
Abstract
While next-generation sequencing (NGS) is used to guide therapy in patients with metastatic lung adenocarcinoma (LUAD), use of NGS to determine pathologic LN metastasis prior to surgery has not been assessed. To bridge this knowledge gap, we performed NGS using MSK-IMPACT in 426 treatment-naive patients with clinical N2-negative LUAD. A multivariable logistic regression model that considered preoperative clinical and genomic variables was constructed. Most patients had cN0 disease (85%) with pN0, pN1, and pN2 rates of 80%, 11%, and 9%, respectively. Genes altered at higher rates in pN-positive than in pN-negative tumors were STK11 (p = 0.024), SMARCA4 (p = 0.006), and SMAD4 (p = 0.011). Fraction of genome altered (p = 0.037), copy number amplifications (p = 0.001), and whole-genome doubling (p = 0.028) were higher in pN-positive tumors. Multivariable analysis revealed solid tumor morphology, tumor SUVmax, clinical stage, SMARCA4 and SMAD4 alterations were independently associated with pathologic LN metastasis. Incorporation of clinical and tumor genomic features can identify patients at risk of pathologic LN metastasis; this may guide therapy decisions before surgical resection.
Similar content being viewed by others


Introduction
Lung adenocarcinoma (LUAD) is the most common histologic subtype of non-small cell lung cancer (NSCLC)1 and is associated with a higher risk of occult lymph node (LN) metastasis than other NSCLC tumors2,3. Combined positron emission tomography/computed tomography (PET/CT) imaging is the standard noninvasive study for LN staging4,5,6; however, it is associated with a high false-negative rate7. In patients with no uptake of fluorodeoxyglucose on PET/CT, the incidence of occult mediastinal LN metastases is 7–18%8,9,10,11. In addition, PET/CT may have reduced accuracy in patients with nodes <1 cm in short-axis diameter12, and false-positives can occur secondary to inflammation or infection13.
Previous studies have identified clinicopathologic variables associated with occult LN metastasis in patients with clinical N2-negative disease. These include centrally located tumors, large tumor size, high primary tumor maximum standardized uptake value (SUVmax), positive N1 nodes on PET/CT, and micropapillary histologic pattern8,9,14,15. In clinical practice, broad-panel next-generation sequencing (NGS) has increasingly been used to elucidate tumor biology, identify targetable driver-gene perturbations, and inform prognoses for patients with NSCLC16. To date, no study has used NGS to determine the risk of pathologic LN metastasis in patients with LUAD. Importantly, it is unknown whether this information could potentially guide the order of first line therapy (i.e., systemic vs. local) in the future. To bridge this knowledge gap, we examined tumor genomic factors in patients with clinically N2-negative (cN0-1) LUAD and assessed their association with pathologic LN metastasis identified at surgery.
Results
Clinicopathologic characteristics
In total, 426 patients met the inclusion criteria; 60% (n = 255) were men, and 74% (n = 316) were current or former smokers. Clinicopathologic features are summarized in Table 1. A majority of patients had clinical stage I disease (n = 334 [78%]); 22% (n = 92) had clinical stage II disease. On pathologic review, 80% of resected tumors (n = 341) were pN0 (pN-negative), and 20% (n = 85) were pN1 or pN2 (pN-positive). On diagnostic CT, 59% (201/341) of pN-negative tumors and 92% (78/85) of pN-positive tumors had solid morphologic appearance (p < 0.001). Median primary tumor SUVmax was 3.5 (IQR, 1.8–6.5) for pN-negative tumors and 7.8 (IQR, 4.6–10.5) for pN-positive tumors (p < 0.001). Invasive staging was performed in 10% of patients with clinical early-stage disease (n = 42)—9% underwent EBUS (n = 38), 0.7% underwent mediastinoscopy (n = 3), and 0.2% underwent both (n = 1).
A median of 3 N1 stations (interquartile range [IQR], 2–3), 3 N2 stations (IQR, 2–3), and 5 total stations (IQR, 4–6) were sampled intraoperatively (Supplementary Table 1). Pathologic stages were as follows: 67% stage I (n = 284), 21% stage II (n = 91), and 12% stage III (n = 51). A total of 362 patients (85%) had cN0 disease, 15% of whom (54/362) had occult pathologic LN metastasis. In addition, 64 patients (15%) had cN1 disease, 48% of whom (31/64) had confirmed pathologic LN involvement. Occult pN2 disease was more frequently identified in patients with cN1 disease than in patients with cN0 disease (20% [13/64] vs. 7% [24/362]).
Genomic factors associated with pathologic LN metastasis
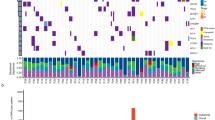
Next, we investigated genomic factors associated with pathologic LN metastasis. Among genes altered in ≥2% of the entire cohort (n = 27), the most frequently altered gene in pN-negative tumors was KRAS (35%)—compared with 34% in pN-positive tumors (Fig. 1). The most frequently altered gene in pN-positive tumors was TP53 (45%)—compared with 34% in pN-negative tumors. Three genes were altered at significantly higher frequencies in pN-positive than in pN-negative tumors: STK11 (22% vs. 12%; p = 0.024), SMARCA4 (8% vs. 1.8%; p = 0.006), and SMAD4 (7% vs. 1.5%; p = 0.011) (Fig. 1). The alteration frequency of RBM10 was significantly higher in pN-negative tumors than in pN-positive tumors (17% vs. 8%; p = 0.044) (Fig. 1). These gene alterations are mainly driven by the ever-smoker cohort, as there is no statistically significant difference between pN-positive never-smokers and pN-negative never-smokers for STK11, RBM10, SMAD4, and SMARCA4 (Supplementary Fig. 1). Interestingly, no significant differences in targetable LUAD alterations (n = 9) were identified (Supplementary Fig. 2).

CT computed tomography, NA not available.
The distribution of tumor mutational burden (TMB) did not significantly differ between pN-negative tumors (median [IQR], 4.9 [2.6–8.8] mutations/megabase [Mb]) and pN-positive tumors (median [IQR], 6.1 [3.5–10.8] mutations/Mb; p = 0.058) (Fig. 2a). However, fraction of genome altered (FGA) was significantly higher in pN-positive tumors (median [IQR], 0.343 [0.171–0.50] vs. 0.269 [0.141–0.443]; p = 0.037) (Fig. 2b).

a Boxplot of tumor mutational burden (mutations/megabase) versus pathologic lymph node status. b Boxplot of fraction of genomic altered versus pathologic lymph node status. c Bar graphs showing percent of total tumor samples with most frequently altered mutational signatures (SBS2, SBS13, SBS4) according to pathologic lymph node metastasis (asterisk (*) signifies p < 0.05). In the boxplots in this figure, the center line represents the median value, the bounds of the box represent the interquartile range, and the whiskers extend to 1.5× the interquartile range on either side of the median.
Additionally, we evaluated the mutational signature profiles in pN-positive and pN-negative tumors. The three most frequent signatures were the smoking signature (SBS4) and the two APOBEC signatures (SBS2 and SBS13). A statistically significantly higher percentage of pN-positive tumors (SBS2 27.3%; SBS13 31.8%) had APOBEC signatures present, compared with pN-negative tumors (SBS2 12.8%; SBS13 14.9%). Both SBS2 (p = 0.03) and SBS13 (p = 0.02) were statistically significantly more enriched in pN-positive tumors. There was no statistically significant difference in smoking signatures between the two groups (Fig. 2c).
Copy number alterations associated with pathologic LN metastasis
To further examine our observation that pN-positive tumors had significantly higher FGA, we next investigated the copy number landscape (Fig. 3). No significant differences in tumor purity were identified between pN-negative and pN-positive tumors (p = 0.059) (Supplementary Fig. 3A). Copy number amplifications were significantly higher in pN-positive tumors than in pN-negative tumors (median [IQR], 0.152 [0.060–0.314] vs. 0.085 [0.037–0.208]; p = 0.001) (Fig. 3a). Copy number deletions were not significantly different between groups (p = 0.299) (Fig. 3b). Significant differences in chromosome arm-level copy number (false-discovery rate [FDR] p < 0.2) between pN-negative and pN-positive tumors were identified at 2p (p = 0.122) and 2q (p = 0.122), where both arms exhibit more copy number changes in pN-positive tumors (Fig. 3c). The copy number changes observed were broad, and no focal copy number changes were observed on chromosome 2 in either pN-positive or pN-negative tumors.

a Boxplot of copy number amplifications versus pathologic lymph node status. b Boxplot of copy number deletions versus pathologic lymph node status. c Comparison of copy number alterations across chromosome arms according to pathologic lymph node status. d Bar plot of the rate of whole-genome doubling versus pathologic lymph node status. In the boxplots in this figure, the center line represents the median value, the bounds of the box represent the interquartile range, and the whiskers extend to 1.5× the interquartile range on either side of the median.
Genes with copy number alterations present in ≥1% of the entire cohort (n = 14 genes; Supplementary Fig. 3B) were also evaluated. Among pN-positive tumors, CDKN2A was the most frequently altered (9%), whereas MDM2 was most frequently altered (10%) among pN-negative tumors. CDK4 was altered significantly more often in pN-negative tumors than in pN-positive tumors (8% vs. 1.2%; p = 0.029) (Supplementary Fig. 3B), and CDKN2A was altered significantly more often in pN-positive tumors than in pN-negative tumors (9% vs. 4%; p = 0.039) (Supplementary Fig. 3B). Finally, pN-positive tumors had a higher rate of whole-genome doubling (WGD; 22% vs. 13%; p = 0.028) (Fig. 3d), further elucidating their chromosomal instability.
Preoperative clinicopathologic and genomic features associated with pathologic LN metastasis
Next, we performed logistic regression analyses to identify clinicopathologic and genomic features associated with pathologic LN metastasis. Univariable logistic regression analysis identified the following preoperative clinicopathologic variables (Supplementary Table 2): solid versus nonsolid tumor morphologic appearance on CT (p < 0.001), tumor size on CT (p = 0.001), tumor SUVmax (p < 0.001), cN1 versus cN0 (p < 0.001), and clinical stage II versus I (p < 0.001). We then performed clinical stage-adjusted univariable logistic regression analyses to identify genomic variables associated with pathologic LN metastasis (Supplementary Table 3). The following genomic variables were considered in the final multivariable model: WGD (p = 0.014), copy number amplifications (p = 0.002), STK11 alteration versus wild-type (p = 0.026), SMARCA4 alteration versus wild-type (p = 0.013), and SMAD4 alteration versus wild-type (p = 0.004).
On multivariable regression analysis combining preoperative clinicopathologic and genomic features, the following variables were independently associated with pathologic LN metastasis (Fig. 4): solid versus nonsolid tumor morphologic appearance on CT (odds ratio [OR], 4.25; 95% confidence interval [CI], 1.81–9.93; p = 0.001), tumor SUVmax (OR, 1.09; 95% CI, 1.03–1.15; p = 0.002), clinical stage II versus I (OR, 3.31; 95% CI, 1.87–5.88; p < 0.001), SMARCA4 alteration versus wild-type (OR, 3.67; 95% CI, 1.02–13.16; p = 0.046), and SMAD4 alteration versus wild-type (OR, 5.01; 95% CI, 1.29–19.45; p = 0.02).

In the forest plot in this figure, the points represent the odds ratio, and the whiskers represent the confidence interval corresponding to the table to the right. CI confidence interval, CT computed tomography, OR odds ratio, SUV standardized uptake value.
Association between copy number amplifications and clinicopathologic characteristics
To further examine the association between copy number amplifications and pathologic LN metastasis in the clinical stage-adjusted analysis of genomic variables above, we investigated the relationship between copy number amplification tertiles (low vs. intermediate vs. high) and select clinicopathologic characteristics. Significant associations between copy number amplification tertiles and preoperative clinical characteristics (tumor size on CT, tumor solid morphologic appearance on CT, and primary tumor SUVmax) were identified; copy number amplification tertiles were also shown to be associated with pathologic characteristics obtained from the surgical specimen (pathologic tumor size, micropapillary- or solid-predominant histologic subtype, visceral pleural invasion, lymphovascular invasion, tumor spread through air spaces, pathologic LN metastasis, and pathologic stage) (Supplementary Fig. 4).
Discussion
The ability to better predict pathologic LN metastasis would allow the identification of subgroups of patients who may benefit from neoadjuvant therapy before tumor resection. In the present study, we found that, of 362 patients with cN0 disease, 15% (n = 54) had pathologic LN metastasis at the time of surgery. This results in a false-negative rate (proportion of pN-positive patients who had cN0 disease) of 64% for preoperative clinical LN staging. To address the limitations of radiographic nodal assessment and staging, we analyzed tumor genomic features as well as clinical and radiographic features to determine variables that are independently associated with pathologic LN metastasis in patients with clinical stage I/II LUAD.
Multiple studies have identified radiographic and pathologic predictors of occult LN metastasis, including SUVmax17, lymphatic invasion17, vascular invasion2, and micropapillary histologic pattern9. Most of these studies used data that can be reliably obtained only from the pathologic specimen; the utility of such data for preoperative or intraoperative decision-making is therefore limited. Our group previously reported that frozen section analysis can be used to detect micropapillary histologic patterns18, the results of which can guide intraoperative decision-making. More recently, we demonstrated that the presence of tumor spread through air spaces on frozen section analysis is an independent predictor of occult pathologic LN metastasis19. However, the challenge with the use of pathologic predictors obtained from intraoperative frozen sections or the surgical specimen is that such features are not applicable to preoperative clinical decision-making. Recently, Verdial and colleagues, in a study of patients with clinical stage I-IIIB NSCLC, reported a prediction model that used only radiographic variables and had a bias-corrected C-index of 0.7815.
During the last decade, NGS has changed the clinician’s approach to the management of NSCLC16. The model developed here combines clinical variables available preoperatively (tumor morphologic appearance on CT, tumor SUVmax, and clinical stage) and genomic data (SMAD4 and SMARCA4 alterations) that can be obtained from a preoperative biopsy specimen to better guide the therapeutic strategy. A similar methodology has recently been used to individualize therapy for patients with melanoma20.
We evaluated various genomic summary metrics, such as TMB and FGA, as well as individual gene alterations for inclusion in the final pathologic LN metastasis prediction model. Alterations in SMAD4 and SMARCA4 were independently associated with pathologic LN metastasis. Both SMAD4 and SMARCA4 were altered at significant rates in tumors from patients with pathologic LN metastasis. SMAD4 mediates signaling of transforming growth factor beta and bone morphogenic protein ligands, and it is a well-defined tumor suppressor in pancreatic and colon cancer21,22. Reduced SMAD4 expression in NSCLC has been associated with increased DNA damage, reduced DNA repair, and increased sensitivity to topoisomerase inhibitors23,24. SMARCA4 is a subunit of the switch/sucrose nonfermentable (SWI/SNF) complex, which plays important roles in chromatin remodeling and, thus, in the regulation of vital cellular processes and functions, such as gene expression, proliferation, and differentiation25. In lung cancer, SMARCA4 inactivation is the most common alteration within the SWI/SNF complex and has been associated with poor oncologic outcomes26,27. Recently, Schoenfeld and colleagues reported that SMARCA4 alterations co-occurred more frequently with KRAS, STK11, and KEAP1 mutations, compared with SMARCA4 wild-type tumors27. Interestingly, they also identified improved outcomes after treatment with immunotherapy in patients with SMARCA4-mutant tumors27,28.
We also investigated the genomic landscape of tumors from patients with pathologic LN metastasis and patients without pathologic LN metastasis. Alteration rates of STK11, SMARCA4, and SMAD4 were significantly higher among tumors from patients with pathologic LN metastasis, consistent with prior findings29. In our cohort, the increased alteration rates are mainly driven by the ever-smoker subgroup, as these genes are not frequently altered in the never-smokers. This is likely caused by the small sample size of never-smokers, especially pN-positive never-smokers, and the lower TMB burden in the never-smokers (Supplementary Fig. 1). A more comprehensive investigation with larger numbers of never-smoker patients is needed to assess differences between pN-positive ever-smokers and never-smokers.
FGA and copy number amplifications were also statistically significantly higher among tumors from patients with pathologic LN metastasis. FGA is a surrogate for chromosome instability, which has been shown to promote tumor metastasis through the activation of the cGAS-STING pathway30,31. FGA has been shown to correlate with survival in other cancers32,33,34,35,36. A higher level of subclonal copy number alterations has been associated with poorer disease-free survival in patients with resected early-stage NSCLC37.
Additionally, we found an enrichment in APOBEC signatures in pN-positive tumors. The APOBEC signatures have been shown to contribute to increased tumor heterogeneity in both primary38 and metastatic NSCLC tumors39. These findings have important clinical implications, as APOBEC signatures have the potential to predict immune response, which may serve as a potential marker for immunotherapy in pN-positive patients40.
Interestingly, we found an association between copy number amplifications and pathologic LN metastasis on univariable analysis, which prompted us to further investigate the relationship between copy number amplification and clinicopathologic variables. We identified a significant relationship between high copy number amplification tertiles and various poor clinicopathologic indicators. Finally, as further evidence of their more aggressive nature, tumors from patients with pathologic LN metastasis were associated with a higher rate of WGD, which has been associated with poor long-term survival across multiple cancers41.
This study has several limitations. NGS was performed using single-region sampling of the primary tumor. As previously noted, intratumoral heterogeneity is intrinsic to LUAD37, and single-region sampling may not accurately capture the complexity of the disease, such as its clonal architecture42. In addition, tumor genomic analyses were performed on the surgical specimen, not on preoperative biopsy specimens. Obtaining high-quality DNA for NGS from biopsy specimens can be a challenge43, but success rates of 80–90% are now being reported for small tumor tissue samples obtained from CT-guided and bronchoscopic biopsies—convincingly demonstrating that acquisition of adequate quality DNA for NGS is possible before surgical resection44. Our final model incorporates SMARCA4 and SMAD4 alterations; however, similar to many other genomic drivers of aggressive tumor biology, the frequency of these alterations is modest, especially in never-smokers45. Finally, external validation is required to evaluate the performance of the model.
In summary, this study highlights the potential importance of genomic data for identification of patients at risk of pathologic LN metastasis. Although clinical stage remains important for identifying patients at risk of pathologic LN metastasis, in isolation it does not perform well in the prediction of nodal disease in this early-stage LUAD cohort. Our final multivariable model comprised preoperative clinical features and SMARCA4 and SMAD4 alteration data that were found to be independently associated with pathologic LN metastasis. The ability to identify patients with stage I/III LUAD who are at high risk of pathologic LN metastasis could potentially guide the therapeutic strategy prior to surgical resection.
Methods
Patient cohort
This study was approved by the institutional review board at Memorial Sloan Kettering Cancer Center. All patients provided written informed consent to participate in the institutional review board–approved protocol. Patients included in the study underwent complete resection for LUAD and had NGS (Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets [MSK-IMPACT]46) performed on their primary tumor between 2010 and 2018. All patients received an anatomic resection (lobectomy, segmentectomy, or pneumonectomy) with LN dissection. Exclusion criteria included induction therapy, wedge resection, microscopic or macroscopic residual disease (R1/R2 resection), and low-quality NGS. Patients included in the study were grouped according to pathologic LN metastasis (see CONSORT diagram, Supplementary Fig. 5). pN1 and pN2 patients were grouped together as there were no statistically significant differences in gene frequency alterations between pN1 and pN2 patients.
Clinical characteristics, preoperative CT and PET, and pathology reports (adjusted according to the 8th edition of the AJCC Cancer Staging Manual) were reviewed. Tumor size, presence of solid tumor morphologic appearance, primary tumor SUVmax, and lymphadenopathy (documented as such or ≥1 cm in short axis on CT scan15) were recorded. Follow-up was performed in accordance with National Comprehensive Cancer Network guidelines47.
Tumor genomic analysis
MSK-IMPACT sequencing was performed and analyzed as previously described46,48. TMB was defined as the fraction of nonsynonymous single-nucleotide or insertion/deletion mutations divided by the length of the coding region (in Mb) sequenced by each panel (0.98, 1.06, and 1.22 Mb in the 341-, 410-, and 468-gene panels, respectively). FGA was computed from the output of Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing (FACETS), which provides accurate, purity- and ploidy-corrected, integer DNA copy number calls from sequenced samples. FGA is defined as the fraction of the genome that differs from the major integer copy number (MCN), which is defined as the integer total copy number spanning the largest portion of the genome49.
Copy number alteration frequency plots were generated using the Integrative Genomics Viewer from Broad Institute. Significant focal copy number alterations were identified from segmented data using GISTIC 2.050. Copy number deletions, amplifications, WGD, and arm-level FGA estimates were calculated from the FACETS method output49. Copy number amplifications were defined as the fraction of the genome that was greater than the MCN, whereas copy number deletions were defined as the fraction of the genome that was less than the MCN. Tumor samples were considered to have undergone WGD if >50% of their autosomal genome had an MCN (the more frequent allele in a given segment) >2. Arm-level FGA was defined as the fraction of the chromosomal arm that differed from the MCN. p-values highlighting differences in arm-level FGA between histologic subtypes were calculated using the Kruskal–Wallis test and were adjusted for multiple comparisons using the FDR method; FDR p < 0.2 was considered significant.
Mutational signatures were computed51 for samples with at least 13.8 Mut/Mb, as described by Zehir et al.52 (pN-positive [n = 44] and pN-negative [n = 141]). We used the most recent version of the single base substitution signatures defined in the Catalogue of Somatic Mutations in Cancer database53. Samples were considered to have a signature present if the mean signature value was greater than 0.1.
Statistical analysis
The Chi-square test and Mann–Whitney U-test were used to compare the categorical and continuous factors between the two patient cohorts (pN-negative [pN0] vs. pN-positive [pN1/pN2]), respectively. Fisher’s exact test was used to compare the alteration frequencies of genes altered in ≥2% of the entire cohort. Univariable logistic regression analysis was performed to quantify the relationships between preoperative clinicopathologic features and pathologic LN metastasis. A separate univariable logistic regression analysis, adjusted for clinical stage, was performed to quantify the relationships between genomic features and pathologic LN metastasis. A multivariable logistic regression model was constructed starting with preoperative clinicopathologic and genomic factors with p < 0.1 in univariable analyses in a backward-selection method. All analyses were two-sided, and p < 0.05 was considered to indicate statistical significance. All analyses were performed using Stata 15.0 (StataCorp, College Station, TX) and R 3.5.3 (R Core Team, Vienna, Austria).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All the genomic and clinical data used in our analyses are publicly available through the cBioPortal for Cancer Genomics54 at https://www.cbioportal.org/study/summary?id=luad_msk_npjpo_2021.
References
Howlader, N. et al. SEER Cancer Statistics Review, 1975–2016. https://seer.cancer.gov/csr/1975_2016/. (2020).
Bille, A., Woo, K. M., Ahmad, U., Rizk, N. P. & Jones, D. R. Incidence of occult pN2 disease following resection and mediastinal lymph node dissection in clinical stage I lung cancer patients. Eur. J. Cardiothorac. Surg. 51, 674–679 (2017).
Suzuki, K. et al. Clinical predictors of N2 disease in the setting of a negative computed tomographic scan in patients with lung cancer. J. Thorac. Cardiovasc. Surg. 117, 593–598 (1999).
Fischer, B. et al. Preoperative staging of lung cancer with combined PET-CT. N. Engl. J. Med. 361, 32–39 (2009).
Lardinois, D. et al. Staging of non-small-cell lung cancer with integrated positron-emission tomography and computed tomography. N. Engl. J. Med. 348, 2500–2507 (2003).
Shim, S. S. et al. Non-small cell lung cancer: prospective comparison of integrated FDG PET/CT and CT alone for preoperative staging. Radiology 236, 1011–1019 (2005).
Lv, Y. L. et al. Diagnostic performance of integrated positron emission tomography/computed tomography for mediastinal lymph node staging in non-small cell lung cancer: a bivariate systematic review and meta-analysis. J. Thorac. Oncol. 6, 1350–1358 (2011).
Kanzaki, R. et al. Occult mediastinal lymph node metastasis in NSCLC patients diagnosed as clinical N0-1 by preoperative integrated FDG-PET/CT and CT: Risk factors, pattern, and histopathological study. Lung Cancer 71, 333–337 (2011).
Yeh, Y. C. et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification predicts occult lymph node metastasis in clinically mediastinal node-negative lung adenocarcinoma. Eur. J. Cardiothorac. Surg. 49, e9–e15 (2016).
Gomez-Caro, A. et al. Incidence of occult mediastinal node involvement in cN0 non-small-cell lung cancer patients after negative uptake of positron emission tomography/computer tomography scan. Eur. J. Cardiothorac. Surg. 37, 1168–1174 (2010).
Veeramachaneni, N. K., Battafarano, R. J., Meyers, B. F., Zoole, J. B. & Patterson, G. A. Risk factors for occult nodal metastasis in clinical T1N0 lung cancer: a negative impact on survival. Eur. J. Cardiothorac. Surg. 33, 466–469 (2008).
Lu, P., Sun, Y., Sun, Y. & Yu, L. The role of (18)F-FDG PET/CT for evaluation of metastatic mediastinal lymph nodes in patients with lung squamous-cell carcinoma or adenocarcinoma. Lung Cancer 85, 53–58 (2014).
Tsim, S., O’Dowd, C. A., Milroy, R. & Davidson, S. Staging of non-small cell lung cancer (NSCLC): a review. Respir. Med. 104, 1767–1774 (2010).
Farjah, F., Lou, F., Sima, C., Rusch, V. W. & Rizk, N. P. A prediction model for pathologic N2 disease in lung cancer patients with a negative mediastinum by positron emission tomography. J. Thorac. Oncol. 8, 1170–1180 (2013).
Verdial, F. C. et al. Prediction model for nodal disease among patients with non-small cell lung cancer. Ann. Thorac. Surg. 107, 1600–1606 (2019).
Korf, B. R. & Rehm, H. L. New approaches to molecular diagnosis. JAMA 309, 1511–1521 (2013).
Moon, Y. et al. Clinicopathologic factors associated with occult lymph node metastasis in patients with clinically diagnosed n0 lung adenocarcinoma. Ann. Thorac. Surg. 101, 1928–1935 (2016).
Yeh, Y. C. et al. Using frozen section to identify histological patterns in stage I lung adenocarcinoma of </= 3 cm: accuracy and interobserver agreement. Histopathology 66, 922–938 (2015).
Vaghjiani, R. G. et al. Tumor spread through air spaces is a predictor of occult lymph node metastasis in clinical stage IA lung adenocarcinoma. J. Thorac. Oncol. 15, 792–802 (2020).
Bellomo, D. et al. Model combining tumor molecular and clinicopathologic risk factors predicts sentinel lymph node metastasis in primary cutaneous melanoma. JCO Precis Oncol. 4, 319–334 (2020).
Malkoski, S. P. & Wang, X. J. Two sides of the story? Smad4 loss in pancreatic cancer versus head-and-neck cancer. FEBS Lett. 586, 1984–1992 (2012).
Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 (2012).
Haeger, S. M. et al. Smad4 loss promotes lung cancer formation but increases sensitivity to DNA topoisomerase inhibitors. Oncogene 35, 577–586 (2016).
Ziemke, M., Patil, T., Nolan, K., Tippimanchai, D. & Malkoski, S. P. Reduced Smad4 expression and DNA topoisomerase inhibitor chemosensitivity in non-small cell lung cancer. Lung Cancer 109, 28–35 (2017).
Wilson, B. G. & Roberts, C. W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492 (2011).
Helming, K. C., Wang, X. & Roberts, C. W. M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 26, 309–317 (2014).
Schoenfeld, A. J. et al. The genomic landscape of SMARCA4 alterations and associations with outcomes in patients with lung cancer. Clin. Cancer Res. 26, 5701–5708 (2020).
Rekhtman, N. et al. SMARCA4-deficient thoracic sarcomatoid tumors represent primarily smoking-related undifferentiated carcinomas rather than primary thoracic sarcomas. J. Thorac. Oncol. 15, 231–247 (2020).
Qiu, T., Guo, H., Zhao, H., Wang, L. & Zhang, Z. Next-generation sequencing for molecular diagnosis of lung adenocarcinoma specimens obtained by fine needle aspiration cytology. Sci. Rep. 5, 1–7 (2015).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018).
Motwani, M., Pesiridis, S. & Fitzgerald, K. A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 20, 657–674 (2019).
Gu, X. et al. Copy number variation: a prognostic marker for young patients with squamous cell carcinoma of the oral tongue. J. Oral. Pathol. Med. 48, 24–30 (2019).
Hieronymus, H. et al. Copy number alteration burden predicts prostate cancer relapse. Proc. Natl Acad. Sci. USA 111, 11139–11144 (2014).
Zhang, L., Feizi, N., Chi, C. & Hu, P. Association analysis of somatic copy number alteration burden with breast cancer survival. Front. Genet. 9, 421 (2018).
Jones, G. D. et al. A genomic-pathologic annotated risk model to predict recurrence in early-stage lung adenocarcinoma: a prospective observational cohort study. JAMA Surg. 156, e205601 (2021).
Caso, R. et al. The underlying tumor genomics of predominant histologic subtypes in lung adenocarcinoma. J. Thorac. Oncol. 15, 1844–1856 (2020).
Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121 (2017).
Swanton, C., McGranahan, N., Starrett, G. J. & Harris, R. S. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 5, 704–712 (2015).
Roper, N. et al. APOBEC mutagenesis and copy-number alterations are drivers of proteogenomic tumor evolution and heterogeneity in metastatic thoracic tumors. Cell Rep. 26, 2651–2666 (2019).
Wang, S., Jia, M., He, Z. & Liu, X.-S. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 37, 3924–3936 (2018).
Bielski, C. M. et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 50, 1189–1195 (2018).
Zhang, J. et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–259 (2014).
Isaka, M. et al. Comparison of clinically relevant mutation profiles between preoperative biopsy and corresponding surgically resected specimens in japanese patients with non–small-cell lung cancer by amplicon-based massively parallel sequencing. Clin. Lung Cancer 18, 519–526 (2017).
Kage, H. et al. Small lung tumor biopsy samples are feasible for high quality targeted next generation sequencing. Cancer Sci. 110, 2652 (2019).
Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120 (2012).
Cheng, D. T. et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 17, 251–264 (2015).
National Comprehensive Cancer Network. Non-Small Cell Lung Cancer (Version 7.2019). https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1450. (2019).
Zhou, J. et al. Analysis of tumor genomic pathway alterations using broad-panel next generation sequencing in surgically resected lung adenocarcinoma. Clin. Cancer Res. 25, 7475–7484 (2019).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Campbell, P. J. & Stratton, M. R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 3, 246–259 (2013).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703 (2017).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Acknowledgements
We gratefully acknowledge the members of the Molecular Diagnostics Service in the Department of Pathology. David B. Sewell, of the MSK Department of Surgery, provided superb editorial assistance. This study was supported by the National Cancer Institute (R01CA217169 and R01CA240472 to D.R.J., R01CA236615 to P.S.A., T32CA009501 to J.G.C., P30 CA008748 to Memorial Sloan Kettering Cancer Center), Hamilton Family Foundation (to D.R.J.), and Department of Defense (LC160212 to P.S.A.). This work was funded in part by the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
Author information
Authors and Affiliations
Contributions
D.R.J. conceived the project and supervised the study. R.C. performed data acquisition, annotation, analysis, interpretation, figure preparation, and manuscript preparation. J.C., B.M., F.S.V., and B.N. assisted with bioinformatics analysis, figure design, and data analysis. K.S.T. performed the statistical analyses. G.J., G.R., D.M., P.A., S.S., and M.B. provided critical feedback and helped to shape and finalize the manuscript. All co-authors agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
G.R. has financial relationships with Scanlan. D.M. is a consultant for Johnson & Johnson, Urogen, Intuitive, and Boston Scientific. P.S.A. has received research funding from ATARA Biotherapeutics and Acea Biosciences, has served on the Scientific Advisory Board or as consultant to ATARA Biotherapeutics, Bayer, Carisma Therapeutics, Imugene, and Takeda Therapeutics, and has patents, royalties, and intellectual property on mesothelin-targeted CARs and other T-cell therapies, method for detection of cancer cells using virus, and pending patent applications on T-cell therapies. M.J.B. serves as a consultant for AstraZeneca. D.R.J. serves as a consultant for AstraZeneca and is on a Clinical Trial Steering Committee for Merck. All other authors have no potential conflicts to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Caso, R., Connolly, J.G., Zhou, J. et al. Preoperative clinical and tumor genomic features associated with pathologic lymph node metastasis in clinical stage I and II lung adenocarcinoma. npj Precis. Onc. 5, 70 (2021). https://doi.org/10.1038/s41698-021-00210-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-021-00210-2
This article is cited by
-
A destabilizing Y891D mutation in activated EGFR impairs sensitivity to kinase inhibition
npj Precision Oncology (2024)
