
Abstract
Activation of MAPK signaling via BRAF mutations may limit the activity of EGFR inhibitors in EGFR-mutant lung cancer patients. However, the impact of BRAF mutations on the selection and fitness of emerging resistant clones during anti-EGFR therapy remains elusive. We tracked the evolution of subclonal mutations by whole-exome sequencing and performed clonal analyses of individual metastases during therapy. Complementary functional analyses of polyclonal EGFR-mutant cell pools showed a dose-dependent enrichment of BRAFV600E and a loss of EGFR inhibitor susceptibility. The clones remain stable and become vulnerable to combined EGFR, RAF, and MEK inhibition. Moreover, only osimertinib/trametinib combination treatment, but not monotherapy with either of these drugs, leads to robust tumor shrinkage in EGFR-driven xenograft models harboring BRAFV600E mutations. These data provide insights into the dynamics of clonal evolution of EGFR-mutant tumors and the therapeutic implications of BRAF co-mutations that may facilitate the development of treatment strategies to improve the prognosis of these patients.
Similar content being viewed by others


Introduction
Targeted treatment of epidermal growth factor receptor (EGFR)-mutant non-small cell lung cancer (NSCLC) is a landmark for rational therapy addressing molecular vulnerabilities1. Treatment with first- and second-generation EGFR tyrosine kinase inhibitors (TKIs) markedly improved the clinical outcome of patients with advanced EGFR-mutant NSCLC2,3,4,5. Currently, osimertinib is the only third-generation EGFR inhibitor approved for the sequential treatment of patients with acquired EGFRT790M resistance mutation occurring after first- and second-generation TKIs6,7. In addition, osimertinib became the new standard-of-care in the first-line treatment of patients with EGFR-mutant NSCLC8,9.
Despite the clinical efficacy of osimertinib in the first- and second-line treatment of EGFR-mutant NSCLC, drug resistance with disease progression is inevitable10,11,12,13,14,15,16,17,18. Various EGFR-dependent and EGFR-independent resistance mechanisms have been identified including EGFRC797S and EGFRG724S mutations, MET/HER2 amplification, activation of the RAS–mitogen-activated protein kinase (MAPK) or RAS–phosphatidylinositol 3-kinase (PI3K) pathways, new fusions, and histological transformation. RAS–MAPK pathway aberrations that are known to confer resistance to osimertinib include BRAF, NRAS, and KRAS mutations10,19,20. BRAF mutations occur in 2–4% of NSCLC patients and the vast majority are localized in the kinase domain, including the most common mutation BRAFV600E. BRAF mutations can be categorized into three classes based on their ability to act as monomers or dimers and based on their kinase activity. BRAFV600E mutations represent class I mutations that, similarly to class II BRAF mutations (RAS-independent), result in activation of the BRAF kinase and the MAPK pathway (gain of function). Class III BRAF mutations (RAS-dependent) result in an impaired BRAF kinase activity and amplify ERK signaling depending on upstream activating signals (e. g. RAS activating mutations, NF1 tumor suppressor deletion)21. All classes of BRAF mutations are recognized as oncogenic driver mutations, yet only BRAFV600E mutations represent clinically actionable drug targets in cancer patients22,23.
BRAFV600E mutations have been identified as a resistance mechanism to osimertinib in roughly 3% of cases with EGFR-mutant lung cancer, with or without concurrent EGFRT790M mutation10,19,20. Several combination therapies have been proposed for BRAF resistance in EGFR-mutant lung cancer, but an integrated genomic analysis of these tumors is lacking and precludes an optimization of therapeutic regimen24,25,26,27. Furthermore, the current understanding of the clonal evolution of EGFR-mutant cells that concomitantly acquire BRAF mutations during anti-EGFR therapy remains limited.
Within the present study, we aimed for a comprehensive and translational approach to systematically characterize the role of co-occurring EGFR/BRAF mutations in patients with advanced lung adenocarcinoma.
Results
Targeting BRAF-driven resistance in EGFR-mutant lung cancer
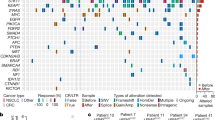
To characterize the role of BRAF mutations in the context of druggable EGFR mutations, data of eligible patients from several centers were analyzed (see “Methods”). This led to the identification of 15 patients with lung adenocarcinoma harboring activating EGFR mutations and co-occurring BRAF mutations (Fig. 1a, Table 1). In five cases, EGFR and BRAF mutations were detected at the time of initial diagnosis, whereas in ten patients, BRAF mutations were acquired after anti-EGFR therapy (Table 1). In eight patients, BRAF mutations occurred after osimertinib treatment, in one patient after gefitinib treatment, and in one patient after afatinib treatment. The treatment history of these ten patients before the detection of acquired BRAF mutations is outlined in Supplementary Fig. 1. The median duration of time elapsed from diagnosis of EGFR-mutant lung cancer to the detection of acquired BRAF mutation was 33.8 months (95% CI: 9.0–99.1 months) (Fig. 1b). Six patients were evaluable for analysis of subsequent treatment and outcome after detection of acquired BRAF mutation (Fig. 1c, Table 2). Median overall survival (OS) for these six patients after detection of BRAF-driven acquired resistance was 7.8 months (95% CI: 5.1–10.5 months; Fig. 1c). Of which, four patients (P12–P15) presenting with acquired activating BRAFV600E and BRAFK601E (gain of function) mutations received either osimertinib and bevacizumab (n = 2), osimertinib and chemotherapy (n = 1), or chemotherapy plus bevacizumab (n = 1) as the next line of treatment after detection of the BRAF mutation (Table 2). In the Cologne cohort, we detected 26/1951 co-occurrences of EGFR and BRAF mutations (1.3%) but narrowed it down to clearly activating EGFR mutations. In the Paris cohort, we detected 4/184 co-occurrences of EGFR and BRAF mutations (2.2%). Overall, our data show that BRAF mutations represent a resistance mechanism in a relevant proportion of EGFR-mutant patients, warranting further investigation of the underlying clinical and evolutionary dynamics.

a Spectrum and distribution of BRAF co-mutations in patients with EGFR-mutant lung adenocarcinoma. b Kaplan–Meier curve of the time elapsed from the detection of the EGFR mutation until the detection of the acquired BRAF mutation (as events) in days. c Kaplan–Meier curve of overall survival for patients P01, P04, P12–P15 that were available for survival analysis. d Overview of the biopsies and key molecular findings by NGS for patient P04. Flow chart (top right) summarizes lines of therapy approaches overtime after the acquisition of BRAFV600E mutation. e, f Clustering of WES-derived mutations based on their CCFs between pairs of tumor biopsies to detect clusters of shared clonal and private mutations. Candidate mutations in EGFR and BRAF are highlighted. g Subclonal composition in individual biopsies indicating two subclones (C1, C3) in the peritoneal metastasis and single clones in the liver metastases. h Clonal evolution of reconstructed cell populations presented as a phylogenetic tree. The computationally inferred most common ancestor C0 is common to all subsequent clones and highlighted mutations are present in descendent clones. (i) Visualization of evolutionary genetic distances between normal tissue and longitudinal biopsies. WES whole-exome sequencing, NGS next-generation sequencing, PD progressive disease, PR partial response, D + T dabrafenib+trametinib, O + D( + T) osimertinib+dabrafenib(+trametinib), O + CTX + B osimertinib+chemotherapy+bevacizumab, O + Tc TACE osimertinib+transarterial chemoembolization, C clone, CCF cancer cell fraction.
Next, we selected two patients (P01, P04) who acquired BRAFV600E mutation under osimertinib treatment to evaluate the safety and efficacy of various drug combinations including EGFR, RAF, MEK, or MET inhibitors, chemotherapy, or bevacizumab (Table 2, Fig. 1d, Supplementary Fig. 2a). We chose functional imaging by FDG-PET for (early) metabolic response evaluation during our investigational conduct (Supplementary Table 1) and monitored treatment-related adverse events that were predominantly of low grade and manageable (Supplementary Table 2). Osimertinib treatment was initiated and carried out for 16 months in P01 and 7 months in P04 before the detection of progressive disease. While in P01 EGFRT790M mutation was sustained, in P04, we observed a loss of EGFRT790M mutation. Both patients started with dabrafenib and trametinib, which in both cases did not lead to a confirmed metabolic response. Both patients underwent a rebiopsy of progressive lesions and started immediately with osimertinib and dabrafenib. In P01, the rebiopsy revealed an EGFRdel19 mutation, loss of EGFRT790M, no BRAFV600E mutation and an intermediate-level MET amplification (GCN 5.58, FISH). Subsequent doublet combinations of osimertinib plus dabrafenib and afatinib plus crizotinib showed either primary refractory disease or metabolic responses that could not be confirmed in the next scans. In contrast, the triple combination of osimertinib, dabrafenib, and trametinib led to a prolonged metabolic response and clinical benefit (Supplementary Table 1, Table 2). In P04, doublet combinations of osimertinib and dabrafenib led only to a short metabolic response not confirmed in the next scan, and with the addition of trametinib, we then observed a marked metabolic response in the primary lung tumor but not in the hepatic metastases. Rebiopsy of the hepatic lesions revealed an EGFRdel19 mutation with T790M and C797S resistance mutations in cis and no BRAF mutation. Treatment was thus changed to osimertinib plus chemotherapy/transarterial chemoembolization due to progressive liver metastases. The patient, unfortunately, died a year after detection of BRAFV600E resistance. Thus, biopsy-guided mutational profiling in conjunction with FDG-PET imaging can guide effective combination therapies to overcome resistance in these patients.
To investigate the clonal dynamics during the development of resistance, we performed whole-exome sequencing (WES) of biopsies from multiple time points and different metastatic sites obtained from P01 to P04 (Fig. 1d–i (P04), Supplementary Fig. 2 (P01)). For patient P01 WES could be performed on the primary tumor and two metastatic samples (Supplementary Fig. 2), while insufficient tissue, unfortunately, precluded analysis of the BRAF-mutant metastasis. Pairwise clustering based on the cancer cell fractions of the mutations (CCFs, i.e. frequency of occurrence in cancer cells after adjustment for purity, ploidy, and copy number (CN)28 revealed a high proportion of private mutations, while only a few mutations (e.g., EGFRdel19) were clonal in all samples (Supplementary Fig. 2b–d). Due to the sequencing quality, an intra-biopsy heterogeneity analysis was not undertaken, but phylogenetic tree analysis between biopsies indicated a branched evolution during resistance development (Supplementary Fig. 2e). Interestingly, a common ancestor gave rise to the pleural upper lung lobe metastasis and clones subsequently developing into the pleural metastasis and the primary tumor. In accordance with this branching model, the CN profiles show shared alterations between all three available samples, but also CN segments exclusive to just one or a pair of samples (Supplementary Fig. 2f). Our data indicate early branching during tumor development and is in accordance with a scenario where resistant cells develop in parallel to the primary tumor even before treatment pressure is applied.
For patient P04 WES was performed on a peritoneal metastasis that occurred during initial inhibitor treatment (EGFRdel19 and EGFRT790M), a liver metastasis 6 months after treatment had been switched to osimertinib (EGFRdel19 and BRAFV600E) and a rebiopsy of the same liver lesion at progressive disease under dabrafenib and trametinib treatment (EGFRdel19 and BRAFV600E) (Fig. 1d). Comparative pairwise CCF-based clustering showed only a few mutations to be shared between the peritoneal biopsy and the first liver biopsy (e.g., EGFRdel19), while the majority was private for each one of metastases (e.g., EGFRT790M and BRAFV600E, respectively) (Fig. 1e). In contrast, almost all mutations were found to be shared between both biopsies of the liver lesion (Fig. 1f). Subclonal composition analysis of the peritoneal metastasis revealed two subclones (C1 60%, C3 40%), while the liver metastases presented with one dominant clone each (Fig. 1g).
For subsequent phylogenetic analyses, a founder clone C0 was derived based on the mutations shared by all biopsies since the material of the primary tumor was unavailable for WES. Tracking the genomic development from C0 indicated a branched evolution diverging towards the peritoneal metastasis carrying EGFRT790M with its first subclone C1, which further spawned a new subclone C3 present in the same biopsy (Fig. 1h). The liver metastasis appeared to have developed from C0 independently of the peritoneal metastasis by acquiring the BRAFV600E mutation (C2). The rebiopsy of that lesion showed a distinguishable clone C4 which only carried one additional non-synonymous mutation of unknown biological relevance indicating a high degree of genetic similarity (Fig. 1h). In addition, genetic similarities between lesions were quantified to gain further insight into the clonal evolution toward therapy resistance (see Supplementary Material for further details). This analysis supports a branched evolution trajectory model with a common ancestor giving rise to the peritoneal metastasis and liver metastases (Fig. 1i). While the peritoneal metastasis and liver metastases are not closely related, only minor changes occurred between the first and second biopsy of the liver lesion (Fig. 1i). Accordingly, the CN landscape is very similar between the peritoneal metastasis and liver metastases, but almost identical between both liver biopsies (Supplementary Fig. 3). This highlights that the different metastases and resistance mechanisms (EGFRT790M and BRAFV600E, respectively) developed independently from a common ancestral clone rather than in a linear relationship. Also, the remarkably high similarity between both liver biopsies indicates that selection pressure did not give rise to a highly distinct new subclone, potentially due to the lack of an EGFR inhibitor in the combination treatment. However, it may also be due to resistance to anti-BRAF therapy already being present in the clone C2 that propagates to C4 or may have a non-genomic basis not detectable by WES.
For a third patient (P14) we obtained a biopsy at the time of progression under osimertinib treatment (Supplementary Fig. 4a). WES of this biopsy showed the presence of several oncogenic EGFR mutations, namely L858R, T790M, and C797S (Supplementary Fig. 4b). All of those mutations were clonal, the T790M and C797S mutations are in agreement with previously described resistance mechanisms to EGFR inhibitors. Interestingly, we also detected a BRAFV600E mutation (Supplementary Fig. 4b, Table 1). In contrast to the EGFR mutations, the BRAF mutation was subclonal and may thus indicate the branching of a newly developing subclone. This further highlights the complexity of disease resistance, which may incorporate several mechanisms of resistance development in parallel. Overall, the clonal analyses highlight that a process of branched evolution underlies resistance to targeted treatments in patients with EGFR-mutant tumors and may give rise to various independent resistance mechanisms.
Resistance through the selection of BRAF V600E-positive clones
To functionally validate our clinical observations, we overexpressed BRAFV600E in EGFRdel19-mutant PC9 cells. To compare BRAFV600E-mediated effects to upstream activation of MAPK signaling, we generated cells expressing NRASQ61K, a mutation reported in preclinical models of acquired EGFR-inhibitor resistance16,19. In a polyclonal pool of PC9 cells stably expressing BRAFV600E or NRASQ61K, only modest activation of the MAPK signaling was detected as determined by immunoblotting of phospho-ERK (Fig. 2a). However, residual phospho-ERK-levels after osimertinib treatment were detected only in cells with BRAFV600E or NRASQ61K overexpression but not in control PC9 empty vector (EV) cells. During 7-14 day treatment the insufficient inhibition of MAPK signaling translated into the outgrowth of osimertinib-resistant clones in cells expressing BRAFV600E or NRASQ61K (Fig. 2b, c). In line with this observation, only ≤0.3% of PC9 (EV) cells were found to be able to give rise to colonies during increasing doses of osimertinib treatment (Fig. 2d). However, overexpression of BRAFV600E or NRASQ61K significantly enhanced the pool of cells with the capacity to outgrow during therapy to 5.2% (BRAF) and 4.5% (NRAS) or less in a dose-dependent manner (Fig. 2d).

a Immunoblotting of PC9 cells expressing the annotated constructs, treated with (+) or without (−) osimertinib (48 h). Hsp90 is used as a loading control. b Clonogenicity assays of PC9 derived cell lines treated with osimertinib for 7 and 14 days or DMSO control for 7 days are displayed. c Quantitative analysis of (b) normalized to PC9 (EV). d Limited dilution assay of PC9-derived cell lines treated for 21 days before analysis. e, f qRT-PCR analysis of mRNA expression in e BRAF and f NRAS in PC9 derived cell lines normalized to EV. g Immunoblotting of PC9 cells expressing the annotated constructs that were treated as in (a). h Viability curves of PC9 cells expressing the annotated constructs treated with osimertinib (72 h) are shown. The relative area under the curve (AUC) in % compared to a theoretical non-responding AUC. Error bars indicate mean ± SD. Two-tailed paired t tests, ***p < 0.001, **p < 0.01, *p ≤ 0.05, n.s.p > 0.05. EGFR epidermal growth factor receptor, BRAF B-rapidly accelerated fibrosarcoma, NRAS neuroblastoma rat sarcoma, EV empty vector.
The next question was whether the enrichment of cells with high BRAFV600E or NRASQ61K expression would have an impact on EGFR inhibitor sensitivity. Therefore, polyclonal PC9BRAF-V600E and PC9NRAS-Q61K cells were preselected either with 10 nM (PC9BRAF/NRAS OS 10 nM) or 100 nM (PC9BRAF/NRAS OS 100 nM) of osimertinib over the course of >30 days. Using RT-PCR a dose-dependent elevation of RNA levels of the respective resistance alleles was found in PC9BRAF OS and PC9NRAS OS cells after osimertinib selection (Fig. 2e, f). Osimertinib-preselected cells exhibited a higher induction of BRAFV600E expression (9.39-fold) than NRASQ61K expression (4.25-fold, p = 0.036). Accordingly, untreated osimertinib-preselected cells with high BRAFV600E expression displayed stronger phospho-ERK staining when compared to NRASQ61K (Fig. 2g). Both osimertinib-preselected PC9BRAF-V600E and PC9NRAS-Q61K cells showed higher levels of sustained phospho-ERK during osimertinib treatment (Fig. 2g) and a higher degree of resistance in viability assays compared to non-selected cells (Fig. 2h). A similar degree of resistance was observed against the EGFR inhibitors erlotinib or afatinib (Supplementary Fig. 5a, b) but not against the non-specific, chemotherapeutic cisplatin (Supplementary Fig. 5c).
To further substantiate our data in an independent model, BRAFV600E was overexpressed in the EGFRdel19-mutant HCC827 cell line. Again, a dose-dependent induction of resistance through osimertinib-preselection was observed in polyclonal HCC827BRAF-V600E cell pools (Supplementary Fig. 5d). These findings are in line with our clinical observations and previous cases that identified BRAF-mediated resistance in EGFR-mutant tumors during anti-EGFR therapy. Our results suggest that BRAF-mutant clones are enriched through EGFR-directed therapy in EGFR-mutant adenocarcinoma.
Overcoming BRAF V600E-mediated resistance in EGFR-mutant cells
Previous studies have found that concomitant KRAS and EGFR mutations may increase the cell death rate of adenocarcinoma cells through hyperactivation of ERK signaling29,30. We tested whether the activation of MAPK signaling via BRAFV600E may have a similar effect in EGFR-mutant PC9 cells. To this end, the cell proliferation of PC9BRAF-V600E and PC9NRAS-Q61K was measured over 5 days, but no major differences were observed compared to EV cells (Fig. 3a). We also did not detect any differences in the basal cell death rate between cell lines (Supplementary Fig. 6a). Consequently, cells with high expression of mutant BRAF/NRAS did not get counter-selected after the withdrawal of osimertinib (Fig. 3b, c).

a Growth series of PC9 derived cell lines counted for 5 days every 24 h (see Methods). b Immunoblotting of PC9BRAF-V600E OS 100 nM, PC9NRAS-Q61K OS 100 nM, and PC9 (EV). Osimertinib-preselected cells were cultured for 0, 7, and 21 days without osimertinib treatment and plated 48 h before lysis. c Cell viability assay of PC9 cells expressing the annotated constructs treated for 72 h with osimertinib is shown. The relative AUC (see Methods) of BRAFV600E OS 100 nM and NRASQ61K OS 100 nM after osimertinib withdrawal for >40 days are shown. d Clonogenicity assay of PC9 cells expressing the annotated constructs treated for 14 days with indicated compounds before staining. e RNA-seq based expression of E2F gene set genes (rows) in PC9 derived cell lines (columns) after 48 h treatment with indicated inhibitors. Expression was normalized as z-score per gene. f Synergy screen of osimertinib and trametinib combination treatment in PC9 derived cell lines for 72 h are displayed. g Immunoblotting of PC9 cells expressing the annotated constructs is shown. Treatment with indicated compounds 48 h before lysis. h Relative tumor volume of xenograft mice injected with PC9BRAF-V600E OS 100 nM cells in % compared to day 0 of the treatment regimen (see Methods). Error bars indicate mean ± SD. Two-tailed paired t tests (all except (h); two-tailed Welch’s t tests with Bonferonni-correction), ***p < 0.001, **p < 0.01, *p < 0.05, n.s.p > 0.05. EGFR epidermal growth factor receptor, BRAF B-rapidly accelerated fibrosarcoma, NRAS neuroblastoma rat sarcoma, EV empty vector.
Next, we tested combination therapies by targeting EGFR and MAPK signaling individually in PC9BRAF-V600E and PC9NRAS-Q61K cells (Fig. 3d). Both MEK inhibition and BRAF inhibition, as monotherapy, had a limited effect on the viability of PC9BRAF-V600E mutant cells (Supplementary Fig. 6b, c). In contrast, the combination of osimertinib and MEK or BRAF inhibition effectively prevented the outgrowth of colonies (Fig. 3d). To further validate our previous findings on a transcriptional level, we performed RNA sequencing of PC9 (EV), PC9BRAF-V600E, and PC9BRAF-V600E OS 100 nM cells treated with osimertinib, trametinib, a combination of both or control for 48 h (see Supplementary Material). As expected, a principal component analysis showed that osimertinib monotherapy had strong effects only on PC9BRAF-V600E cells, while trametinib plus osimertinib comparably impacted both PC9BRAF-V600E and PC9BRAF-V600E OS 100 nM cells (Supplementary Fig. 6d). We next clustered samples based on the expression of E2F target genes to assess the impact on cell cycle-related gene expression as a surrogate marker for the cytotoxic effects of the given perturbation (Fig. 3e). In this analysis, the strongest downregulation of E2F genes was present in the group of cell line/treatment combinations that led to reduced cell numbers in crystal violet assays (Fig. 3d). Repression of E2F target genes was lower in unselected PC9BRAF-V600E cells with osimertinib compared to PC9 EV cells or compared to combination treatment (Fig. 3d). This indicates the limited efficacy of osimertinib monotherapy treatment if a BRAF mutation is present even without prior selection and supports the use of combination treatment. Furthermore, in PC9BRAF-V600E OS 100 nM cells the expression of MAPK pathway responsive genes was only perturbed during osimertinib and trametinib treatment (Supplementary Fig. 7a)31. Next, we assessed the synergy between osimertinib and trametinib, using ZIP-based synergy analysis, and found a strong synergy that correlated with the expression of BRAF-V600E in PC9 cells (Fig. 3f, see Methods). The calculated synergy score for osimertinib and vemurafenib was limited and we found an antagonism for the combination of trametinib and vemurafenib inhibition in PC9BRAF-V600E mutant cells (Supplementary Fig. 7b, c). Finally, using a three-fold titration matrix, we observed a considerably low synergy for osimertinib and vemurafenib treatment compared to osimertinib and trametinib treatment, which was not further increased in a triple combination by adding vemurafenib (Supplementary Fig. 7d). In accordance with the synergy results, osimertinib with trametinib in contrast to osimertinib alone resulted in full inhibition of phospho-ERK signaling. Vemurafenib did not fully abrogate the sustained phospho-ERK signaling, as it also hyperactivated phospho-ERK as monotherapy, most likely due to the paradoxical effect on the endogenous wild type BRAF kinase (Fig. 3g)32. To further validate our in vitro results, we performed an in vivo study with xenografts implanted with PC9BRAF-V600E cells that were preselected for high BRAFV600E expression. Once the mice developed tumors, we started with the treatment regimen consisting of vehicle, osimertinib, trametinib or the combination of osimertinib and trametinib for 21 days (Fig. 3h). Compared to vehicle treatment, trametinib did not significantly decrease tumor volume, and osimertinib monotherapy led to a measurable tumor growth reduction. However, only, combination therapy led to robust tumor shrinkage in these xenografts (Fig. 3h). Thus, our in vivo data largely reflects our in vitro findings and suggests that combination therapy is necessary to induce substantial tumor shrinkage in tumors harboring activating EGFR and BRAF mutations. Of importance, none of the mice in the individual treatment arms experienced weight loss (Supplementary Fig. 7e) or any other severe treatment-associated side effects.
Discussion
Osimertinib replaced other EGFR inhibitors in the early lines of therapy. This development had a major impact on the resistance profiles and development of effective salvage therapies10,11,12,13,14,15,18,33. The activation of MAPK signaling seems to play a more prominent role in patients' progressive on third-generation EGFR inhibitors when compared to first- and second-generation EGFR inhibitors11,16,19,20,34. Our comprehensive genomics study of EGFR-mutant patients with co-occurring BRAF mutations provides insights into the evolution of MAPK-driven resistance and its impact on EGFR-directed treatment.
Our combination of longitudinal clinical and genomic analyses provides insight into the subclonal heterogeneity of the individual tumors and corresponding metastases during resistance evolution. Our clonality analyses revealed that resistance to osimertinib (initiated at detection of EGFRT790M mutation) and subsequent combination of dabrafenib plus trametinib (initiated at detection of BRAFV600E mutation) was driven by an evolutionary branching process rather than a linear trajectory of one clone that continues to acquire additional resistance mutations. Moreover, in both patients, the different metastases are genetically distinct from each other but arise from common ancestors that do not carry a resistance mutation. Even within our limited cohort, we observe different patterns of clonal evolution: while for P04 a common ancestor most likely from the primary tumor gave rise to the different metastases, for patient P01 phylogenetic analyses are in accordance with a model supporting much earlier branching. Of note, resistance mutations such as EGFRT790M and BRAFV600E were not detected by either panel sequencing or WES in samples still sensitive to the respective inhibitors. This may indicate that they developed either de novo during treatment or were pre-existent, but at a frequency to low to be detected without selection pressure.
Overall, our results demonstrate the presence and further development of tumor heterogeneity that can give rise to multiple resistance mechanisms due to treatment selection pressure. Moreover, our genomic analysis emphasizes that we are faced with a complex mutational landscape based on intra-tumoral, inter-tumoral, and inter-patient heterogeneity. It thus constitutes a major clinical challenge for the development of an efficient treatment strategy to counteract tumor progression. Based on the present findings a diagnostic strategy aiming to address the multilayered heterogeneity e.g. using liquid biopsies or multiple re-biopsies appears warranted to optimize treatment schedules. Our data suggest that one promising treatment strategy for patients with concurrent EGFR and MAPK pathway activation may require alternating treatment regimens with intermittent changes between drug combinations based upon observed heterogenic tumor response and emerging resistance patterns. To facilitate this strategy FDG-PET can be quite useful for rapid treatment evaluation and hence, dynamic clinical management as demonstrated by our investigational approach. However, we are aware that more patients need to be profiled in the future to compliment our results.
We and others have previously found that acquired resistance through activation of MAPK signaling via KRAS mutations can be detected in patients receiving third-generation EGFR inhibitors11,12. BRAF mutations and BRAF rearrangements have also been shown to play a similar role like KRAS in the resistance setting of EGFR-mutant adenocarcinoma16,19,34. This is surprising as previous functional analyses indicated that mutant KRAS mutations may augment the cell death rate of EGFR-mutant cells and thereby limit the outgrowth of resistant clones29. Our cell line models indicate that concomitant MAPK pathway signaling is tolerated when BRAF or NRAS are activated. This corresponds with our clinical observation that BRAF mutations can co-occur with EGFR mutations even before anti-EGFR therapy. Interestingly, the levels of phospho-ERK activation differ strongly between BRAF- and NRAS-mutant cells but we did not observe major differences in the ability of these alleles to promote resistance or cell death in EGFR-mutant cells. These functional observations are also in line with our finding that BRAF/EGFR-mutant lung tumors are recurrently found across different cancer centers, indicating a basis for the co-existence of BRAF/EGFR mutations without selection pressure. Future studies are required to fully decipher the potential differences between MAPK signaling activation at different levels of the pathway in the context of EGFR-mutant lung adenocarcinoma. Nevertheless, our in vitro and in vivo findings fully support the notion that EGFR/MEK combination might be a viable option to overcome BRAF-driven resistance in patients with EGFR-mutant lung adenocarcinoma.
In summary, our data uncover basic principles of drug-induced evolutionary paths underlying BRAF-driven resistance in patients with lung adenocarcinoma. The integrated analyses support a model in which concomitant activation of EGFR and BRAF is selected through anti-EGFR therapy that combines well with EGFR, BRAF, and MEK inhibitors to overcome resistance. Our systematic exploration of clinically relevant drug combinations may offer additional avenues for follow-up investigations into novel targeted treatment strategies for patients with co-occurring EGFR and BRAF mutations.
Methods
Patients
We compiled a cohort of 15 patients with lung adenocarcinoma and activating EGFR mutations that harbored co-occurring BRAF mutations with and without prior anti-EGFR treatment. Patients were identified within the Network Genomic Medicine (NGM) Lung Cancer in Cologne, Germany, Institute Gustave Roussy in Paris, France, and Cantonal Hospital of Lucerne, Switzerland. Treatment, genetic findings, and survival of these patients were evaluated. All patients consented to be analyzed. The study was conducted in concordance with local ethical guidelines and was reviewed by the institutional ethics committee. Selected patients were treated with different lines of therapy including combinations of osimertinib, dabrafenib, and trametinib. These patients provided written informed consent for a prospective investigational molecular- and imaging-guided personalized treatment approach. Rebiopsies were acquired at disease progression. Tissue biopsy was performed through core needle biopsy according to local standard procedures. Survival of all patients was calculated using the Kaplan Meier method.
Molecular analyses
The vast majority of the specimens analyzed in our study consisted of tumor tissue (n = 13). For two patients, liquid biopsies were evaluated. (Table 1). Next-generation sequencing (NGS)-based molecular profiling was performed for each patient either on tumor tissue or on circulating tumor DNA (ctDNA). For patients, P01 and P04 whole-exome sequencing was additionally performed on the tumor tissue. For patients P01 and P04, we obtained longitudinally serial repeated tissue biopsies of the leading tumor lesions at each time of progression during treatment with different combinations of osimertinib, dabrafenib and trametinib, and other therapies. NGS of tumor tissue was performed as previously described35,36,37,38. Plasma analysis of ctDNA was performed as previously reported39.
PET-CT assessments
The efficacy of treatment was evaluated by positron emission tomography (PET)/computed tomography (CT) scans using radiolabeled 18F-2-fluoro-2-deoxy-d-glucose (FDG). Scans were acquired at baseline, and as early as 2 weeks (early assessment) and again at regular intervals roughly every 6 or more weeks (late assessments) after initiation or change of therapy to capture early metabolic response (measured by standard uptake value (SUV)max) and morphologic response over time. Scans were conducted as previously described and performed on a Biograph mCT Flow-Edge 128 PET/CT-system (Siemens Medical Solutions) with a 128-slice spiral CT component from the base of the skull to the mid-thigh40. We followed Positron Emission Response Criteria in Solid Tumors version 1.0 guidelines, assuming that response is characterized by an SUV reduction of at least 30% in the hottest lesion41.
Whole-exome sequencing (WES)
WES was performed on FFPE-derived DNA from serial tumor tissue rebiopsies obtained at the time of tumor progression during treatment of patients P01 and P04. In addition, for one patient DNA was extracted from the primary tumor using the truXTRAC FFPE DNA extraction kit (Covaris, USA, Cat. No. 520307). Exomes were individually prepared using 200 ng of DNA using standard protocol SureSelectXT Automated Target Enrichment for Illumina Paired-End Multiplexed Sequencing and Agilent Bravo automated liquid handling platform. As for patient P14, there was only a post-osimertinib tumor biopsy available for WES, which was enriched using the Agilent SureSelect CR kit (Agilent, USA). After validation (2200 TapeStation, Agilent Technologies) and quantification (Qubit System, Invitrogen, Waltham, USA) pools of libraries were generated. The pools were quantified using the KAPA Library Quantification kit (Peqlab, Germany, KAPBKK4854) and 7900HT Sequence Detection System (Applied Biosystems, Waltham, USA) and subsequently sequenced at 140× mean coverage on an Illumina NovaSeq6000 sequencing instrument using a paired-end 2 × 100 bp protocol.
WES and clonality analysis
Analysis of raw sequencing data and clonality analyses were performed using an established pipeline42: After alignment of the raw sequencing data to the hg19 reference genome in total 137–325 million reads could successfully be mapped per sample corresponding to a mean coverage of 93×–200× per sample and covering all intended exonic target region with ≥20× coverage for 90–98% of those. In summary, of the 42.3 megabases of exonic regions as defined by the GRCh 37/hg19 RefSeq genome annotation, 39–40 megabases of exons were sufficiently covered for mutation calling and subsequent analyses. Thus, allelic fractions of somatic mutations were corrected for purity and CN changes to determine cancer cell fractions (CCF). The distribution of CCFs was then searched for distinct subpopulations by using a nonparametric method to deconvolute the noise in the CCFs. This allows for the identification of genetically distinct tumor subclones and the reconstruction of tumor evolutionary histories.
Cell culture and functional analyses
Human NSCLC cell lines were verified by STR profiling at the Institute for Forensic Medicine of the University Hospital of Cologne. PC9, HCC827, and HEK293T cell lines were obtained from ATCC. PC9 and HCC827 cells and their osimertinib-preselected derivatives were cultured in RPMI (Fisher Scientific, USA, Cat. No. 12004997) HEK293T cells were cultured in DMEM (Fisher Scientific, USA, Cat. No. 61965-026). All media were supplemented with 10% fetal bovine serum (Fisher Scientific, USA, Cat. No. 10270-106) and 1% penicillin/streptomycin (Fisher Scientific, USA, Cat. No. 15070-063). All cells were grown at 37 °C in a humidified atmosphere with 5% CO2.
Reagents
For cell culture studies, osimertinib (LC Laboratories, USA, Cat. No. 1421373-65-0), trametinib (LC Laboratories, USA, Cat. No. 871700-17-3), and vemurafenib (LC Laboratories, USA, Cat. No. 918504-65-1) were dissolved in dimethyl sulfoxide (DMSO) (Carl Roth, Germany, Cat. No. 4720.4) to a final stock concentration of 10 mM. Cisplatin (pharmacy of University Hospital of Cologne) was diluted to 3.33 mM in 0.9% NaCl.
Crystal violet assay
Totally, 105 cells were plated into one well of a 6-well plate and treated with DMSO (control), 300 nM osimertinib, 100 nM trametinib, 1 µM vemurafenib, and combinations osimertinib plus trametinib and osimertinib plus vemurafenib. Seven or 14 days after treatment, cells were fixed in 4% paraformaldehyde (Carl Roth, Germany, Cat. No. CP10.1) in phosphate-buffered saline (PBS) (Fisher Scientific, USA, Cat. No. 14190144), stained with 0.1% crystal violet (Sigma Aldrich, USA, Cat. No. C3886-25G) in PBS, and rinsed in PBS before image acquisition. For quantification, the Crystal Violet dye was dissolved in 2 ml methanol (Carl Roth, Germany, Cat. No. CP43.4) in the 6-well plate. Twenty microlitres of this solution were diluted 1:10 with methanol and injected into 96-well plates. The read-out was the absorption at 560 nm wavelength. The results per cell line were normalized against their DMSO-controls and then against the empty vector (EV) control cell line.
Protein overexpression experiments
Vectors pBABE puro, pBABE-puro-BRAFV600E, and pBABE-NRASQ61K were cotransfected with a helper plasmid into HEK 293T cells using TransIT-LT1 reagent (Mirus, USA, Cat. No. MIR2300). Forty-eight hour post transfection, replication-incompetent retroviruses were collected from the supernatant for infection of PC9 and HCC827 in the presence of 8 μg/ml polybrene (Merck Millipore, USA, Cat. No. TR1003-G). Twenty-four hour after infection, the growth medium was changed and 3 μg/ml (PC9) or 2 μg/ml (HCC827) puromycin (Sigma Aldrich, USA, Cat. No. p8833) was added for selection for 7 days. After selection, cells were analyzed for protein expression.
pBABE-puro was a gift from Hartmut Land & Jay Morgenstern & Bob Weinberg (Addgene plasmid # 1764; RRID:Addgene_1764).
pBabe-Puro-BRAFV600E was a gift from William Hahn (Addgene plasmid # 15269; RRID:Addgene_15269).
pBabe-NRASQ61K was a gift from Channing Der (Addgene plasmid # 12543; RRID:Addgene_12543).
Cell viability screening
To assess cell viability, cells were plated in 96-well plates in triplicates, and compounds were added at 9 decreasing compound concentrations 24 h after seeding. Seventy-two hours later, cell viability was measured via Cell Titer-Glo (CTG) assay (Promega, USA, Cat. No. g7573) and was normalized to DMSO-treated controls. Resistance in % was calculated as the area under the curve (AUC), calculated via Gauss’s trapezoid area formula and then divided by a theoretical non-responding AUC, all calculated in R. Data are represented as mean ± standard error of the mean and significance was calculated by paired Student’s t tests.
RNA isolation and qRT-PCR
Totally, 5 × 105 cells were plated into one well of a 6-well plate and harvested after 24 h. Total RNA was isolated using the RNeasy-kit (Qiagen, Germany, Cat. No. 74106) according to the manufacturer’s instructions, including DNAse I digestion (Qiagen, Germany, Cat. No. 79256). In all, 1.5 μg of total RNA was reverse transcribed using Super-script II (Thermo Fisher Scientific, USA, Cat. No. 18064022) with random hexamer primers. Quantitative real-time PCR (qPCR) was performed using the QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) and Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, USA, Cat. No. 4309155). Data were normalized to GAPDH RNA levels and are presented as mean ± SD and significance was calculated by paired Student’s t tests.
Flow cytometry
Cell lines were seeded into 6-well plates (1 × 105 cells/well). Twenty-four-hour later Staurosporine (Sigma Aldrich, USA, S4400) or DMSO control was added to the medium. Twenty-four-hour later supernatant was collected, cells were trypsinized (Fisher Scientific, USA, Cat. No. 11560626), washed with ice-cold PBS, and resuspended in antibody-binding buffer (10 mM HEPES pH 7.4 (Fisher Scientific, USA, Cat. No. 15630080), 140 mM NaCl; 2.5 mM CaCl2). Cells were stained for Annexin-V (BD Biosciences, USA, Cat. No. 556420) and 50 µg/mL propidium iodide (Carl Roth, Germany, Cat. No. CN74). After 20 min of incubation in the dark, samples were analyzed on a FACS Gallios Flow Cytometer (Beckman Coulter). We used FACS Kaluza software (Beckman Coulter) to quantify populations. At least 5 × 104 events were assessed per measurement. All measurements were performed as duplicates. Gates used can be found in Supplementary Fig. 8. Data are presented as mean ± SD.
Immunoblot
Cell lysates were prepared using RIPA buffer supplemented with protease inhibitors (Complete Mini Protease Inhibitor Cocktail, Roche, Switzerland, Cat. No. 11836170001). Protein concentration was determined by BCA assay (Thermo Fisher Scientific, USA, Cat. No. 23225) and equal amounts of protein (20 µg) were separated on 4–12% Tris-glycine sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels (Thermo Fisher Scientific, USA, Cat. No. XP04125BOX) and transferred to PVDF-FL membrane (Sigma Aldrich, USA, Cat. No. IPFL00010). Membranes were blocked in 5% milk (Carl Roth, Germany, Cat. No. T145.1) blocking buffer in Tris-buffered saline (TBS), incubated with primary antibodies, washed, and incubated with fluorescently labeled secondary antibodies before detection with Odyssey CLx imaging system (LI-COR Biosciences). Images were processed using the Image Studio Software (LI-COR Biosciences). Primary antibodies are EGFR (Cell Signaling, USA, Cat. No. CS-4267), p-EGFR (Cell Signaling, USA, Cat. No. CS-3777), BRAF-V600E (Spring Bioscience, USA, Cat. No. E-19290), BRAF (Santa Cruz Biotechnology, USA, Cat. No. SC-5284), ERK (Cell Signaling, USA, Cat. No. CS-9102), p-ERK (Cell Signaling, USA, Cat. No. CS-4370), Akt (Cell Signaling, USA, Cat. No. CS-2920), p-Akt (Cell Signaling, USA, Cat. No. CS-9271) and Hsp90 (Cell Signaling, USA, Cat. No. CS-4877). All primary antibodies were diluted 1:1000 in 5% milk blocking buffer in TBS with 0.2% Tween®20 (Sigma Aldrich, USA, Cat. No. P7949-500ML). Secondary antibodies are goat anti-rabbit 800CW (LI-COR Biosciences, USA, Cat. No. 926-32211), goat anti-mouse 800CW (LI-COR Biosciences, USA, Cat. No. 926-3220), goat anti-rabbit 680LT (LI-COR Biosciences, USA, Cat. No. #926-68021), and goat anti-mouse 680LT (LI-COR Biosciences, USA, Cat. No. 926-68020). All secondary antibodies were diluted 1:20,000 in 2.5% milk blocking buffer in TBS with 0.2% Tween®20 and 0.01% sodium dodecyl sulfate (SDS) (Carl Roth, Germany, Cat. No. 8029.4).
All blots derive from the same experiment and were processed in parallel. Uncropped blots can be found in Supplementary Figs. 9–13.
Synergy screen
Cells were plated in a 6 × 6 wells matrix in 96-well plates. After 24 h cells were treated with five decreasing concentrations of compound A plus DMSO control starting from right to left. Cells were also treated at the same time with five decreasing concentrations of compound B plus DMSO control starting from the bottom to the top. The topmost left well is only treated with DMSO, while the bottommost right well is treated with the highest concentration of both compounds. The following starting concentrations were used: 300 nM of osimertinib, 100 nM of trametinib, and 1 µM of vemurafenib. Seventy-two hours after treatment, cell viability was measured via CTG assay and was normalized to DMSO-treated controls. Synergy scores were then calculated in R using the SynergyFinder Package and the Zero Interaction Potency (ZIP) reference model as implemented in the package. The mean of the nine highest synergy scores from each matrix is presented ±SD and significance were calculated by paired Student’s t tests.
3D Synergy screen
Cells were plated as described in “Synergy screen”, just on six plates, each with a set concentration of vemurafenib to add a third dimension. Cells were treated for the same time and with the same concentration as in “Synergy screen”. Cell viability was measured the same way as in “Synergy screen”. The expected drug combination responses were calculated based on ZIP reference model using SynergyFinder43. Deviations between observed and expected responses with positive and negative values denote synergy and antagonism, respectively.
Growth series
Totally, 1 × 105 cells per well were plated 5 times in triplicates per cell line in 6-well plates. For 5 days, always after 24 h, one triplicate of each cell line was trypsinized and counted via Z Series Coulter Counter (Beckmann Coulter). Results were normalized to day 1 and were anticipated from the slope of a best-fitting line through each data set Data are presented as mean ± SD.
Generating osimertinib selected cell lines
PC9BRAF-V600E, PC9NRAS-Q61K, and HCC827BRAF-V600E cell lines were each treated with 10 nM or 100 nM osimertinib respectively for >30 days. After that cells were labeled osimertinib selected (OS) 10 nM or 100 nM, respectively, and experiments were performed. Even after >30 days osimertinib treatment in cell culture, cells were continuously kept under osimertinib treatment.
3′UTR-RNA sequencing
For each cell line (PC9 pBABE EV, PC9 pBABE BRAFV600E, and PC9 pBABE BRAFV600E OS 100) 5 × 105 cells were plated and left to adhere overnight. The next day they were treated with 300 nM osimertinib, 100 nM trametinib, a combination of both, or DMSO control for 48 h. RNA extraction and sequencing were performed using the Qiagen RNeasy Mini kit following the manufacturer’s instruction. Totally, 500 ng total RNA were used to prepare 3′ UTR mRNA libraries using the Lexogen QuantSeq kit (Lexogen, Austria, Cat. No. 015.96) according to the standard protocol44. Quality controlled cDNA pools were quantified with the KAPA Library Quantification kit and sequenced on a NovaSeq sequencer (Illumina, USA) with a 1 × 100 bp protocol. Raw data were aligned to the human genome reference GRCh38 using STAR aligner45 and gene expression was quantified with RSEM46 prior to downstream analysis with the R package DESeq247. E2F target genes were obtained from the MSigDB Hallmark collection and MAPK feedback genes from a recently published MAPK activity score31.
In vivo xenograft model
The local authorities and the animal protection committee approved all animal procedures of this study.
PC9BRAF-V600E OS100 nM cells (5 × 106) were resuspended in 100 µL PBS and then inoculated subcutaneously in both flanks of 8- to 12-week-old female nude mice (RJ:NMRI-FOXN1 NU, Janvier Labs) and treatment was initiated when tumors reached a mean volume of approximately 50 mm3. Mice were treated daily for 21 days orally with vehicle solution (1% DMSO, 30% PEG300, 0.5% hydroxypropyl methylcellulose, 0.2% Tween-80, ddH2O) QD, osimertinib (5 mg/kg in 1% DMSO + 30% PEG300 + ddH2O) QD, trametinib (1 mg/kg in 0.5% hydroxypropyl methylcellulose, 0.2% Tween-80, ddH2O) QD or combination (osimertinib as described before and trametinib as described before) Tumor volumes were measured daily in two dimensions using a caliper, and the volume was expressed in mm3 using the formula: V = 0.5 × (length × width2), where V is tumor volume, length is the longest tumor dimension and width is the longest tumor dimension perpendicular to the length.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data generated and analyzed during this study are described in the manuscript and Supplementary Material. WES data were uploaded to The European Genome-phenome Archive (EGA) with the accession code EGAS00001005614. RNA sequencing data were uploaded to ArrayExpress with the accession code E-MTAB-11004. All relevant data and materials that support the findings of this work are available from the corresponding authors upon reasonable request.
Code availability
No custom code or algorithms were created.
References
Skoulidis, F. & Heymach, J. V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 19, 495–509 (2019).
Mok, T. S. et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 (2009).
Rosell, R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 13, 239–246 (2012).
Sequist, L. V. et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 31, 3327–3334 (2013).
Wu, Y.-L. et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 18, 1454–1466 (2017).
Cross, D. A. E. et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4, 1046–1061 (2014).
Jänne, P. A. et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 372, 1689–1699 (2015).
Soria, J.-C. et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 378, 113–125 (2018).
Ramalingam, S. S. et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med https://doi.org/10.1056/nejmoa1913662 (2019).
Leonetti, A. et al. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 121, 725–737 (2019).
Ortiz-Cuaran, S. et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.ccr-15-1915 (2016).
Michels, S. et al. Genomic profiling identifies outcome-relevant mechanisms of innate and acquired resistance to third-generation epidermal growth factor receptor tyrosine kinase inhibitor therapy in lung cancer. JCO Precis. Oncol. https://doi.org/10.1200/po.18.00210 (2019).
Tumbrink, H. L., Heimsoeth, A. & Sos, M. L. The next tier of EGFR resistance mutations in lung cancer. Oncogene https://doi.org/10.1038/s41388-020-01510-w (2020).
Thress, K. S. et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. https://doi.org/10.1038/nm.3854 (2015).
Chabon, J. J. et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 7, 11815 (2016).
Eberlein, C. A. et al. Acquired resistance to mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. https://doi.org/10.1158/0008-5472.can-14-3167 (2015).
Ricordel, C., Friboulet, L., Facchinetti, F. & Soria, J.-C. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann. Oncol. 29, i28–i37 (2018).
Fassunke, J. et al. Overcoming EGFRG724S-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat. Commun. 9, 4655 (2018).
Ohashi, K. et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl Acad. Sci. USA 109, E2127–E2133 (2012).
Ho, C.-C. et al. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J. Thorac. Oncol. 12, 567–572 (2017).
Bracht, J. W. P. et al. BRAF mutations classes I, II, and III in NSCLC patients included in the SLLIP trial: the need for a new pre-clinical treatment rationale. Cancers 11, 1381 (2019).
Planchard, D. et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 17, 984–993 (2016).
Planchard, D. et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 18, 1307–1316 (2017).
Ding, H. et al. Durable clinical response of advanced lung adenocarcinoma harboring EGFR-19del/T790M/BRAFV600E mutations after treating with osimertinib and dabrafenib plus trametinib: a case report. Oncotargets Ther. ume 13, 7933–7939 (2020).
Huang, Y., Gan, J., Guo, K., Deng, Y. & Fang, W. Acquired BRAF V600E mutation mediated resistance to osimertinib and responded to osimertinib, dabrafenib, and trametinib combination therapy. J. Thorac. Oncol. 14, e236–e237 (2019).
Meng, P. et al. Combined osimertinib, dabrafenib and trametinib treatment for advanced non-small-cell lung cancer patients with an osimertinib-induced BRAF V600E mutation. Lung Cancer 146, 358–361 (2020).
Xie, Z. et al. Lung adenocarcinoma harboring concomitant EGFR mutations and BRAF V600E responds to a combination of osimertinib and vemurafenib to overcome osimertinib resistance. Clin. Lung Cancer https://doi.org/10.1016/j.cllc.2020.06.008 (2020).
Cun, Y., Yang, T.-P., Achter, V., Lang, U. & Peifer, M. Copy-number analysis and inference of subclonal populations in cancer genomes using Sclust. Nat. Protoc. 13, 1488–1501 (2018).
Unni, A. M. et al. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife 7, e33718 (2018).
McFadden, D. G. et al. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc. Natl Acad. Sci. USA 113, E6409–E6417 (2016).
Wagle, M.-C. et al. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. Npj Precis. Oncol. 2, 7 (2018).
Poulikakos, P. I., Zhang, C., Bollag, G., Shokat, K. M. & Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 (2010).
Oxnard, G. R. et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 4, 1527–1534 (2018).
Vojnic, M. et al. Acquired BRAF rearrangements induce secondary resistance to EGFR therapy in EGFR-mutated lung cancers. J. Thorac. Oncol. 14, 802–815 (2019).
König, K. et al. Implementation of amplicon parallel sequencing leads to improvement of diagnosis and therapy of lung cancer patients. J. Thorac. Oncol. 10, 1049–1057 (2015).
Peifer, M. et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet 44, 1104–1110 (2012).
George, J. et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015).
Recondo, G. et al. Feasibility and first reports of the MATCH-R repeated biopsy trial at Gustave Roussy. Npj Precis. Oncol. 4, 27 (2020).
Remon, J. et al. Real-world utility of an amplicon-based next-generation sequencing liquid biopsy for broad molecular profiling in patients with advanced non–small-cell lung cancer. JCO Precis. Oncol. 3, 1–14 (2019).
Kuhnert, G. et al. Impact of PET/CT image reconstruction methods and liver uptake normalization strategies on quantitative image analysis. Eur. J. Nucl. Med. Mol. Imaging 43, 249–258 (2016).
Wahl, R. L., Jacene, H., Kasamon, Y. & Lodge, M. A. From RECIST to PERCIST: evolving considerations for PET response criteria in solid tumors. J. Nucl. Med. 50, 122S–150S (2009).
Herling, C. D. et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 9, 727 (2018).
Ianevski, A., Giri, K. A. & Aittokallio, T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 48, W488–W493 (2020).
Brägelmann, J. et al. Systematic kinase inhibitor profiling identifies CDK9 as a synthetic lethal target in NUT midline carcinoma. Cell Rep. 20, 2833–2845 (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
We thank all members of the Sos lab and Peifer lab for their fruitful comments, especially Katia Garbert. We furthermore thank the Regional Computing Center of the University of Cologne (RRZK) for providing computing time on the DFG-funded HPC cluster CHEOPS as well as support and Graziella Bosco for data archiving at EGA. This work was supported by the Bundesministerium für Bildung und Forschung [e:Med initiative; 01ZX1901A to M.L.S., M.P. and R.K.T.]; a research grant by the Thyssen Foundation [10.19.2.025MN to M.L.S.] and the Deutsche Krebshilfe [70112888 to M.L.S., Mildred Scheel Nachwuchszentrum Grant 70113307 to J.B. and D.F.A.]; and the Else Kröner Fresenius Stiftung [Memorial Grant 2018_EKMS.35 to J.B.]. The funding sources had no involvement in the conceptual or executive process of this study.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Each author confirms that he or she has made substantial contributions to the conception or design of the work or the acquisition, analysis, interpretation, and assembly of the data; contributed to paper writing; all authors approved the final paper; all authors are accountable for all aspects of the work. D.S. and D.F.A. contributed equally to this work as co-first authors. J.B., M.S., and M.L.S. contributed equally to this work as co-senior authors.
Corresponding authors
Ethics declarations
Competing interests
D.S. has received honoraria for advisory roles or lectures from BMS, Boehringer-Ingelheim, MSD, Novartis, Roche, Healthcare Consulting Cologne, Abbvie; and has received travel and accommodation support from AstraZeneca, BMS, Boehringer-Ingelheim, MSD, Novartis, Roche, Abbvie. J.F. has received honoraria from AstraZeneca. B.B. has received sponsored research at Gustave Roussy Cancer Center from Abbvie, Amgen, AstraZeneca, BioGen, Blueprint Medicines, BMS, Boehringer Ingelheim, Celgene, Cristal Therapeutics, Daiichi-Sankyo, Eli Lilly, GSK, Ignyta, IPSEN, Inivata, Janssen, Merck KGaA, MSD, Nektar, Onxeo, OSE immunotherapeutics, Pfizer, Pharma Mar, Roche-Genentech, Sanofi, Servier, Spectrum Pharmaceuticals, Takeda, Tiziana Pharma, Tolero Pharmaceuticals. D.P. has had consulting or advisory roles for AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Daiichi Sankyo, Eli Lilly, Merck, Novartis, Pfizer, prIME Oncology, Peer CME, Roche, Samsung Bioepis; and has received honoraria from AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Eli Lilly, Merck, Novartis, Pfizer, prIME Oncology, Peer CME, Roche, Samsung Bioepis; and has done clinical trials research as principal or co-investigator (institutional financial interests) with support from AstraZeneca, BMS, Boehringer Ingelheim, Eli Lilly, Merck, Novartis, Pfizer, Roche, Medimmun, Sanofi-Aventis, Taiho Pharma, Novocure, Inivata, Takeda, Pharma Mar, Janssen, Daiichi Sankyo; and has received travel and accommodation support from AstraZeneca, Roche, Novartis, Pfizer. L.N. has received honoraria from Pfizer, Celgene, Novartis, Roche, Boehringer Ingelheim, Janssen, BMS, Takeda, Bayer; and has had consulting or advisory roles for Novartis, Boehringer Ingelheim, BMS, Roche, Janssen, Pfizer, Takeda, Bayer; and has received research funding from Pfizer, Novartis, MSD, Janssen, Amgen; and has received travel and accommodation support from Novartis, Pfizer, Celgene, Boehringer Ingelheim, Janssen. S.M. has received honoraria from Pfizer, Novartis, AstraZeneca; and has received research support from Novartis, Pfizer, BMS, Janssen. R.K.T. is the founder/shareholder and has had a consultancy/advisory role of/for PearlRiver Bio GmbH; is the founder/shareholder and has had a consultancy/advisory role of/for Epiphanes Inc; and is the founder/shareholder and has had a consultancy/advisory role of/for CDL Therapeutics GmbH; and has a stock or other ownership and has had a consultancy/advisory role of/for Merck, J&J, AstraZeneca, Bayer; and has a stock or other ownership and has had a consultancy/advisory role of/for Roche; and has a commercial research grant from Roche; and has a stock or other ownership of Novartis, GSK; and has had a consultancy/advisory role for New Oncology AG, Clovis, Daiichi-Sankyo, Boehringer Ingelheim, MSD, Lilly, Sanofi-Aventis, Puma. S.M.-B. has received honoraria for advisory boards or lectures from BMS, Novartis, Roche, Pfizer, Bayer, Molecular Health, Targos, Astra Zeneca; and has received non-financial support from BMS, Janssen. O.G. has received honoraria for advisory boards from AMGEN, LILLY, BAYER. L.M. has had consulting or advisory roles for Roche Diagnostics, Takeda, Roche; and has had a lecture or educational activities for BMS, Tecnofarma, Roche; and has received travel and accommodation support from BMS, Roche; and has received Mentorship program with key opinion leaders from AstraZeneca; and has received sponsored research from Amgen, BMS, Boehringer Ingelheim. T.W. has had an advisory role for Lilly; and has received speakers honoraria from Novartis; and has received travel support from Roche, Pfizer. R.B. is co-founder and chief scientific officer of Targos Mol Inc., Kassel Germany; and has received personal fees and others for advisory boards or lectures from BMS, MSD, Novartis, Roche, Lilly, AstraZeneca, Illumina, AbbVie, Amgen, Boehringer-Ingelheim, Merk-Serono, Qiagen, Pfizer. J.W. has received honoraria for advisory roles or lectures from Amgen, AstraZeneca, Blueprint, BMS, Boehringer Ingelheim, Chugai, Daiichi Sankyo, Ignyta, Janssen, Lilly, Loxo, MSD, Novartis, Pfizer, Roche, Seattle Genetics, Takeda; and has received research support from BMS, Janssen Pharmaceutica, Novartis, Pfizer. M.S. has received honoraria for advisory roles from Pfizer, Roche, AMGEN, Novartis, Takeda, Boehringer Ingelheim; and has received institutional research support from AMGEN, Dracen Pharmaceuticals. M.L.S. is a founder, shareholder and advisor of PearlRiver Bio – a Centessa Pharmaceuticals company - and MLS has a commercial research grant from PearlRiver Bio - a Centessa company. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schaufler, D., Ast, D.F., Tumbrink, H.L. et al. Clonal dynamics of BRAF-driven drug resistance in EGFR-mutant lung cancer. npj Precis. Onc. 5, 102 (2021). https://doi.org/10.1038/s41698-021-00241-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-021-00241-9
This article is cited by
-
ERK pathway agonism for cancer therapy: evidence, insights, and a target discovery framework
npj Precision Oncology (2024)
-
Synchronous intracellular delivery of EGFR-targeted antibody–drug conjugates by p38-mediated non-canonical endocytosis
Scientific Reports (2022)
