologies. We find that the devices exhibit an outstanding photo-response resulting in responsivity (R) > 103 A/W, time responses (rise time, tr and fall time tf) ≈ 0.1 s, and broadband sensitivity under light illumination from the UV (365 nm) to the NIR (940 nm). These features outperform previously reported photo-response of heterostructures based on graphene directly grown on insulators16,36,37,38,39. Finally, we investigate the charge transfer mechanism involved between the graphene and MoS2, measuring the surface potential (SP) of the heterostructure by Kelvin probe force microscopy (KPFM)40,41,42,43.
Results and discussion
Figure 1 summarizes the processing steps followed to prepare 2D heterostructures. Figure 1a depicts the low T synthesis of graphene on sapphire by the plasma-assisted activation and dissociation of gas precursor (C2H2) and diluent (H2) to radicals (CHx, C2Hx, Hx). The plasma CVD system and the growth scheme are described in Supplementary Figs. 1 and 2, respectively. Figure 1b illustrates how the graphene layer is mechanically micropatterned to define a channel in a simple process. First, we use a diamond tip to coarsely eliminate graphene by scratching, and then we use a needle tip, fixed to a mobile support (see Methods). Figure 1c depicts the micromechanical exfoliation of monolayer MoS2 gently peeled off from bulk material by using a PDMS Gel-Film. Then a single-layer MoS2 is deterministically transferred on top of the graphene channel, as shown in Fig. 1d (see Methods).

a Plasma-assisted CVD growth of graphene. Activation and dissociation of radicals. b Two-step micropatterning of graphene channel and contacts. c Micromechanical exfoliation of MoS2 with Gel-film. d Deterministic transfer of MoS2 on top of graphene channel and the scheme of the final 2D architecture for the device.
Direct graphene synthesis by plasma-assisted CVD
We first minimize the graphene nuclei density on sapphire and optimize the material graphitic quality preventing amorphous deposits varying the hydrogen/carbon ratio (H/C) at a given T26. The complete list of experiments performed is shown in Supplementary Fig. 3. Initially, we start the nucleation at 600 °C. The characterization results are shown in Supplementary Fig. 4. However, the corresponding Raman analysis discloses that the grown nuclei do not meet the graphitic quality required to continue growing the films. Figure 2 present the results of increasing the T at 700 °C. Figure 2a shows the Raman spectra of a series of nucleation experiments as a function of H/C. Supplementary Fig. 5 complements this analysis with AFM characterization and thickness measurement. At constant nucleation time (t1), the thickness decreases as the H/C increases due to the lower C2H2 concentration. Our material follows faithfully the three-stage classification model of carbon disorder44,45, leading from amorphous carbon to nanocrystalline graphite. The width of the D peak and the G peak position of each spectrum are used to assess the graphitic quality44. Figure 2b shows how the width of the D peak decreases to less than 30 cm−1 as the H/C increases, indicating better quality nuclei45. The G peak position shifts from 1600 to 1580 cm−1, as we see in Fig. 2c, pointing out the high crystallinity of the deposited materials46. Along with the D peak narrowing and G peak shifting, a key feature that indicates the transition to nanocrystalline graphene is the emergence of the D′ peak. In partially amorphous carbon, G and D′ peaks are so wide that it is more convenient to consider them as a single, upshifted, wide G band at ~1600 cm−1 and the ID/IG and I2D/IG ratios are low, as we see at low H/C in Fig. 2a–c and in Supplementary Fig. 5. When amorphous carbon evolves to nanocrystalline graphite, as we see at high H/C in Fig. 2a–c and Supplementary Fig. 5, the G + D′ peak doublet appears due to the narrowing of the peaks, as we also see in Fig. 2b for the D peak, and the G peak position shifts to 1580 cm−1. We also see in Supplementary Fig. 5, where we include the spectra of Fig. 2a separately, that ID/IG increases and ID/ID′∼ 4, being these effects mostly related to the grain boundaries of high-density small grains47, and that I2D/IG ratio increases due to graphitization level being its final value an indication of the thickness46. Figure 2d displays the AFM topographic image of the nanocrystals grown with high H/C = 250. Despite the high structural quality of the nanocrystals, the nucleation density, around 800 nuclei/µm2 after image analysis, is very high due to the long nucleation time (t1 = 90 min) used in these experiments required for the initial structural characterization25. The next goal within optimization is to decrease the nuclei density. First, t1 is progressively shortened to t1 = 20 min and finally to t1 = 5 min (H/C = 250, 700 °C). The corresponding AFM topographic images are shown in Fig. 2e, f) respectively. At t1 = 20 min, it can be observed how the density decreases to 448 nuclei/µm2, after image analysis. However, in Fig. 2f, the nucleation density assessment is not reliable. The thickness measurements and Raman analysis of these samples included in Supplementary Fig. 6, confirms that the thickness is around 1 nm and the graphitic quality for t1 = 20 min is similar to the one observed in the previous experiment at t1 = 90 min. In order to study the density and crystallinity at short nucleation time, in the following experiments, we apply in each run a second growth step (t2 = 120′), increasing slightly H/C = 275. This approach allows us to study the nuclei density with short t1 if we control the sub-monolayer coverage with t2. Figure 2g shows the AFM image of the nanocrystals grown in two steps (t1 = 5 min, H/C = 250; t2 = 120 min, H/C = 275). The hexagonal shape of the grown crystals and how the nucleation density decreases, around 250 nuclei/µm2, is revealed in the image. The grain size increases compared with the previous experiment, as the material accumulates from the edges during t2. The corresponding line profile in Fig. 2j, from the green line in Fig. 2g, shows that the thickness of the hexagonal crystals is in the nanometer scale that would correspond to few-layer graphene. It is important to point out that with these parameters, if we decrease further t1, a plateau in the nucleation density is observed first and, finally, the absence of nuclei as nucleation does not occur (not shown). To finish the optimization, we run a series of experiments decreasing the total flow of gas, and so the chamber pressure48, leaving the H/C, t1, t2, and T constant. We find how the nucleation density decreases to a minimum value, Fig. 2h, i. Figure 2h shows the nuclei grown with H2 flow = 25 sccm and C2H2 flow = 0.1 sccm. The hexagonal graphene nuclei present a low nucleation density of 64 nuclei/ µm2, that drastically drops to 4 nuclei/ µm2 for H2 flow = 15 sccm and C2H2 flow = 0.06 sccm, as seen Fig. 2i. The decrease in nucleation density is accompanied by an increase in the thickness of the nuclei as can be observed in the line profiles in Fig. 2j–l). The corresponding Raman analysis of the samples in Fig. 2g–i and the graphical evolution of nucleation density and thickness is presented in Supplementary Fig. 7.

a Raman spectra of graphene nuclei grown at 700 °C as a function of the H/C ratio. H2 flow = 50 sccm, C2H2 flow ÷ 0.4–0.2 sccm. Analysis of the FWHM of D peak (b) and IG position (c) from the previous spectra. d–f AFM topographic images of the nuclei grown with H/C = 250, and variable t1; d t1 = 90 min. e t1 = 20 min. f t1 = 5 min. g–i AFM topographic images of the nuclei grown with H/C = 250, t1 = 5 min, H/C = 275, t2 = 120 min and variable flow. g C2H2 flow = 0.2 sccm, H2 flow = 50 sccm. h C2H2 flow = 0.1 sccm, H2 flow = 25 sccm. i C2H2 flow = 0.06 sccm, H2 flow = 15 sccm. j–l Line profiles that correspond to the green lines in images (g–i). Z scale of AFM images indicated in the profiles.
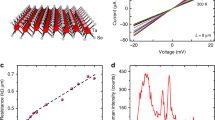
Supplementary Fig. 8 presents a comprehensive graphical description of the growth kinetics and reaction mechanism proposed to explain the nucleation and subsequent growth of graphene. During the heating step, we first anneal the sample with plasma-activated and dissociated Hx species and radicals, promoting surface cleaning. The number of radicals depends on the plasma power used (200 W). As a result of cleaning, volatile H2O or CHx can be desorbed. Then, we introduce the C2H2 in the plasma and radicals CHx, C2Hx, and Hx are produced that initially adsorb physically on the substrate. The dissociation of precursor in the gas phase is the main advantage of plasma-assisted CVD in comparison with catalytic processes. As dissociation is an endothermic process, high T is needed even with catalysis. The adsorption is a function of the T and H/C ratio and is enhanced at higher T and low H/C26. Next, the diffusion of species on the surface occurs. No bulk diffusion of radicals is expected on insulators19. The diffusion is a direct function of T and surface morphology. As we grow at low T, low diffusion occurs before the nucleation resulting in high nuclei density. That’s the reason to carefully study the H/C, t1, flow and to implement the second step, edge growth, already discussed in Fig. 2. The nucleation—and subsequent growth- of graphitic carbon from radicals is an exothermic process and can take place at low T, once the radicals are available from plasma activation25. At the same time, side reactions are taking place. The most important is the effect of H during the nucleation that allows to stabilize a crystalline carbon phase avoiding the nucleation of amorphous carbon (etching amorphous phases) when the H/C and T are properly selected. Other side reactions, as reduction of the substrate, are likely to appear at high H/C and low flow, since the process develops more slowly, producing a prolonged substrate exposure to radicals. The reduction of the substrate could enhance its roughness, promoting the nucleation of thicker seeds, as observed in Fig. 2h, i. Once the nucleation density is suitable, we proceed with the second growth step, slightly increasing H/C. When we increase the H/C ratio, the chemical absorption of the plasma radicals to the edge of the nuclei deposited during nucleation is, in principle, more favorable, from an energetic point of view, than further nucleation in the substrate. Continuous films have been grown in the second step to complete the substrate coverage (t2 = >240‘). Two recipes, with different flow, are used to grow continuous films with variable thickness at 700 °C. The AFM topographic image of a thin film in Fig. 3a grown with H2 flow = 50 sccm and C2H2 flow = 0.2 sccm demonstrates that as-grown films are continuous, with good coalescence of grains, without the wrinkles reported in the high T pyrolytic growth15 and without evidence of vertical growth observed in previous substrates26. The corresponding line profile in Fig. 3b confirms the extremely low roughness, RMS = 4.3 Å, of the sample. The typical Raman spectrum of the sample in Fig. 3c shows the D, G, and 2D peaks of graphitic carbon with a 2D/G ratio related to few-layer graphene and with a D peak ascribed to grain boundaries46. Figure 3d shows the AFM topographic image of another thin film grown with H2 flow = 25 sccm and C2H2 flow = 0.1 sccm. The film is continuous, with good coalescence and without wrinkles. The growth evolution for this film and the nucleation density as a function of t2 is presented in Supplementary Fig. 9. It is confirmed that the nucleation density does not notably increase with time and that the final grain size is around 200 nm. However, the corresponding line profile in Fig. 3e reveals that the roughness, RMS = 9.9 Å, of the sample increases as expected due to the initial nuclei thickness observed in Fig. 2k. We relate this effect to the partial reduction and roughness increase of the substrate at low flow discussed above. The corresponding Raman spectrum in Fig. 3f shows the D, G, and 2D peaks of graphitic carbon with a 2D/G ratio that decreases in comparison with Fig. 3c this is related again to the higher thickness. The optical transmittance analysis of both films in Fig. 3g confirms again the thickness difference. The graphene formation with both recipes is further verified through XPS C 1s spectra, as shown in Fig. 3h, which also confirms the dominant contribution of sp2 carbon. The intensity observed in the spectra is thickness dependent. We also measure the sheet resistance (R□) of the films and the results show low R□ = 0.95–1 kΩ in as-grown material that drops to R□ = 0.75–0.83 kΩ after high T annealing (~1000 °C) depending inversely on thickness, rivaling with recent reports on high T graphene grown on sapphire15,28. As we consider that the high smoothness observed in the graphene layers grown with the high flow is the most critical feature of the films, we select them to fabricate heterostructures. It is expected that the flatness translates into good interfacial contact when we transfer another material on top, as we will demonstrate below.

a AFM topographic image of the graphene film grown with H2 flow = 50 sccm and C2H2 flow = 0.2 sccm. The green line indicates the place of the corresponding line profile in (b). c Corresponding Raman spectrum of the sample. d AFM topographic image of the graphene film grown with H2 flow = 25 sccm and C2H2 flow = 0.1 sccm. The green line indicates the place of the corresponding line profile in (e). f Raman spectrum of the graphene film. g Optical transmittance of the graphene films. h XPS C 1 s core level spectra of the thin films.
Preparation of 2D heterostructures
The continuous films are micropatterned; first, with a diamond tip, we scratch both sides of our pristine samples, as shown in the scheme of Fig. 1b and Supplementary Fig. 10b. Then, with a needle tip, we micro-wear the remaining area, defining the graphene channel and contacts, as observed in the optical image of Figs. 4a, 1b and Supplementary Fig. 10d. The contacts are oriented in the perpendicular direction, as can be observed in Supplementary Fig. 10d. We confirm the complete graphene elimination by Raman analysis on the scratched area, (not shown). We then isolate thin MoS2 flakes from parent crystals by micromechanical exfoliation, Fig. 1c and characterize them. As a representative example, we can see in Fig. 4b and Supplementary Fig. 10c the optical microscope image of the Gel-Film surface used in the last step of micromechanical exfoliation from parent crystals with thin flakes, one of them containing 1 L MoS2 area, that can be observed on the left side of Fig. 4b and highlighted into black dashed line in Supplementary Fig. 10c. Figure 4c displays the optical microscopy image of the 2D heterostructure made with the corresponding flake and channel by the deterministic transfer method explained previously in Fig. 1d. Note that, in order to assess the performance of our graphene films regardless of the MoS2 thickness, we are also interested in multilayer area that is known to enhance the IR absorption49, so, there is no need to further trimming the few-layer MoS250. Figure 4d depicts the differential micro-reflectance spectrum acquired from the 1 L area of the MoS2 flake in Fig. 4b. The spectrum confirms that the maximum of the A = 1.90 eV and B = 2.04 eV exciton peaks from MoS2 are in the position that corresponds to a monolayer51. We also double-check the monolayer thickness by Raman spectroscopy in Fig. 4e. We see that the Raman phonon peaks, E12G = 384 cm−1 and A1G = 403 cm−1 from MoS2 appear at the position that corresponds to monolayer MoS2, with the Raman shift difference between peaks of 19 cm−1 52. Prior to the all-dry viscoelastic deterministic transfer53 of the MoS2 monolayer, we anneal our graphene samples in a vacuum (10−6 mbar) at 500 °C for 2 h.

a Optical microscope image of the graphene channel after the micro-wear process with a needle tip. The contacts are oriented perpendicularly. b Optical microscope image of the Gel-Film surface with a MoS2 flake with portions of different thickness. 1 L MoS2 portion can be observed on the left side. c Optical microscope image of the final heterostructure with 1 L MoS2 highlighted with a blue dashed line and graphene channel highlighted with a black dotted line. d Differential micro-reflectance spectrum of 1 L MoS2. e Raman spectrum of 1 L MoS2.
Photo-response of 2D heterostructure
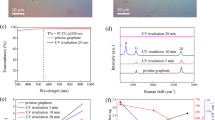
Before the photo-response is measured, the heterostructure shown in Fig. 4c is annealed in a vacuum (10−6 mbar) at 200 °C for 2 h to remove atmospheric adsorbates, improving, therefore, the interfacial contact between graphene and MoS2. We illuminate the device using optic fiber coupled to light-emitting diodes (LEDs) with wavelengths from the UV (405 nm) to the NIR (1100 nm)54. We test the photo-response of the samples in a simple two terminal device configuration, as shown in Fig. 5a. Before heterostructure processing, we also analyse the photo-response of the graphene channel individually as a control measurement. We confirm that the photocurrent obtained is negligible, as observed in Supplementary Fig. 11. Figure 5b depicts the total current (I) in absolute value as a function of the applied bias voltage (VDS) (IVDS curves hereafter) in dark conditions, black dotted line, and under different illumination wavelengths (λ) at a constant incident LED optical power = 1 mW. The spot size or illumination area (AL) ≈ 1.25 × 107 µm2 and the device effective area (AD) ≈ 250 µm2 estimated from the overlapping of materials in Fig. 4c. The response is linear as a function of VDS (V) and symmetric in all curves pointing towards the ohmic behavior due to the continuity of the graphene channel and cleanness of the contacts. We can see that the current (I) increases upon illumination for all λ (nm), as previously reported in similar heterostructures55,56,57,58. Note that for the same graphene-MoS2 architecture photodetectors used here, some works reported negative photocurrent35,36,38,59. This disagreement has been recently unraveled in terms of the adsorbate density and their photo-induced desorption upon illumination16. Adsorbate desorption can lead to a change from a negative photocurrent at high adsorbate density to a positive one in cleaner interfaces16. In our case, as we anneal the samples before and after the transfer, an adsorbate-free and sharp interface is expected, rendering positive photocurrent in several devices made with different MoS2 flakes and graphene samples. Figure 5c depicts the calculated positive photocurrent (IP hereafter, subtracting the dark current from I) generated as a function of VDS (V) and λ (nm). We see that the magnitude of the response is quite similar for all λ (nm) from the UV (λ = 405 nm) to NIR (λ = 660 nm), it decreases in the IR (λ = 850 nm, λ = 940 nm) and is absent for larger λ (nm) (we illuminate the sample with λ = 1100 nm, without photoexcitation). The photo-response observed in the IR is not expected for 1 L MoS2 due to its intrinsic bandgap60, and we ascribe it to the multilayer MoS249,61 part attached to the monolayer as seen in gray and white in Fig. 4c. Figure 5d, e show the IP as a function of time (s) and the λ (nm) upon illumination (ON) and in dark (OFF) conditions (IPt curves hereafter). We use a constant VDS = 2 V and LED optical power = 1 mW. We confirm the larger sensitivity in the UV-NIR and its reduction in the IR that we ascribe to the bandgap transition from direct in 1 L MoS260 to indirect in FL MoS261. A detailed analysis of the response at λ = 660 nm in Fig. 5e, shows that the rise time (tr) and fall time (tf) are in the millisecond regime, tr = 120 ms and tf = 122 ms. This response time is much faster than observed in previous works using graphene grown on insulators at high T16,36,38,39. This dynamic response is similar for all λ (nm) from the UV (λ = 405 nm) to IR (λ = 940 nm). Next, we measure IPt curves for all λ (nm) while the LED optical power is swept from 1 µw to 1.17 mW-2.5 mW (the upper limit depends on the λ (nm)) at different VDS. Figure 5f depicts an example of the typical curves, λ = 595 nm, VDS = 2 V. The results for other λ (nm), VDS from 2 to 5 V and low LED optical power = 1 µW can be seen in Supplementary Figs. 12, 13. A nonlinear increase of the photocurrent is observed as a function of the optical power. The sensitivity is much higher for low power, as previously reported in similar architectures62,63. The responsivity R (A/W) of the devices as a function of the LED Optical Power, λ (nm) and VDS is calculated using the following equation,
where IP is the photocurrent, AD (µm2) is the effective area of the device, and Ps (W/µm2) = LED optical power (W)/AL (µm2) is the power density of the light source. The calculation results in Fig. 5g exhibit an outstanding R > 103 (A/W) for all λ (nm) at low LED optical power = 1 µW and VDS = 5 V (see Supplementary Fig. 13 for a detailed view of R (A/W) at low LED optical power = 1 µW). We relate this good performance to the sharp interface between materials and the ohmic behavior of the graphene contacts used. We see that R decreases as a function of the LED optical power, as previously reported in photoconductor-based sensors40,62,63. The final R (A/W) values decrease in the IR, mainly due to the indirect bandgap of FL MoS2. Regarding the IR illumination (λ = 850 nm, λ = 940 nm), we note in Fig. 5g that the signal is missing at LED optical power <10 µW due to extremely low signal-to-noise ratio during acquisition. We finally perform some complementary analysis of the photo-response of the proposed heterostructure in two devices, as can be seen in Supplementary Figs. 14,15. We first fabricate a device with a MoS2 flake with a complete monolayer area, Supplementary Fig. 14, in order to confirm the sensitivity of the device as a function of the number of layers. The R of the device decreases slightly in the UV-Vis in comparison with the device in Fig. 5, but it is in the same order of magnitude. The main difference is the lack of photo-response in the IR due to the monolayer bandgap. The device detects the light at low optical power with low noise. Supplementary Fig. 15 presents a device fabricated with a few-layer MoS2 flake. In this case, the heterostructure shows photo-response in the UV-Vis-IR, although the R decreases in comparison with the initial device in Fig. 5 (same order of magnitude). This device does not detect the light at low optical power in UV-Vis-IR, as observed in the IR detection in Fig. 5. These results, overall, allow us to ascertain the importance of the semiconductor thickness for sensitivity (monolayer) and broadband photodetection (multilayer).

a Scheme of the photo-response measuring set-up. b IVDS curves with current in absolute value under different (λ) at LED optical power = 1 mW. c The corresponding IPVDS curves from (b) with photocurrent in absolute value. d, e IPt curves at a constant VDS = 2 V and optical power = 1 mW. f IPt curve at λ = 595 nm, with optical power swept from 80 µw to 1.7 mW, VDS = 2 V. g Responsivity R(A/W) of the devices as a function of the LED optical power (W), λ (nm), and VDS (V).
Charge transfer mechanism
In order to understand more in-depth the observed positive photocurrent, we study the surface potential, SP, of the device area by KPFM imaging with an Au tip40. Prior to SP acquisition, we check our system with a KPFM standard (with Au, Si, and Al) and double-check the measurement with an Au pad evaporated on top of our graphene sample, near the device area. The calibration data can be checked in Supplementary Fig. 16. We also assume the standard value of the work function (WF) of Au ≈ 5.1 eV40. Figure 6a shows the AFM topography of the device heterostructure area. The inset in the right upper corner corresponds to the optical image of the heterostructure with the graphene channel and the MoS2 flake on top. The few layers area is composed of different portions with a variable number of layers. We see on the right side of the channel a bare graphene area that enables us to measure the intrinsic graphene SP. Figure 6b shows the corresponding SP map, including the average SP calculated from the acquired values in the areas marked with a dashed black/white line square. Note that in our system, the higher the SP, the smaller the WF (SP of Al = 1271 ± 27 mV with standard Al WF ≈ 3.9 eV, see Supplementary Fig. 16), and the SP relative value of Au in contact with Au tip should be near zero (SP of Au standard = 11 ± 9 mV, SP Au pad = 11 ± 20 mV, see Supplementary Fig. 16). From the values in Fig. 6b, the calculated Graphene WF ≈ 5.205 eV (Au WF ≈ 5.1 eV - bare graphene SP = −105 mV in Fig. 6b), pointing out the p-doping of graphene64,65. The electrical transfer curve, measured to further prove the p-type doping of the present graphene layer and validate this conclusion, is included in Supplementary Fig. 17. Following the same procedure, taking the SP values of the MoS2 flake on top of the graphene channel, we calculate the 1 L MoS2 WF ≈ 4.868 eV and FL MoS2 WF ≈ 4.973 eV. Figure 6c shows the relative electronic band diagram of graphene and 1 L MoS2 with the calculated values. Figure 6d displays the fermi level alignment and the direction of the built-in Efield generated in the interface upon contact with the measured WF mismatch between MoS2 and graphene. These results allow us to propose that the charge transfer mechanism involved under illumination is based on the e- transfer from p-doped graphene to the MoS2, holes in the opposite direction as depicted in Fig. 6d. This mechanism increases the p-doping of graphene under illumination that explains the enhancement of the current observed, as previously reported on positive photocurrent49,62,64,66.

a AFM topographic image and b KPFM potential map of the MoS2-graphene heterostructure area. The surface potential average values of the corresponding areas in dashed black/white line squares are included. c Band diagram of the heterostructure before contact and d after contact, with bias and under illumination.
In summary, this study demonstrates the successful integration of directly grown graphene at low T in functional optoelectronics. We demonstrate the direct growth of good-quality graphene on sapphire by plasma-assisted CVD at low T (700 °C). The optimization of the nucleation density enables the growth of continuous, uniform, wrinkle-free, and smooth graphene films with high crystallinity and conductivity. The as-grown graphene film properties are demonstrated to be suitable for optoelectronic device applications. The micromechanical patterning of the graphene channel and contacts to prepare 2D heterostructures with MoS2 has been shown to be an efficient method to construct a photodetector architecture. The graphene contact-channel and MoS2 sensitizer heterostructures exhibit positive photocurrent with an outstanding photo-response, R > 103 A/W, low time responses ≈0.1 s, and broadband sensitivity. These results are due to the good interfacial contact between layers and very clean contacts. Finally, surface potential measurements of the heterostructures shared light onto the charge transfer mechanism involved. It can be concluded that the grown graphene onto sapphire is p-doped and transfers electrons to MoS2 upon illumination, increasing efficiently both the doping level and the total current flow through the device.
Methods
Direct graphene synthesis
Electron cyclotron resonance plasma-assisted chemical vapor deposition (ECR-CVD) is used for the growth of graphene material from C2H2/H2 gas mixtures. A description of the plasma CVD ASTEX AX 4500 system can be found in Supplementary Fig. 1 and ref. 25. Sapphire (c-Al2O3, University Wafer Inc.) is used as a substrate for graphene growth. We include the detailed scheme of the growth with the process steps in Supplementary Fig. 2. The synthesis consists of two steps. In the first step, high-quality graphene seeds nucleate. In the second step, by means of parameter modification, the growth is promoted from the nucleated dots up to the layer formation. The plasma power (200 W) is constant throughout the study. The distance between the plasma chamber bottom part and the sample is around 30 cm. The substrate temperature from plasma heating is below 120 °C. We use a dedicated heating system to control substrate T. The background pressure is 5.4 × 10−5 mbar. During synthesis, the total pressure changes in the range of 5.4 × 10−2 mbar (flow: C2H2 = 1.65 sccm and H2 = 50 sccm) and 10−3 mbar (flow: C2H2 = 0.06 sccm and H2 = 15 sccm) and T in the range of 600–700 °C. At low pressure, He gas is used to sustain the plasma. In Supplementary Fig. 3, we include all the experiments done during the systematic study of the growth evolution.
Graphene patterning
Figure 1b illustrates how the graphene layer is mechanically micropatterned in a simple process. First, we use a diamond tip to coarsely eliminate graphene by scratching and then we use a needle tip, fixed to the mobile support in our deterministic placement set-up53, that is scanned over the sample to eliminate with major precision the undesired graphene areas. The resulting graphene architecture consists of two large graphene contacts separated by a continuous graphene channel ~10 µm-width and ~ 50–100 µm length. Finally, we anneal the samples in a vacuum (10−6 mbar) at 500 °C for 2 h.
Deterministic transfer of MoS2 and heterostructure formation
MoS2 micromechanically exfoliated from bulk natural mineral (Molly Hill Mine, Quebec, Canada)67 is cleaved with Nitto SPV 224 tape. Then, the tape containing the cleaved bulk micro crystals is put in contact with a Gel-Film (Gel Pak®) surface and gently peeled off to leave some thin flakes on the Gel-Film substrate, as depicted in Fig. 1c. The surface of the Gel-Film is then inspected under an optical microscope (Motic BA 310 MET-T) in transmission mode to identify suitable MoS2 monolayers (see Fig. 4b). The flake thickness is assessed first by their apparent transmittance68, then double-checked by micro-reflectance spectroscopy51 to verify the single-layer thickness and, finally, analyzed by Raman spectroscopy (S&I Monovista CRS+) to cross-check the monolayer thickness. Once a suitable single-layer MoS2 flake is identified, it is transferred on top of the graphene channel with the all-dry viscoelastic deterministic placement method summarized in Fig. 1d53.
Characterization
Room T Atomic Force Microscopy (AFM) measurements are performed with a commercial instrument and software from Nanotec Electrónica S.L69. Two different operation modes are employed: dynamic mode, exciting the tip at its resonance frequency (~75 kHz) to acquire topographic information of the samples, and contact mode, to remove mechanically the carbon deposits and measure the graphene thickness70. The surface potential of the heterostructure is characterized by Kelvin probe force microscopy (KPFM) using a MikroMasch HQ:NSC18/Cr-Au (Fr = 75 KHz; k = 2.8 N/m) conductive tip. The structures of our graphene films and the micromechanically exfoliated MoS2 flakes are assessed by Raman spectroscopy using a confocal Raman microscope (S&I Monovista CRS +). Raman spectra have been obtained using a 532 nm excitation laser, a 100x objective lens (NA = 0.9), and an incident laser power of 6 mW for graphene and much less power of 0.045 mW for MoS2 characterization. X-ray photoelectron spectroscopy (XPS) is carried out using a PHOIBOS 100 1D electron/ion analyzer with a one-dimensional delay line detector and a monochromatic Al Kα anode (1486.6 eV) with a pass energy of 15 eV. The binding energy (BE) scale was calibrated with respect to the C 1s core level peak at 285 eV. The sheet resistance of the continuous films is characterized by four-point probe measurements (JANDEL RMS2 Universal Probe). Fiber Optic coupled LEDs (Thorlabs MXXXF)54 with wavelengths from 405 to 1100 nm are used as light sources and a source-meter unit (Keithley® 2450) is used for performing the acquisition of electrical signals during the photo-response analysis. The electrical transfer curve is measured to prove the doping of the graphene layer (see Supplementary Fig. 17). In order to efficiently gate the graphene channel, an ionic gel (hydrogel) of 1 mm thickness is attached on top of the graphene sample and further connected with conductive copper tape. The hydrogel has a capacitance per area of 8.3 × 10−3 Fm−2 and is advantageous for applying high local gate bias using low external gate voltage due to the electrical double-layer effect.
Data availability
The authors declare that the comprehensive data supporting the findings of this study are available within the paper and its supplementary information.
References
Fang, M., Wang, K., Lu, H., Yang, Y. & Nutt, S. Covalent polymer functionalization of graphene nanosheets and mechanical properties of composites. J. Mater. Chem. 19, 7098–7105 (2009).
Bae, S. et al. Roll-to-roll production of 30-inch graphene films for transparent electrodes. Nat. Nanotech. 5, 574–578 (2010).
Reina, A. et al. Large area, few-layer graphene films on arbitrary substrates by chemical vapor deposition. Nano Lett. 9, 30–35 (2009).
Geim, A. K. Graphene: status and prospects. Science 324, 1530–1534 (2009).
Wu, T. et al. Fast growth of inch-sized single-crystalline graphene from a controlled single nucleus on Cu–Ni alloys. Nat. Mater. 15, 43–47 (2016).
Xu, X. et al. Ultrafast epitaxial growth of metre-sized single-crystal graphene on industrial Cu foil. Sci. Bull. 62, 1074–1080 (2017).
Miseikis, V. et al. Deterministic patterned growth of high-mobility large-crystal graphene: a path towards wafer scale integration. 2D Mater. 4, 021004 (2017).
Liu, F. et al. Achievements and challenges of graphene chemical vapor deposition growth. Adv. Func. Mater. 32, 2203191 (2022).
Zhao, C., Liu, F., Kong, X., Yan, T. & Ding, F. The wrinkle formation in graphene on transition metal substrate: a molecular dynamics study. Int. J. Smart Nano Mater. 11, 277–287 (2020).
Deng, B. et al. Wrinkle-free single-crystal graphene wafer grown on strain-engineered substrates. ACS Nano 11, 12337–12345 (2017).
Nguyen, V. L. et al. Layer-controlled single-crystalline graphene film with stacking order via Cu–Si alloy formation. Nat. Nanotech. 15, 861–867 (2020).
Lupina, G. et al. Residual metallic contamination of transferred chemical vapor deposited graphene. ACS Nano 9, 4776–4785 (2015).
Novoselov, K. S. et al. A roadmap for graphene. Nature 490, 192–200 (2012).
Saito, K. & Ogino, T. Direct growth of graphene films on sapphire (0001) and (112̅0) surfaces by self-catalytic chemical vapor deposition. J. Phys. Chem. C. 118, 5523–5529 (2014).
Mishra, N. et al. Wafer-scale synthesis of graphene on sapphire: toward Fab-compatible graphene. Small 15, 1904906 (2019).
Beckmann, Y. et al. Role of surface adsorbates on the photoresponse of (MO)CVD-grown graphene–MoS2 heterostructure photodetectors. ACS Appl. Mater. Interfaces 14, 35184–35193 (2022).
Zhai, Z., Shen, H., Chen, J., Li, X. & Jiang, Y. Direct growth of nitrogen-doped graphene films on glass by plasma-assisted hot filament CVD for enhanced electricity generation. J. Mater. Chem. A 7, 12038–12049 (2019).
Vishwakarma, R. et al. Direct synthesis of large-area graphene on insulating substrates at low temperature using microwave plasma CVD. ACS Omega 4, 11263–11270 (2019).
Muñoz, R. & Gómez-Aleixandre, C. Fast and non-catalytic growth of transparent and conductive graphene-like carbon films on glass at low temperature. J. Phys. D Appl. Phys. 47, 045305 (2014).
Luo, A. et al. Evolution of morphology and defects of graphene with growth parameters by PECVD. Mater. Res. Express 7, 035025 (2020).
Kulczyk-Malecka, J. et al. Low-temperature synthesis of vertically aligned graphene through microwave-assisted chemical vapour deposition. Thin Solid Films 733, 138801 (2021).
Chang, C.-J. et al. Layered graphene growth directly on sapphire substrates for applications. ACS Omega 7, 13128–13133 (2022).
Chen, Z. et al. High-brightness blue light-emitting diodes enabled by a directly grown graphene buffer layer. Adv. Mater. 30, 1801608 (2018).
Ci, H. et al. Enhancement of heat dissipation in ultraviolet light-emitting diodes by a vertically oriented graphene nanowall buffer layer. Adv. Mater. 31, 1901624 (2019).
Muñoz, R. et al. Direct synthesis of graphene on silicon oxide by low temperature plasma enhanced chemical vapor deposition. Nanoscale 10, 12779–12787 (2018).
Muñoz, R. et al. Low temperature metal free growth of graphene on insulating substrates by plasma assisted chemical vapor deposition. 2D Mater. 4, 015009 (2017).
Yen, C.-C., Chang, Y.-C., Tsai, H.-C. & Woon, W.-Y. Nucleation and growth dynamics of graphene grown through low power capacitive coupled radio frequency plasma enhanced chemical vapor deposition. Carbon 154, 420–427 (2019).
Jiang, B. et al. Batch synthesis of transfer-free graphene with wafer-scale uniformity. Nano Res. 13, 1564–1570 (2020).
Geim, A. K. & Grigorieva, I. V. Van der Waals heterostructures. Nature 499, 419–425 (2013).
Liu, Y. et al. Van der Waals heterostructures and devices. Nat. Rev. Mater. 1, 16042 (2016).
Castellanos-Gomez, A. et al. Van der Waals heterostructures. Nat. Rev. Methods Prim. 2, 58 (2022).
Novoselov, K. S., Mishchenko, A., Carvalho, A. & Castro Neto, A. H. 2D materials and van der Waals heterostructures. Science 353, aac9439 (2016).
Britnell, L. et al. Strong light-matter interactions in heterostructures of atomically thin films. Science 340, 1311–1314 (2013).
Bertolazzi, S., Krasnozhon, D. & Kis, A. Nonvolatile memory cells based on MoS2/graphene heterostructures. ACS Nano 7, 3246–3252 (2013).
Roy, K. et al. Graphene–MoS2 hybrid structures for multifunctional photoresponsive memory devices. Nat. Nanotechnol. 8, 826–830 (2013).
Hoang, A. T. et al. Epitaxial growth of wafer-scale molybdenum disulfide/graphene heterostructures by metal–organic vapor-phase epitaxy and their application in photodetectors. ACS Appl. Mater. Interfaces 12, 44335–44344 (2020).
Lin, Y.-C. et al. Direct synthesis of van der Waals solids. ACS Nano 8, 3715–3723 (2014).
Chen, C. et al. Large-scale synthesis of a uniform film of bilayer MoS2 on graphene for 2D heterostructure phototransistors. ACS Appl. Mater. Interfaces 8, 19004–19011 (2016).
Xu, H. et al. High detectivity and transparent few-layer MoS2/glassy-graphene heterostructure photodetectors. Adv. Mater. 30, 1706561 (2018).
Gao, S. et al. Graphene/MoS2/graphene vertical heterostructure-based broadband photodetector with high performance. Adv. Mater. Interfaces 8, 2001730 (2021).
Nonnenmacher, M., O’Boyle, M. P. & Wickramasinghe, H. K. Kelvin probe force microscopy. Appl. Phys. Lett. 58, 2921–2923 (1991).
Melitz, W., Shen, J., Kummel, A. C. & Lee, S. Kelvin probe force microscopy and its application. Surf. Sci. Rep. 66, 1–27 (2011).
Escasain, E., Lopez-Elvira, E., Baro, A. M., Colchero, J. & Palacios-Lidon, E. Nanoscale surface photovoltage of organic semiconductors with two pass Kelvin probe microscopy. Nanotechnology 22, 375704 (2011).
Ferrari, A. C. & Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 61, 14095–14107 (2000).
Ferrari, A. C. & Robertson, J. Resonant Raman spectroscopy of disordered, amorphous, and diamondlike carbon. Phys. Rev. B 64, 075414 (2001).
Ferrari, A. C. & Basko, D. M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 8, 235–246 (2013).
Eckmann, A. et al. Probing the nature of defects in graphene by Raman spectroscopy. Nano Lett. 12, 3925–3930 (2012).
Ueda, Y., Maruyama, T. & Naritsuka, S. Effect of growth pressure on graphene direct growth on an A-plane sapphire substrate: implications for graphene-based electronic devices. ACS Appl. Nano Mater. 4, 343–351 (2021).
Buscema, M. et al. Photocurrent generation with two-dimensional van der Waals semiconductors. Chem. Soc. Rev. 44, 3691–3718 (2015).
Yong, X. et al. Laser trimming for lithography-free fabrications of MoS2 devices. Nano Res. 16, 5042–5046 (2022).
Frisenda, R. et al. Micro-reflectance and transmittance spectroscopy: a versatile and powerful tool to characterize 2D materials. J. Phys. D Appl. Phys. 50, 074002 (2017).
Li, H. et al. From bulk to monolayer MoS2: evolution of Raman scattering. Adv. Func. Mater 22, 1385–1390 (2012).
Castellanos-Gomez, A. et al. Deterministic transfer of two-dimensional materials by all-dry viscoelastic stamping. 2D Mater. 1, 011002 (2014).
Quereda J, Z. Q., Diez, E., Frisenda, R. & Castellanos-Gómez, A. Fiber-coupled light-emitting diodes (LEDs) as safe and convenient light sources for the characterization of optoelectronic devices. Open Res. Europe 1, 98 (2022).
Li, X. et al. A self-powered graphene–MoS2 hybrid phototransistor with fast response rate and high on–off ratio. Carbon 92, 126–132 (2015).
Lim, Y. R. et al. Rational design of multifunctional devices based on molybdenum disulfide and graphene hybrid nanostructures. Appl. Surf. Sci. 392, 557–561 (2017).
Sun, B. et al. Large-area flexible photodetector based on atomically thin MoS2/graphene film. Mater. Des. 154, 1–7 (2018).
Pierucci, D. et al. Large area molybdenum disulphide- epitaxial graphene vertical Van der Waals heterostructures. Sci. Rep. 6, 26656 (2016).
Ma, D. et al. A universal etching-free transfer of MoS2 films for applications in photodetectors. Nano Res. 8, 3662–3672 (2015).
Mak, K. F., Lee, C., Hone, J., Shan, J. & Heinz, T. F. Atomically thin MoS2: a novel direct-gap semiconductor. Phys. Rev. Lett. 105, 136805 (2010).
Ellis, J. K., Lucero, M. J. & Scuseria, G. E. The indirect to direct band gap transition in multilayered MoS2 as predicted by screened hybrid density functional theory. Appl. Phys. Lett. 99, 261908 (2011).
Xu, H. et al. High responsivity and gate tunable graphene-MoS2 hybrid phototransistor. Small 10, 2300–2306 (2014).
Zhang, W. et al. Ultrahigh-gain photodetectors based on atomically thin graphene-MoS2 heterostructures. Sci. Rep. 4, 3826 (2014).
Koppens, F. H. L. et al. Photodetectors based on graphene, other two-dimensional materials and hybrid systems. Nat. Nanotechnol. 9, 780–793 (2014).
Yu, Y.-J. et al. Tuning the graphene work function by electric field effect. Nano Lett. 9, 3430–3434 (2009).
Liu, R. et al. Band alignment engineering in two-dimensional transition metal dichalcogenide-based heterostructures for photodetectors. Small Struct. 2, 2000136 (2021).
Frisenda, R., Niu, Y., Gant, P., Muñoz, M. & Castellanos-Gomez, A. Naturally occurring van der Waals materials. npj 2D Mater. Appl. 4, 38 (2020).
Taghavi, N. S. et al. Thickness determination of MoS2, MoSe2, WS2 and WSe2 on transparent stamps used for deterministic transfer of 2D materials. Nano Res. 12, 1691–1695 (2019).
Horcas, I. et al. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 78, 013705 (2007).
Kim, D.-G., Lee, S. & Kim, K. Mechanical removal of surface residues on graphene for TEM characterizations. Appl. Microsc. 50, 28 (2020).
Acknowledgements
We acknowledge funding from the innovation program under grant agreements No. 785219 (Graphene Core2-Graphene-based disruptive technologies) and No. 881603 (Graphene Core3-Graphene-based disruptive technologies). A.C.-G. acknowledges to grant agreement n° 755655, ECR-StG 2017 project 2D-TOPSENSE. O.C. acknowledges The European Union’s Horizon 2020 research and innovation program under the grant agreement 956813 (2Exciting). S.P. acknowledges the Spanish Ministry of Science and Innovation (Grant PRE2018-084818). Y.X. acknowledges the Key Research and Development Program of Shaanxi (Program No.2021KW-02).
Author information
Authors and Affiliations
Contributions
R.M.: Conceptualization, methodology, validation, formal analysis, investigation (synthesis, deterministic transference, Raman acquisition, photoresponse acquisition) resources, writing original draft, and writing—review & editing. E.L.-E.: Investigation (KPFM measurements, Raman spectroscopy), formal analysis, and writing—review & editing. C.M.: Investigation (AFM analysis), formal analysis, resources, and writing—review & editing. F.C.: Investigation (contact preparation, photo-response acquisition) and writing—review & editing. Y.X.: Investigation (photo-response acquisition), formal analysis, and writing—review & editing. O.Ç.: Resources, photo-response acquisition & programming, and writing—review & editing. T.P.: Investigation (contact preparation, deterministic transference, electrical response analysis) and writing—review & editing. S.P.: Investigation (deterministic transference, micro-reflectance analysis) and writing—review & editing. A.C.-G.: Resources, funding acquisition, and writing—review & editing. M.G.H.: Conceptualization, resources, writing—review & editing, funding acquisition, and project administration.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muñoz, R., López-Elvira, E., Munuera, C. et al. Low T direct plasma assisted growth of graphene on sapphire and its integration in graphene/MoS2 heterostructure-based photodetectors. npj 2D Mater Appl 7, 57 (2023). https://doi.org/10.1038/s41699-023-00419-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-023-00419-8
