
Abstract
High-latitude explosive volcanic eruptions can cause substantial hemispheric cooling. Here, we use a whole-atmosphere chemistry-climate model to simulate Northern Hemisphere (NH) high-latitude volcanic eruptions of magnitude similar to the 1991 Mt. Pinatubo eruption. Our simulations reveal that the initial stability of the polar vortex strongly influences sulphur dioxide lifetime and aerosol growth by controlling the dispersion of injected gases after such eruptions in winter. Consequently, atmospheric variability introduces a spread in the cumulative aerosol radiative forcing of more than 20%. We test the aerosol evolution’s sensitivity to co-injection of sulphur and halogens, injection season, and altitude, and show how aerosol processes impact radiative forcing. Several of these sensitivities are of similar magnitude to the variability stemming from initial conditions, highlighting the significant influence of atmospheric variability. We compare the modelled volcanic sulphate deposition over the Greenland ice sheet with the relationship assumed in reconstructions of past NH eruptions. Our analysis yields an estimate of the Greenland transfer function for NH extratropical eruptions that, when applied to ice core data, produces volcanic stratospheric sulphur injections from NH extratropical eruptions 23% smaller than in currently used volcanic forcing reconstructions. Furthermore, the transfer function’s uncertainty, which propagates into the estimate of sulphur release, needs to be at least doubled to account for atmospheric variability and unknown eruption parameters. Our results offer insights into the processes shaping the climatic impacts of NH high-latitude eruptions and highlight the need for more accurate representation of these events in volcanic forcing reconstructions.
Similar content being viewed by others
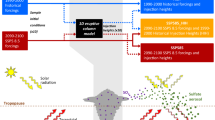


Introduction
Explosive volcanic eruptions are a major driver of natural climate variability1,2. Strong explosive eruptions can directly inject large amounts of SO2 into the stratosphere, where it undergoes oxidation to form sulphuric acid and subsequently forms sulphate aerosols. These aerosols scatter and absorb incoming solar radiation and absorb and emit thermal longwave radiation. The magnitude of this radiative perturbation depends on the mass, lifetime, and microphysical properties of the volcanic aerosols. Understanding the processes controlling these factors is crucial for accurately reconstructing the climate variability of the past3,4 and predicting the impacts of future eruptions5,6.
To date, most studies of explosive volcanic eruptions have focused on tropical eruptions, while high-latitude eruptions have received comparatively less attention. One plausible reason for the emphasis on tropical eruptions could be that the largest volcanic event observed with satellite imagery and modern in situ instruments was the tropical eruption of Mt. Pinatubo in 1991. This event stimulated studies aiming to understand its atmospheric impacts and validate aerosol-climate model simulations of tropical eruptions. It has also been commonly believed that high-latitude eruptions are less climate-relevant compared to tropical eruptions (e.g., refs. 7,8,9) but recent work has questioned this notion10,11, instead suggesting that high-latitude stratospheric sulphur injections lead to stronger hemispheric cooling than tropical injections, given the same sulphur mass. While there exists a handful of modelling studies of high-latitude eruptions, these have either focused on the climatic impacts of specific historical eruptions such as Laki 1783−1784 and Katmai 191212,13,14,15,16, or used models that prescribe the volcanic aerosols and/or relevant atmospheric chemistry7,10,13,14,15,17,18,19. Interactive aerosol and chemistry simulation, especially interactive simulation of atmospheric hydroxyl (OH), has been indicated to be important for the accurate representation of volcanic aerosol formation20,21,22. Additionally, many past studies have employed models lacking a fully resolved stratospheric circulation or an internally generated quasi-biennial oscillation, thus neglecting intricacies of the atmospheric response23. The dispersion of volcanic material injected into the stratosphere has also been found to be highly sensitive to meteorological conditions24. In the NH high latitudes, where the stratospheric polar vortex exhibits strong seasonal and interannual variability25, this effect can be particularly significant. Yet, the impact of atmospheric variability on the evolution of volcanic aerosols following high-latitude eruptions suffers from a lack of systematic investigation utilising the potential of atmospheric models with prognostic aerosols, comprehensive chemistry throughout the atmosphere, and resolved middle atmosphere transport.
The climatic impacts of a volcanic eruption depend on factors such as the erupted mass, injection height, eruption season, and plume composition, collectively referred to as eruption source parameters. A long-standing question is the potential effects of gases and material besides SO2 in the volcanic plume, in particular halogen gases, which are contained in magmas at both high and low latitudes26,27,28,29,30. Petrologic analyses show that past eruptions have emitted chlorine (Cl) and bromine (Br) in amounts with potential for substantial ozone destruction31,32,33. Scavenging processes within volcanic plumes make it uncertain how much of the halogens reach the stratosphere34,35,36. However, plume modelling suggests an injection efficiency over 25% for Plinian eruptions37, and in-situ measurements of the high-latitude eruption of Hekla in 2000 showed little evidence of halogen scavenging at all38,39. In chemistry-climate model studies of tropical eruptions, it has been found that stratospheric injection of volcanic halogens significantly alters the evolution of co-injected sulphur, the stratospheric composition, and the strength and duration of the volcanic forcing40,41,42. However, the impacts of stratospheric co-injection of sulphur and halogens from high-latitude eruptions have so far not been considered with comprehensive chemistry-climate models.
Ice core-derived reconstructions of the volcanic forcing of high-latitude eruptions carry large uncertainties3,4. Much of this uncertainty stems from the so-called “transfer function" used to infer the volcanic stratospheric sulphur injection (VSSI) from ice core sulphate records. Sulphate deposition measured in Greenland in units of mass per unit area need to be multiplied by some area to calculate the total mass that was in the stratosphere. If the Greenland deposition was equal to the global average, then this transfer function would be simply the area of Earth, but this must be modified to account for unequal settling of sulphate from the stratosphere to the troposphere, atmospheric circulation, and varying precipitation processes. The eVolv2k3 and HolVol v.1.04 volcanic forcing time series are used as boundary conditions in transient simulations within the Paleoclimate Modelling Intercomparison Project phase 4 (PMIP443) and Holocene simulations44. Both time series rely on a transfer function for NH extratropical eruptions estimated by Gao et al.45 which was derived from model simulations with the aerosol-climate model GISS ModelE14. The wide range of possible eruption styles, especially at Iceland46 with its close proximity to the Greenland ice sheet, warrants reevaluating the applicability of a single transfer function for the entire record of NH extratropical eruptions. Additionally, unknown eruption source parameters and initial atmospheric conditions for historical eruptions should be reflected in the uncertainty of the transfer function in order to achieve accurate reconstructions of past climate variability.
To comprehensively investigate high-latitude eruptions, it is necessary to consider the potential influence of the strong variability of the high-latitude stratosphere as well as eruption source parameters such as the co-injection of sulphur and halogens on the volcanic aerosol evolution. In this study, we use the chemistry-climate model Community Earth System Model (CESM 2.1.3) with a fully resolved troposphere, stratosphere, and mesosphere, prognostic aerosols, and full-atmosphere chemistry to simulate high-latitude explosive eruptions. We investigate the sensitivities to the winter variability of the NH stratosphere as well as a set of eruption source parameters consisting of plume composition, eruption season, and plume height, and evaluate the impacts on sulphate aerosol evolution and radiative forcing. Finally, in light of our modelled Greenland sulphate deposition, we revisit the assumptions underlying the recommended volcanic forcing reconstructions for PMIP4 and the Holocene.
Results
Sensitivity to the initial polar vortex state
To evaluate the effects of atmospheric variability on volcanic aerosol evolution, we compare the formation and lifetime of aerosols, their mean size, and radiative forcing across ensemble members of a baseline NH high-latitude eruption scenario with different initial states of the NH polar vortex (Table 1, Methods). For such eruptions occurring in winter, the strength and location of the polar vortex influence the dispersion of the injected SO2.
Figure 1 shows the evolution of aerosols following the volcanic injection in each ensemble member. The injected SO2 undergoes gradual oxidation with OH, transforming into H2SO4 over the course of the first 5−6 months, as indicated by the decline in SO2 burden shown in Fig. 1a. The ensemble members (labelled 1−6) are ordered based on the e-folding time of the SO2 burden. This e-folding time ranges from 2.3 to 3.9 months, with a difference of 1.6 months between ensemble members 1 and 6.

a Global SO2 burden anomaly and its e-folding time. The ensemble members (labelled 1−6) are ordered based on the SO2 e-folding time. b Global SO4 burden anomaly. The grey dashed lines in (a, b) indicate the injected sulphur mass divided by e. c Global mean SO4-mass-weighted mean effective radius (Reff). The grey dashed line indicates the peak scattering efficiency for volcanic SO4 as a function of Reff47. d Global mean SAOD at 550 nm anomaly (left axis, solid lines) and cumulative global mean SAOD at 550 nm anomaly (right axis, dashed lines). Note the different time axes.
The difference in SO2 lifetime is reflected in the initiation of sulphate aerosol (SO4) formation, as illustrated in Fig. 1b. Across the ensemble members, the timing of maximum SO4 burden correlates with the initiation of aerosol formation.
The global mean SO4-mass-weighted mean effective radius (Reff) responds to new sulphate aerosol formation and condensation of sulphate onto existing aerosols. In each ensemble member, the onset of aerosol growth coincides with the onset of aerosol formation (Fig. 1c). The growth rate also depends on the initial condition. The ensemble members with a long SO2 lifetime, or equally a delay in aerosol formation, display a higher aerosol growth rate and reach the highest maximum Reff. Therefore, member 6 exhibits the largest maximum Reff (0.34 μm) and members 1 and 2 the smallest (0.31 μm). This range (0.03 μm) is about ten times larger than the 2σ-variability in the unperturbed control run, depending on the season.
The variation in aerosol growth across the ensemble members affects the radiative forcing. The global mean stratospheric optical depth (SAOD) at 550 nm wavelength primarily scales with the SO4 burden, but it is also influenced by the scattering efficiency of the aerosols. According to Mie scattering theory, volcanic SO4 exhibits the highest scattering efficiency for aerosols with Reff close to 0.25 μm47. The SAOD therefore attains a larger maximum value (Fig. 1d, left axis) in the ensemble members where Reff is closer to 0.25 μm during the peak SO4 burden. Ensemble member 1 thus attains a maximum SAOD 12% larger than that in member 6. As a result of the higher maximum SAOD, and the reduced gravitational settling experienced by smaller aerosols, the ensemble members with lower maximum Reff also generally display the largest cumulative SAOD (Fig. 1d, right axis).
In summary, the polar vortex initial condition can considerably change the SO2 lifetime, which, in turn, influences the evolution of aerosol burden and sizes, as well as the resulting radiative forcing, including both the peak and cumulative SAOD. Hence, we now turn to investigate the underlying mechanisms and determine which specific property of the initial polar vortex state controls the subsequent aerosol evolution.
The initial and rate-limiting stage of stratospheric sulphate aerosol formation is the reaction of SO2 with OH, in the oxidation chain:
which ends with H2SO4 condensing with stratospheric water vapour to form sulphate aerosols20. The main source of stratospheric OH is the photolysis of O3 by solar ultraviolet radiation. Advection of OH is inhibited by its extremely short lifetime. As a result, stratospheric OH concentrations are very low during the Arctic polar night, exhibiting weak interannual variability and a strong gradient towards lower latitudes (Supplementary Fig. 1).
The state of the polar vortex, which is closely associated with atmospheric wave activity, can control the exposure to OH through modulating the equatorward transport of the injected SO2, and hence the rate of Reaction (1). Figure 2a shows the varying extent of dispersion of injected SO2 during the initial month in each ensemble member. Depending on the polar vortex initial condition, the SO2 can either be strongly dispersed to lower latitudes (member 1), exclusively confined to the high latitudes (member 6), or exhibit an intermediate pattern. This distribution is more clearly seen in the corresponding zonal mean meridional profiles of SO2 (Fig. 2b, top axis), which are to varying degrees skewed towards the lower latitudes depending on the transport. As apparent from the climatological OH at the injection level (Fig. 2b, bottom axis), the ensemble members with a polar vortex initial condition favourable for equatorward SO2 transport expose the SO2 to higher OH concentrations. As a result, these members display expedited SO2 oxidation, as indicated by the non-linear correlation between the SO2 latitude-centre of mass and the SO2 e-folding time (Fig. 2b, inset axis).

a January mean column SO2 in the six ensemble members of the baseline experiment. Triangles indicate the injection location. b Meridional distribution of January mean zonal mean column SO2 for the ensemble members (top axis) and the model’s January mean zonal mean OH climatology at 24 km (bottom axis). The shading indicates two standard deviations of the climatology. The inset axis displays the relationship between the latitude-centre of mass of January mean zonal mean column SO2 and the SO2 e-folding time (also shown in Fig. 1a). c Distribution of DJF daily mean zonal mean zonal wind at 60°N, 10 hPa and minimum 60−90°N zonal mean temperature at 10 hPa in the control run (black circles/histograms) with the initial polar vortex state (January 1−20) for each ensemble member highlighted (coloured circles/triangles). Error bars represent one standard deviation for each 20-day cluster centred on the means. The two-dimensional spread of each 20-day cluster is expressed by σU,T (legend), the sum of the standard deviations of wind and temperature normalised by the DJF standard deviations (Equation (2)).
To investigate the factors contributing to the varied equatorward transport, it is necessary to go beyond evaluating the initial polar vortex’s strength or location alone. Figure 2c provides a closer examination of the initial states by showing the distribution of polar vortex wind (represented by the zonal mean zonal wind at 10 hPa, 60°N) and polar vortex temperature (represented by the minimum zonal mean temperature between 60 and 90°N at 10 hPa) in the control run, and highlights the different initial states used in the six ensemble members. We here define the “initial state" as the period from January 1 to January 20 of the year used to initialise each ensemble member. During this time window, the polar vortex transports the volcanic injection while itself remaining unaffected by it due to system inertia, that is, the polar vortex evolution is virtually identical to that in the unperturbed control run.
Inspecting the mean zonal wind and temperature of the 20-day clusters (Fig. 2c), no discernible relationship is apparent between these variables and the degree of SO2 dispersion depicted in Fig. 2a, b. However, the combined variability of both quantities within each cluster plays a significant role. The variability within cluster n, denoted as σU,T(n), can be quantified as the added standard deviations of wind, σU(n), and temperature, σT(n), normalised by the standard deviations of the December-January-February (DJF) background distribution, σU(DJF) and σT(DJF):
where U represents the zonal mean zonal wind at 60°N, 10 hPa and T the minimum 60−90°N zonal mean temperature at 10 hPa. The legend of Fig. 2c displays the corresponding values of σU,T for each initial condition, quantifying the variability indicated by the error bars. These values exhibit a positive correlation with the meridional transport of SO2 transport depicted in Fig. 2a, b and, consequently, a negative correlation with the SO2 e-folding time.
The variability within each cluster serves as an indicator of the instability of the polar vortex, representing the extent of changes of the vortex characteristics during the initial 20 days after injection. As illustrated in Fig. 1, the effect of this instability on the dispersion of the SO2 injection impacts not only the oxidation of the SO2 but also the evolution of aerosol burden and size, and consequently the radiative forcing. The reduction of aerosol size can, like that of SO2 lifetime, be attributed to increased dispersion in the ensemble members with high σU,T values. A more confined and concentrated aerosol cloud, as in the ensemble members with a more stable polar vortex, exhibits higher aerosol growth rates, such as in member 6, while more diluted aerosols undergo less growth.
In conclusion, our ensemble of simulations with different initial polar vortex conditions demonstrates that the level of OH exposure experienced by the injected SO2, which is modulated by the initial polar vortex state, considerably affects the radiative forcing resulting from a high-latitude stratospheric volcanic injection in winter. By strengthening the dispersion of the injected SO2, an unstable initial polar vortex reduces the SO2 lifetime, accelerating the peak in volcanic forcing, and limits the growth of aerosols, amplifying the forcing’s magnitude.
Sensitivity to eruption source parameters
To evaluate the sensitivity of volcanic aerosol evolution to eruption source parameters, we analyse the aerosol properties discussed above in a set of sensitivity experiments with varied plume composition, injection height, and injection season (Table 1, Methods). The sensitivity experiments are labelled as in Table 1 and the ensemble mean of the baseline scenario will henceforth be referred to as H-24km, unless stated otherwise.
In H-24km, the SO2 e-folding time is 3 months (Fig. 3a). In all sensitivity experiments, the SO2 e-folding time is reduced relative to this, with the shortest e-folding time of approximately 1.3 months in H-16km-jul and S-16km-jul. Like for varying polar vortex initial conditions, the availability of OH is the primary control on the SO2 lifetime. Changing the injection season and height implicitly changes OH availability due to pronounced seasonal and vertical variations in stratospheric OH concentrations at high latitudes (Supplementary Fig. 1). Consequently, July injections, exposed to higher concentrations of OH, generally exhibit shorter SO2 lifetimes compared to corresponding January injections.

The shading indicates two standard deviations of the H-24km ensemble. a Global SO2 burden anomaly. b Global SO4 burden anomaly and the e-folding times of total sulphur (S, SO2 + SO4). The grey dashed lines in (a, b) indicate the injected sulphur mass divided by e. c Global mean SO4-mass-weighted mean effective radius (Reff). The grey dashed line indicates the peak scattering efficiency for volcanic SO4 as a function of Reff47. d Global mean SAOD550nm anomaly (left axis). The markers indicate the cumulative global mean SAOD550nm anomaly after 5 years, and the error bars represent two standard deviations of the H-24km ensemble (right axis). Note the different time axes.
The co-injection of sulphur and halogens does not result in a significantly slower SO2 oxidation compared to the injection of sulphur alone (Fig. 3a). The SO2 e-folding time in S-24km is within the 2σ-variability of the H-24km ensemble. Similarly, when comparing H-16km and H-16km-jul to their respective sulphur-only counterparts, S-16km and S-16km-jul, there is no noticeable variation in the SO2 e-folding time. This contrasts with previous modelling studies conducted for eruptions at lower latitudes, which suggested that the presence of halogens in conjunction with sulphur can potentially extend the SO2 lifetime due to OH depletion through oxidation of the halides41,42,48. A sensitivity to co-injecting halogens appears between H-24km-jul and S-24km-jul, with a more than 1 month longer SO2 e-folding time in H-24km-jul. However, rather than a local OH depletion slowing SO2 oxidation in H-24km-jul, this difference can be attributed to a dynamical response. In S-24km-jul, the SO2 rapidly ascends to the middle stratosphere due to local heating from longwave and near-infra-red absorption by newly formed SO4 aerosols (e.g., ref. 49) or shortwave absorption by locally enhanced ozone concentrations (Supplementary Fig. 2). This self-lofting effect accelerates SO4 formation due to the strong vertical OH gradient in the high-latitude summer stratosphere (Supplementary Fig. 1), resulting in a considerably shorter SO2 e-folding time. In contrast, self-lofting is prevented in H-24km-jul by the simultaneous stratospheric cooling due to ozone destruction by the volcanic halogens (Supplementary Fig. 2), causing the SO2 to remain approximately at the injection altitude until it is converted into SO4.
The global burden of SO4 reaches its peak between 3 and 8 months after SO2 injection, depending on the eruption source parameters (Fig. 3b). In H-24km, the total sulphur (S, SO2+ SO4) has an e-folding time of 13.8 months, ranging from 12.9 to 14.5 months within the two standard deviations interval. This range is comparable to the sensitivity of the S e-folding time to the injection season and composition, with a range spanning 12.7 months in H-24km-jul to 15.3 months in S-24km. The SO4 lifetime is most strongly reduced in the 16 km injections due to the proximity to the tropopause where removal processes work on the order of days to weeks. Consequently, H-16km, S-16km, H-16km-jul, and S-16km-jul exhibit S e-folding times 38% to 62% shorter than their respective 24 km counterparts, H-24km, S-24km, H-24km-jul, and S-24km-jul.
Sulphur-only injections invariably result in longer S e-folding times compared to sulphur-and-halogen co-injections, for unchanged injection season and altitude. This difference is attributed to self-lofting extending the SO4 lifetime, which in the case of sulphur and halogen co-injections is offset by cooling caused by ozone loss (Supplementary Fig. 2). As a result, the S e-folding time in S-24km is 11% longer compared to H-24km. This effect is also evident in the other sensitivity experiments, where S e-folding times range from 4% to 23% longer in the sulphur-only injections compared to the sulphur-and-halogen co-injections.
After the initial nucleation of SO4 aerosols, Reff increases as the aerosols grow through condensation and coagulation, as shown in Fig. 3c. In H-24km, Reff thus increases during the first year after injection, attaining a maximum value of 0.32 μm after 9 months. Reff thereafter decreases monotonically until it returns to the background mean effective radius of ~0.15 μm.
Co-injection of sulphur and halogens decreases Reff compared to sulphur-only injections. This reduction is a consequence of the shorter SO4 lifetime in the co-injections. Consequently, the difference in Reff between S-24km and H-24km is significant only in the second and third years after injection, when the higher SO4 burden in S-24km has allowed for more aerosol growth by coagulation. The effect is particularly pronounced between S-24km-jul and H-24km-jul, where the maximum Reff values are 0.33 μm and 0.29 μm, respectively, since the aforementioned accelerated aerosol formation in S-24km-jul further adds to the prolonged atmospheric residence time of the SO4. This dependence of Reff on the atmospheric residence time of the SO4 is also evident in the other sensitivity experiments, where the maximum global mean Reff is on average 4% lower in the injections with sulphur and halogen co-injection compared to the sulphur-only injections. For the same reason, the injections at 16 km exhibit an average of ~21% lower maximum global mean Reff compared to the injections at 24 km, with Reff values ranging from 0.23 μm in H-16km to 0.26 μm in S-16km-jul.
The global mean SAOD generally follows the evolution of SO4 burden in each of the sensitivity experiments (Fig. 3d, left axis). However, deviations arise due to differences in Reff, which can amplify or reduce the SAOD depending on its value relative to the radius of maximum scattering efficiency. Furthermore, since the SO4 lifetime, compared to the maximum SO4 burden, is more sensitive to varying eruption source parameters, the differences in cumulative global mean SAOD (Fig. 3d, right axis) are more pronounced than the differences in the maximum values.
The maximum global mean SAOD is largely insensitive to the composition of the injection (Fig. 3d, left axis), since co-injection of sulphur and halogens, compared to sulphur-only injection, primarily acts to reduce the SO4 lifetime and reduce Reff after the time of the maximum SO4 burden. However, in all instances, the sulphur-only experiments exhibit a stronger cumulative forcing because of the prolonged SO4 lifetime (Fig. 3d, right axis). On average, the sulphur-only experiments result in a cumulative SAOD 27% higher than their counterparts with sulphur and halogen co-injection, slightly higher than the 21% 2σ-variability of the H-24km ensemble (error bars).
Injection altitude has the strongest effect on maximum global mean SAOD. The values range from 0.14 in H-16km and S-16km to 0.19 in H-16km-jul and S-16km-jul, compared to 0.20 in S-24km-jul to 0.21 ± 0.02 in H-24km. For the 24 km injections, where the global mean Reff exceeds 0.25 μm during the peak SO4 burden, co-injecting halogens brings Reff closer to the peak scattering efficiency (Fig. 3c). This partially offsets the decrease in global mean SAOD caused by shorter SO4 lifetimes. For the 16 km injections, however, where the global mean Reff is below 0.25 μm during the peak SO4 burden, the reduction in Reff resulting from halogen co-injection further contributes to a decrease in SAOD.
In summary, the sensitivity to eruption source parameters for a high-latitude volcanic stratospheric injection is for several aspects of the aerosol evolution comparable in scale to the variance arising from atmospheric variability. The SO2 lifetime varies by up to 1.7 months for the tested eruption source parameters, which is similar in magnitude to the range within the baseline experiment ensemble (1.6 months). The composition of the volcanic injection has a considerable impact on aerosol evolution, qualitatively independent of varying other eruption source parameters: Compared to sulphur and halogen co-injection, sulphur-only injection results in longer aerosol lifetimes, leading to larger cumulative SAOD. It also results in increased aerosol size. However, this effect is primarily due to the extended aerosol lifetime, and does not typically coincide with the peak aerosol burden. Among the tested combinations of eruption source parameters, injection altitude has the strongest impact on the peak and cumulative SAOD. Despite higher aerosol scattering efficiencies in these experiments, the reduced lifetime due to the proximity to the tropopause leads to considerably smaller cumulative SAOD.
Modelled sulphate deposition and implications for volcanic forcing reconstructions
Surface deposition is the ultimate fate of volcanic aerosols. We here examine the timing, magnitude, and spatial distribution of sulphate deposition over the Greenland ice sheet (GrIS) in our simulations. Additionally, we revisit the assumptions regarding sulphate deposition from extratropical eruptions that existing reconstructions of past volcanic forcing are based on.
The eVolv2k3 and HolVol v.1.04 time series of stratospheric sulphur injections serve as the foundation for volcanic forcing in transient paleoclimate simulations conducted as part of the PMIP4 framework43 and Holocene simulations44. For extratropical NH eruptions (>25°N), the eVolv2k dataset utilises cumulative sulphate deposition rates inferred from three Greenland ice cores, NEEM, NGRIP, and GISP2, to reconstruct stratospheric sulphur injections. HolVol v.1.0 uses a similar approach for extratropical eruptions but is based solely on GISP2, which provides a Holocene-long record.
The eVolv2k and HolVol v.1.0 datasets employ the Greenland transfer function for NH extratropical eruptions45 (LG) to infer the VSSI of these eruptions (MS) from the mean Greenland SO4 flux (fG) obtained from the deposition at the ice core sites listed above. This simple relationship is expressed as
where the factor 3 is the ratio between the molecular mass of SO4 and the atomic mass of S.
Since a large NH extratropical eruption with both a well-observed stratospheric sulphur injection and recorded ice core sulphate deposition is unavailable, LG is currently based on aerosol-climate model simulations of two high-latitude eruptions performed by Oman et al.14,45. As emphasised by the authors of the study, GISS ModelE may not accurately represent all the relevant processes associated with volcanic sulphate deposition. Consequently, the value of LG is poorly constrained and is considered a primary source of uncertainty in the reconstructions3,4. The simulations we present in the current study can potentially improve the estimation of LG, as CESM2-WACCM6 offers a more accurate stratospheric circulation and stratosphere-troposphere-exchange, as well as improved and more finely resolved dry and wet deposition processes and orographic effects over the Greenland ice sheet50,51.
Our simulations can provide insights about and estimates of the uncertainties associated with LG which originate from both atmospheric variability and unknown eruption source parameters. Our estimates build upon previous assessments of uncertainties52,53, aiming to offer additional insight as well as improve our understanding of intermodel disagreements.
Figure 4 a displays the averaged SO4 deposition over the GrIS across all model experiments listed in Table 1. Individual experiments exhibit the same general features of the spatial distribution, with the strongest SO4 deposition along the southern slopes (Supplementary Fig. 3). Being dominated by wet deposition, the SO4 deposition aligns with the spatial pattern of annual precipitation, which is well-represented in the interior of the ice sheet51. Because GrIS precipitation is predominantly determined by orography and the location of the Atlantic storm track, the magnitude of SO4 deposition over the ice sheet is strongly non-uniform. Still, the modelled SO4 deposition at the three coring sites aligns in terms of magnitude, consistent with the NGRIP, NEEM, and GISP2 ice-core SO4 records (Supplementary Fig. 4 in Toohey & Sigl3).

a Average modelled GrIS deposition across all model experiments. The NEEM, NGRIP, and GISP2 ice core drilling sites are indicated, along with the domain used by Gao et al.45 to derive the Greenland transfer function for NH extratropical eruptions employed in eVolv2k and HolVol v.1.0. b Transfer functions derived from the mean deposition at the locations of the three ice cores (circles), over the Gao et al.45 domain (squares), and over the ice sheet (triangles) for each model experiment. The ensemble means are displayed for H-24km and error bars represent one standard deviation. The transfer function derived by Gao et al.45 used in reconstructions (eVolv2k and HolVol v.1.0) and the mean transfer function across all model experiments in this study, derived from the mean deposition at the locations of the three ice cores, are indicated by the horizontal lines. The shaded areas represent one standard deviation. c The mean SO4 deposition rate over the GrIS in the model experiments, using the ice sheet mask displayed in (a). The individual ensemble members of the baseline experiment are shown in Supplementary Fig. 4.
However, the uneven distribution of GrIS sulphate deposition implies that consistency is needed between the methods used to estimate fG and LG. This consistency is currently lacking in eVolv2k and HolVol v.1.0. In Gao et al.45, LG was derived from Eq. (3) using the area-weighted average in the domain spanning 66-82°N latitude and 50-35°W longitude (outlined in Fig. 4a) to calculate the modelled “Greenland ice sheet average deposition", fG. On the other hand, eVolv2k and HolVol v.1.0 average the SO4 deposition across ice cores. As a result, when Eq. (3) is applied in the reconstructions, these different averaging methods may lead to an incorrect assumption that the areas capture the same magnitude of SO4 deposition.
In Fig. 4b, the transfer functions derived from the modelled SO4 deposition in each of our experiments are presented. The transfer functions based on three different methods for calculating fG are displayed: using the average over the three ice core sites (circles), the domain used by Gao et al.45 (squares), and all model grid points over the GrIS (triangles). The choice of method significantly affects the value of the resulting transfer functions, with an average range of 0.15 × 109 km2 across the model experiments and a maximum range of 0.30 × 109 km2 for H-16 km-jul. As a reference of scale, the random error (1σ) attached to the Gao et al.45 estimate of LG in eVolv2k and HolVol v.1.0 was estimated to 0.09 × 109 km2 (illustrated by shading in Fig. 4b). This uncertainty was derived from simulated atmospheric variability in a separate model study of tropical eruptions using the aerosol-climate model MAECHAM5-HAM52.
Variations in the magnitude and spatial pattern of SO4 deposition over the ice sheet between our model experiments result in a range of values for LG. When using the average SO4 deposition values at the three ice core sites, the mean transfer function of all experiments is 0.44 × 109 km2, as indicated by the dashed line in Fig. 4b. This value is 23% smaller than the LG used in eVolv2k and HolVol v.1.0.
We also estimate an uncertainty of LG when averaging over ice core sites which amounts to 0.14 × 109 km2 at the 1σ level. In relative terms, this uncertainty (32%), which accounts for both unknown eruption source parameters and atmospheric variability combined, is twice the magnitude of that currently estimated in eVolv2k and HolVol v.1.0, which only accounts for atmospheric variability. Furthermore, by considering the variance of the baseline experiment ensemble (H-24km) as an estimate of the uncertainty due to atmospheric variability alone, we quantify this at the 1σ level to 12%. This estimate is slightly lower than the previous estimate by Toohey et al.52.
In addition to affecting the magnitude of GrIS SO4 deposition, variations in eruption source parameters also alter the timing of deposition. When considering the average SO4 deposition over the GrIS (Fig. 4c), the differences between the sensitivity experiments are notably larger compared to the variability within the baseline experiment ensemble. For example, in H-24km-jul/S-24km-jul, the SO4 deposition starts to rise approximately 9 months after the injection, while in H-16km/S-16km, it only takes 1−3 months. This variable delay between sulphur injection and GrIS deposition can introduce uncertainty when attempting to precisely date unknown eruptions based on highly resolved ice-core records, particularly for eruptions with no tropospheric component of emissions.
Discussion
We have investigated the evolution and radiative forcing of SO4 aerosols following Northern Hemisphere high-latitude volcanic stratospheric injections of Pinatubo magnitude in a pre-industrial atmosphere, using a high-top chemistry-climate model with prognostic aerosols and full-atmosphere chemistry.
The first part of our analysis focused on the influence of the highly variable Northern Hemisphere stratosphere on these injections during winter. We have identified that the initial condition of the polar vortex plays a crucial role in controlling the lifetime of SO2 and the growth of aerosols, ultimately affecting the SAOD. An unstable polar vortex is associated with enhanced equatorward advection of SO2, resulting in accelerated SO2 oxidation due to increased exposure to OH, as well as reduced aerosol growth due to decreased condensation and coagulation in the diluted aerosol cloud. Conversely, a stable initial polar vortex is associated with a confinement of SO2 to polar latitudes, where low OH levels persist throughout the winter months, resulting in a prolonged SO2 lifetime as well as higher aerosol growth rates. These mechanisms lead to SO2 e-folding times ranging from 2.3 to 3.9 months within the ensemble, and a 2σ-variability of the cumulative radiative forcing of 21%.
In addition, we have tested the aerosol evolution’s dependence on eruption source parameters, including composition of volatiles, injection season, and injection altitude. The impacts of varying composition and season on SO2 lifetime and SAOD are comparable in magnitude to the variability caused by initial conditions, underlining the important role of atmospheric variability in the high-latitude stratosphere of the Northern Hemisphere. The co-injection of sulphur and halogens, compared to sulphur-only injection, results in an average reduction of 21% in cumulative SAOD by inhibiting aerosol self-lofting through ozone depletion-induced stratospheric cooling.
We have revisited the reconstruction of past volcanic forcing of high-latitude eruptions from ice core records of SO4 deposition in light of our modelled Greenland ice sheet deposition. In this regard, we draw attention to challenges associated with the assumptions underlying volcanic forcing reconstructions used in simulations of the last two millennia in PMIP4 and the Holocene. We emphasise the need for consistency in the averaging methods used to derive the Greenland transfer function and calculate the Greenland SO4 flux, given the non-uniform spatial deposition pattern over the Greenland Ice Sheet, and demonstrate how eruption source parameters change the temporal evolution of the ice sheet SO4 deposition. Finally, using the modelled SO4 deposition from our simulations, we derive an estimate of the Greenland transfer function used to interpret ice core deposition from extratropical explosive eruptions in the Northern Hemisphere. Our transfer function estimate is 23% smaller than that used in current volcanic forcing reconstructions3,4 based on earlier model simulations45. Importantly, our results offer a model-derived estimate of the random error associated with the transfer function, which accounts for both atmospheric variability and unknown eruption source parameters. This uncertainty is quantified to 64% at the 2σ level, double the uncertainty assigned to the Greenland transfer function in the eVolv2k and HolVol v.1.0 reconstructions. However, the considerable intermodel disagreement concerning SO4 aerosol evolution and deposition calls for further efforts to constrain the uncertainty.
The findings presented in this study contribute to advancing our understanding of the complex dynamics associated with large high-latitude volcanic eruptions. Specifically, the mechanisms demonstrated here offer insights that can be instrumental in improving predictions of such eruptions’ impacts in the future, based on the atmospheric state during the early stages in combination with the eruption source parameters.
Furthermore, the identified mechanism of the polar vortex’s impact on aerosol dispersion and radiative forcing has implications beyond natural eruptions. It also sheds light on the potential use of deliberate sulphur injections at high latitudes. Understanding the role of the initial polar vortex state in governing the fate of volcanic emissions provides valuable insights into how intentional sulphur injections might interact with atmospheric dynamics in high-latitude regions. This knowledge can inform future research and assessments related to climate intervention strategies, specifically the use of stratospheric aerosol injection in the Arctic.
It is important to consider our model results in the context of the significant intermodel disagreement that exists in simulating volcanic eruptions. The Model Intercomparison Project on the climatic response to Volcanic forcing (VolMIP54) revealed a large variation among models in the SAOD response to large tropical eruptions. Specifically, a previous version of CESM2-WACCM6 exhibited the highest global mean SAOD among the participating models, whereas the aerosol-climate model MAECHAM5-HAM yielded the smallest SAOD22,55. Therefore, in lack of a complete model intercomparison focusing on high-latitude explosive eruptions, we turn to simulations with MAECHAM5-HAM as an indicator of intermodel disagreement. Two 17 Tg SO2, 64°N, 24 km altitude point-injection experiments were run with MAECHAM5-HAM, varying the injection date between January 1 and July 1. These simulations are analogous to our simulations S-24km and S-24km-jul, with the exceptions that MAECHAM5-HAM was run in an atmosphere-only configuration with modern day background conditions (see Toohey et al.10 for details). Comparing the CESM2-WACCM6 and MAECHAM5-HAM simulations (Supplementary Fig. 5) suggests that the model disagreement in the SAOD response to high-latitude eruptions is at least as great as that for tropical eruptions. The model disagreement depends on the season of eruption, with small disagreement between the two models for a January eruption, but substantial differences for a July eruption. The latter is likely attributed to the absence of interactive OH chemistry in MAECHAM5-HAM leading to the formation of very large aerosols10. This seasonality highlights that the atmospheric response to high-latitude eruptions involves processes that are distinctively different from those associated with tropical eruptions. Closing the knowledge gap regarding high-latitude explosive eruptions would therefore benefit greatly from a systematic model intercomparison specifically focusing on high-latitude eruptions, with a primary focus on the source-to-forcing processes related to stratospheric chemistry and aerosol microphysics. Given the importance of aerosol formation and growth for the modelled volcanic forcing, it is worth noting that model parameterisations may have biases in these processes56,57.
Model disagreements also extend to volcanic SO4 deposition, which has important implications for model-derived estimates of transfer functions. Marshall et al.55 showed that the timing and spatial distribution of volcanic SO4 deposition following large tropical eruptions varied considerably among models. In this model ensemble, a previous version of CESM2-WACCM6 simulated one of the lowest magnitudes of GrIS volcanic SO4 deposition. It is therefore possible that the estimate of LG presented in the current work could be among the upper estimates of what the current generation of aerosol-climate models can be expected to produce. However, Marshall et al.53, using statistical emulation based on simulations with the UM-UKCA model, inferred on average almost 50% higher SO2 emissions for the ten largest bi-polar ice core deposition signals compared to the eVolv2k SO2 estimates. A systematic model intercomparison including high-latitude eruptions would shed light on the processes that control volcanic SO4 deposition and enable a better quantification of the intermodel spread in transfer function estimates.
Both the eVolv2k and HolVol v.1.0 datasets supply time series of SAOD in addition to VSSI. The SAOD datasets are provided for modelling groups that seek to avoid the computationally expensive interactive simulation of aerosols. SAOD estimates are generated using the Easy Volcanic Aerosol forcing generator (EVA58), which takes the VSSI estimates as input. EVA is a simplified model designed to achieve agreement with the observed distribution and optical properties of aerosols following the 1991 Mt. Pinatubo eruption. However, discrepancies arise when comparing the SAOD produced by EVA for a 17 Tg SO2 eruption in January at 64°N to the simulations carried out using CESM2-WACCM6 and MAECHAM5-HAM (Supplementary Fig. 5). If CESM2-WACCM6 and MAECHAM5-HAM represent the upper and lower bounds of modelled SAOD estimates, it suggests that EVA may underestimate the magnitude of SAOD for high-latitude eruptions when compared to aerosol-climate models (Supplementary Fig. 5). Additionally, EVA does not replicate the timing and meridional profile of SAOD produced in the aerosol-climate models. While satellite observations indicate that EVA may overestimate SAOD for small extratropical injections near the tropopause58, our results suggest that, for high-altitude injections, EVA may underestimate SAOD. Consequently, for high-latitude eruptions represented in eVolv2k and HolVol v.1.0 with high plume heights, the potential underestimation of SAOD by EVA could partly or completely counterbalance a positive bias in the VSSI estimates resulting from an inflated Greenland transfer function. Therefore, efforts to improve the representation of high-latitude eruptions in paleoclimate simulations should also aim to improve the interpretation of SAOD derived from VSSI for use in models without interactive aerosol simulation59.
The number of eruption source parameters tested in our sensitivity experiments in Section 2.2 is limited by computational cost. Similarly, conducting ensemble runs with different initial conditions for each eruption scenario to assess significance was not feasible with the desired model configuration. Future modelling studies of high-latitude eruptions can help address remaining gaps in the eruption source parameter space. Recent attention has in particular been given to plume composition, including variations in halogen injection efficiencies40,41, volcanic ash60, water vapour61, and the radiative effects of volcanic gases and ash18,62,63,64. These factors have been shown to significantly influence the aerosol evolution after tropical eruptions. In addition, further exploration of the influence of eruption month on the evolution of volcanic volatiles and aerosols could provide valuable insights into the climate effects of high-latitude eruptions8,17 and is worth including in future model intercomparisons54,65.
Continued efforts should also focus on how eruption source parameters control sulphate ice sheet deposition. The magnitude of SO2 injection, which was not included in our set of eruption source parameters, could be particularly important in this regard, as there are indications that dynamical responses can modify deposition patterns in response to strong volcanic forcing52. To obtain a reliable estimate of the random error of LG resulting from unknown eruption source parameters, it is necessary to sample a wide distribution of eruption source parameters that are representative of real volcanic eruptions. Recent studies applying statistical emulation53,66 have demonstrated the potential of alternative approaches for evaluating how eruption source parameters impact both radiative forcing and sulphate deposition.
The primary objective of this article has been to explore the potential impact of the variability prevailing in the high-latitude NH stratosphere on volcanic injections. However, it is important to note that several modes of variability exist within the Earth system that can significantly influence both the distribution of volcanic material and the magnitude of the impacts. For an evaluation of the control exerted by initial conditions on the impacts of both high- and low-latitude volcanic eruptions, we point to a related paper by Zhuo et al.67 with a detailed analysis of how various initial conditions, beyond the ones considered in this article, contribute to shaping volcanic forcing and response.
In summary, our results demonstrate that atmospheric variability and eruption source parameters can strongly modulate the evolution of SO2 and aerosols, the radiative forcing, and SO4 deposition following high-latitude volcanic injections. This must be taken into account in reconstructions of past volcanic forcing as well as future scenarios involving stratospheric sulphur injections, and highlights the considerable potential for improving our understanding of high-latitude volcanic eruptions.
Methods
Earth system model
We use the fully coupled Earth system model Community Earth System Model version 2 (CESM2) with the high-top atmospheric component Whole Atmosphere Community Climate Model version 6 (WACCM6), coupled interactively to CESM2’s ocean, land, and sea-ice models. This fully coupled configuration allows for the accurate representation of Earth system features such as El Niño Southern Oscillation-related teleconnections, sea-ice distribution, biogeochemical cycles, and land-ice surface mass balances51.
WACCM6 uses 70 vertical levels spanning from the Earth’s surface to 6 × 10−6 hPa (~140 km, the lower thermosphere), and is run at the standard horizontal resolution of 0.9° latitude by 1.25° longitude50.
Lower boundary conditions, including concentrations of greenhouse gases and ozone depleting substances, anthropogenic emissions of aerosols and precursor gases, and surface land conditions are fixed at annually repeating 1850 conditions.
The background volcanic forcing in this configuration is accounted for with constant emissions of SO2 and SO4. Explosive volcanic eruptions are represented by three-dimensional SO2 emissions time-averaged over the historical period (1850−2014), while continuously degassing volcanoes are represented by time-averaged observation-based localised surface emissions of SO2 and SO450.
We employ the full-atmosphere chemistry configuration, including reactions relevant for the troposphere, stratosphere, mesosphere, and lower thermosphere. This includes the SOx, ClOx, BrOx, Ox, HOx, and NOx chemical families, totalling 231 solution species and 583 chemical reactions consisting of gas-phase, photolytic, and heterogeneous reactions50,68. WACCM6 thus explicitly accounts for the interactive modelling of ozone and oxidants, including halogens (Br, Cl, F). Activation of halogens results from heterogeneous reactions on aerosols, including stratospheric sulphate, nitric acid trihydrate, and ice as described in Gettelman et al.50, Kinnison et al.69, and Solomon et al.70. Polar stratospheric clouds form interactively and cause denitrification and dehydration in very cold regions. WACCM6 accurately reproduces the observed climatology of ozone as well as its evolution in the 20th and 21st centuries50. Photolysis rates are based on a combination of inline parameterisations and a lookup table approach as described in Kinnison et al.69. The radiatively active gases comprise H2O, O2, CO2, O3, N2O, CH4, and CFCs/HFCs.
In WACCM6, the formation, growth, shrinkage, and deposition of aerosols are simulated interactively in both the stratosphere and troposphere. These processes are represented by the four-mode Modal Aerosol Model (MAM471) which expresses the aerosol size distribution using lognormal modes for Aitken, accumulation, coarse, and primary carbon modes. MAM4 has been specifically adapted to modelling stratospheric sulphate aerosols67, which form through homogeneous nucleation of sulphuric acid and water, and evolve through condensation, evaporation, and coagulation. The model accounts for both the direct effects of aerosols on radiation and the indirect effects, which involve modifications to cloud properties through cloud microphysics. The prognostic treatment of gas-phase volcanic emissions of SO2 into sulphate accurately captures the observed evolution of stratospheric aerosols and their radiative impacts following the 1991 eruption of Mt. Pinatubo21 as well as more recent small-to-moderate eruptions50.
WACCM6 effectively captures middle-atmosphere modes of variability50. It accurately simulates NH stratospheric winter variability, including occurrences of stratospheric sudden warmings, and internally generates a quasi-biennial oscillation in the tropics, demonstrating its capability to replicate important dynamical features.
Experimental setup and simulations
We performed simulations of high-latitude explosive volcanic eruptions using the CESM2-WACCM6 model. We defined a baseline high-latitude explosive volcanic eruption scenario located at the Katla volcanic system in Iceland (64°N, 19°W). In this scenario, 17 Tg of SO2 is co-injected with 2.93 Tg of HCl and 9.5 Gg of HBr on January 1, at 24 km altitude. The chosen SO2 mass and injection altitude are based on observations of the tropical 1991 Mt. Pinatubo eruption72,73. Our baseline scenario is thus a high-latitude analogue to Pinatubo in these aspects. High-latitude eruptions of this magnitude appear to be rare in the past 2500 years3, but are associated with some of the strongest NH climate anomalies10,11.
The selection of Iceland as the eruption location is motivated by its status as the most active volcanic region in the high-latitude Northern Hemisphere, with the Katla system in particular exhibiting a high frequency of explosive eruptions74,75. Additionally, simulating Icelandic eruptions can help inform the interpretation of Greenland ice core SO4 deposition signals from historical Icelandic eruptions, which due to Iceland’s proximity to Greenland often stem from combined stratospheric and tropospheric volcanic sulphur emissions.
The co-injected halogens and their magnitudes are derived from average Central American Volcanic Arc petrological estimates of vent emissions31,32, assuming an injection efficiency of 10% as motivated in Brenna et al.40,41. The injection efficiency is an estimation of the extent of scavenging of the highly soluble halogen gases within the volcanic plume. It is thus worth noting that a scavenging efficiency of 10% may be overly conservative when applied to high-latitude eruptions due to the drier atmospheric column in this region, as also indicated by in-situ observatons38,39. The limited constraints on halogen emissions from high-latitude volcanoes make the Central American Volcanic Arc estimate a suitable option for our idealised eruption scenario.
The eruption date of January 1 is chosen since the NH high-latitude stratosphere displays the strongest variability during winter, which is characterised by variations in the strength and location of the polar vortex. By initialising ensemble members from January 1 of six different years in the control run, we can sample an ensemble of the baseline eruption scenario with six different polar vortex initial states (Table 1). These years are selected based on the zonal mean zonal wind at 10 hPa, 60°N, a commonly used metric for the state of the NH polar vortex76,77. The January mean of this quantity ranges from −1 ms−1 to 39 ms−1 among the selected years (Supplementary Fig. 6), reflecting the climatological distribution. In all members, the injection location (64°N, 19°W) is situated close to the edge of the polar vortex at the injection height (Supplementary Fig. 7). The baseline scenario ensemble allows us to quantify the uncertainty arising from atmospheric variability and identify potential relationships between the polar vortex state and volcanic aerosol evolution.
In addition to the baseline scenario ensemble, we performed a set of sensitivity simulations with varying eruption source parameters (Table 1). We test the sensitivity to the composition of volcanic volatiles, motivated by the results of previous modelling studies on sulphur and halogen co-injection in tropical eruptions, as well as the varying concentrations of halogens in high-latitude magmas and the uncertainty of the stratospheric injection efficiency. Additionally, we test the sensitivity to injection season and injection altitude, considering the strong seasonal and vertical variations in circulation and chemistry of the high-latitude stratosphere.
To carry out the simulations, we used the model configuration described in Section 4.1 and ran a control simulation for 56 years, disregarding the first 20 years as spin-up. The baseline experiment ensemble and sensitivity experiments were initialised from selected dates in the control simulation, and anomalies were calculated as the differences from the control climatology. In all experiments, the volcanic injection is represented as a single-grid box, 1-hour emission starting at midday on either January or July 1. The different model realisations and their corresponding polar vortex initial states and eruption source parameters are summarised in Table 1.
Data availability
The CESM2-WACCM6 data generated and analysed in this paper are archived and available in the NIRD Research Data Archive (https://doi.org/10.11582/2023.00127).
References
Crowley, T. J. Causes of climate change over the past 1000 years. Science 289, 270–277 (2000).
Forster, P. et al. The Earth’s energy budget, climate feedbacks, and climate sensitivity. In Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, 923-1054 (Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2021).
Toohey, M. & Sigl, M. Volcanic stratospheric sulfur injections and aerosol optical depth from 500 BCE to 1900 CE. Earth Syst. Sci. Data 9, 809–831 (2017).
Sigl, M., Toohey, M., McConnell, J. R., Cole-Dai, J. & Severi, M. Volcanic stratospheric sulfur injections and aerosol optical depth during the Holocene (past 11,500 years) from a bipolar ice core array. Earth Syst. Sci. Data 14, 3167–3196 (2022).
Bethke, I. et al. Potential volcanic impacts on future climate variability. Nat. Clim. Change 7, 799–805 (2017).
Man, W. et al. Potential influences of volcanic eruptions on future global land monsoon precipitation changes. Earth’s Future 9, e2020EF001803 (2021).
Schneider, D. P., Ammann, C. M., Otto-Bliesner, B. L. & Kaufman, D. S. Climate response to large, high-latitude and lowlatitude volcanic eruptions in the community climate system model. J. Geophys. Res. Atmos. 114, D15101 (2009).
Toohey, M., Krüger, K., Niemeier, U. & Timmreck, C. The influence of eruption season on the global aerosol evolution and radiative impact of tropical volcanic eruptions. Atmos. Chem. Phys. 11, 12351–12367 (2011).
Wunderlich, F. & Mitchell, D. M. Revisiting the observed surface climate response to large volcanic eruptions. Atmos. Chem. Phys. 17, 485–499 (2017).
Toohey, M. et al. Disproportionately strong climate forcing from extratropical explosive volcanic eruptions. Nat. Geosci. 12, 100–107 (2019).
Burke, A. et al. High sensitivity of summer temperatures to stratospheric sulfur loading from volcanoes in the Northern Hemisphere. Proc. Natl Acad. Sci. USA. 120, e2221810120 (2023).
Stevenson, D. et al. Atmospheric impact of the 1783–1784 Laki eruption: Part I chemistry modelling. Atmos. Chem. Phys. 3, 487–507 (2003).
Oman, L., Robock, A., Stenchikov, G., Schmidt, G. A. & Ruedy, R. Climatic response to high-latitude volcanic eruptions. J. Geophys. Res. Atmos. 110, D13103 (2005).
Oman, L. et al. Modeling the distribution of the volcanic aerosol cloud from the 1783–1784 Laki eruption. J. Geophys. Res. Atmos. 111, D12209 (2006).
Oman, L., Robock, A., Stenchikov, G. L. & Thordarson, T. High-latitude eruptions cast shadow over the African monsoon and the flow of the Nile. Geophys. Res. Lett. 33, L18711 (2006).
Zambri, B., Robock, A., Mills, M. J. & Schmidt, A. Modeling the 1783–1784 Laki eruption in Iceland: 2. climate impacts. J. Geophys. Res. Atmos. 124, 6770–6790 (2019).
Kravitz, B. & Robock, A. Climate effects of high-latitude volcanic eruptions: role of the time of year. J. Geophys. Res. Atmos. 116, D01105 (2011).
Niemeier, U. et al. Initial fate of fine ash and sulfur from large volcanic eruptions. Atmos. Chem. Phys. 9, 9043–9057 (2009).
McConnell, J. R. et al. Extreme climate after massive eruption of Alaska’s Okmok volcano in 43 BCE and effects on the late Roman Republic and Ptolemaic kingdom. Proc. Natl Acad. Sci. USA 117, 15443–15449 (2020).
Bekki, S. Oxidation of volcanic SO2 : a sink for stratospheric OH and H2O. Geophys. Res. Lett. 22, 913–916 (1995).
Mills, M. J. et al. Radiative and chemical response to interactive stratospheric sulfate aerosols in fully coupled CESM1 (WACCM). J. Geophys. Res. Atmos. 122, 13–061 (2017).
Clyne, M. et al. Model physics and chemistry causing intermodel disagreement within the VolMIP-Tambora interactive stratospheric aerosol ensemble. Atmos. Chem. Phys. 21, 3317–3343 (2021).
Brenna, H. et al. Decadal disruption of the QBO by tropical volcanic supereruptions. Geophys. Res. Lett. 48, e2020GL089687 (2021).
Jones, A. C., Haywood, J. M., Jones, A. & Aquila, V. Sensitivity of volcanic aerosol dispersion to meteorological conditions: a Pinatubo case study. J. Geophys. Res. Atmos. 121, 6892–6908 (2016).
Labitzke, K. G. & Van Loon, H. The Stratosphere: Phenomena, History, and Relevance (Springer., 1999).
Symonds, R. et al. Volatiles in magmas. Rev. Mineral. Geochem. 30, 1–66 (1994).
Bureau, H., Keppler, H. & Métrich, N. Volcanic degassing of bromine and iodine: experimental fluid/melt partitioning data and applications to stratospheric chemistry. Earth Planet. Sci. Lett. 183, 51–60 (2000).
Thordarson, T., Self, S., Oskarsson, N. & Hulsebosch, T. Sulfur, chlorine, and fluorine degassing and atmospheric loading by the 1783–1784 AD Laki (Skaftár Fires) eruption in Iceland. Bull. Volcanol. 58, 205–225 (1996).
Heue, K.-P. et al. SO2 and BrO observation in the plume of the Eyjafjallajökull volcano 2010: CARIBIC and COME-2 retrievals. Atmos. Chem. Phys. 11, 2973–2989 (2011).
Augustin, T. D. Les éléments halogènes dans les magmas, du traçage des conditions de stockage aux flux éruptifs. Ph.D. thesis, Sorbonne université (2021).
Kutterolf, S. et al. Combined bromine and chlorine release from large explosive volcanic eruptions: a threat to stratospheric ozone? Geology 41, 707–710 (2013).
Kutterolf, S. et al. Bromine and chlorine emissions from Plinian eruptions along the Central American Volcanic Arc: From source to atmosphere. Earth Planet. Sci. Lett. 429, 234–246 (2015).
Krüger, K., Kutterolf, S. & Hansteen, T. H. Halogen release from Plinian eruptions and depletion of stratospheric ozone. In Volcanism and Global Environmental Change, 244–259 (Cambridge Univ. Press, 2015).
Tabazadeh, A. & Turco, R. P. Stratospheric chlorine injection by volcanic eruptions: HCl scavenging and implications for ozone. Science 260, 1082–1086 (1993).
Halmer, M. M., Schmincke, H.-U. & Graf, H.-F. The annual volcanic gas input into the atmosphere, in particular into the stratosphere: a global data set for the past 100 years. J. Volcanol. Geotherm. Res. 115, 511–528 (2002).
Mather, T. A. Volcanoes and the environment: Lessons for understanding Earth’s past and future from studies of present-day volcanic emissions. J. Volcanol. Geotherm. Res. 304, 160–179 (2015).
Textor, C., Graf, H.-F., Herzog, M. & Oberhuber, J. Injection of gases into the stratosphere by explosive volcanic eruptions. J. Geophys. Res. Atmos. 108, 4606 (2003).
Hunton, D. E. et al. In-situ aircraft observations of the 2000 Mt. Hekla volcanic cloud: Composition and chemical evolution in the Arctic lower stratosphere. J. Volcanol. Geotherm. Res. 145, 23–34 (2005).
Rose, W. I. et al. Atmospheric chemistry of a 33–34 hour old volcanic cloud from Hekla volcano (Iceland): Insights from direct sampling and the application of chemical box modeling. J. Geophys. Res. Atmos. 111 (2006).
Brenna, H., Kutterolf, S. & Krüger, K. Global ozone depletion and increase of UV radiation caused by pre-industrial tropical volcanic eruptions. Sci. Rep. 9, 1–14 (2019).
Brenna, H., Kutterolf, S., Mills, M. J. & Krüger, K. The potential impacts of a sulfur-and halogen-rich supereruption such as Los Chocoyos on the atmosphere and climate. Atmos. Chem. Phys. 20, 6521–6539 (2020).
Staunton-Sykes, J. et al. Co-emission of volcanic sulfur and halogens amplifies volcanic effective radiative forcing. Atmos. Chem. Phys. 21, 9009–9029 (2021).
Jungclaus, J. H. et al. The PMIP4 contribution to CMIP6–part 3: the last millennium, scientific objective, and experimental design for the PMIP4 past1000 simulations. Geosci. Model Dev. 10, 4005–4033 (2017).
Dallmeyer, A. et al. Holocene vegetation transitions and their climatic drivers in MPI-ESM1. 2. Clim. Past. 17, 2481–2513 (2021).
Gao, C., Oman, L., Robock, A. & Stenchikov, G. L. Atmospheric volcanic loading derived from bipolar ice cores: Accounting for the spatial distribution of volcanic deposition. J. Geophys. Res. Atmos. 112, D09109 (2007).
Thordarson, T. & Larsen, G. Volcanism in Iceland in historical time: volcano types, eruption styles and eruptive history. J. Geodyn. 43, 118–152 (2007).
Lacis, A. Volcanic aerosol radiative properties. PAGES Newsl. 23, 50–51 (2015).
Lurton, T. et al. Model simulations of the chemical and aerosol microphysical evolution of the Sarychev Peak 2009 eruption cloud compared to in situ and satellite observations. Atmos. Chem. Phys. 18, 3223–3247 (2018).
Aquila, V., Oman, L. D., Stolarski, R. S., Colarco, P. R. & Newman, P. A. Dispersion of the volcanic sulfate cloud from a Mount Pinatubo–like eruption. J. Geophys. Res. Atmos. 117, D06216 (2012).
Gettelman, A. et al. The whole atmosphere community climate model version 6 (WACCM6). J. Geophys. Res. Atmos. 124, 12380–12403 (2019).
Danabasoglu, G. et al. The Community Earth System Model version 2 (CESM2). J. Adv. Model Earth Syst. 12, e2019MS001916 (2020).
Toohey, M., Krüger, K. & Timmreck, C. Volcanic sulfate deposition to Greenland and Antarctica: a modeling sensitivity study. J. Geophys. Res. Atmos. 118, 4788–4800 (2013).
Marshall, L. R. et al. Unknown eruption source parameters cause large uncertainty in historical volcanic radiative forcing reconstructions. J. Geophys. Res. Atmos. 126, e2020JD033578 (2021).
Zanchettin, D. et al. The model intercomparison project on the climatic response to volcanic forcing (VolMIP): experimental design and forcing input data for CMIP6. Geosci. Model Dev. 9, 2701–2719 (2016).
Marshall, L. et al. Multi-model comparison of the volcanic sulfate deposition from the 1815 eruption of Mt. Tambora. Atmos. Chem. Phys. 18, 2307–2328 (2018).
Rose, C. et al. New particle formation in the volcanic eruption plume of the Piton de la Fournaise: specific features from a long-term dataset. Atmos. Chem. Phys. 19, 13243–13265 (2019).
Sahyoun, M. et al. Evidence of new particle formation within Etna and Stromboli volcanic plumes and its parameterization from airborne in situ measurements. J. Geophys. Res. Atmos. 124, 5650–5668 (2019).
Toohey, M., Stevens, B., Schmidt, H. & Timmreck, C. Easy volcanic aerosol (EVA v1. 0): an idealized forcing generator for climate simulations. Geosci. Model Dev. 9, 4049–4070 (2016).
Aubry, T. J., Toohey, M., Marshall, L., Schmidt, A. & Jellinek, A. M. A new volcanic stratospheric sulfate aerosol forcing emulator (EVA_H): comparison with interactive stratospheric aerosol models. J. Geophys. Res. Atmos. 125, e2019JD031303 (2020).
Zhu, Y. et al. Persisting volcanic ash particles impact stratospheric SO2 lifetime and aerosol optical properties. Nat. Commun. 11, 1–11 (2020).
Millan, L. et al. The Hunga Tonga-Hunga Ha’apai hydration of the stratosphere. Geophys. Res. Lett. 49, e2022GL099381 (2022).
Osipov, S., Stenchikov, G., Tsigaridis, K., LeGrande, A. N. & Bauer, S. E. The role of the SO2 radiative effect in sustaining the volcanic winter and soothing the Toba impact on climate. J. Geophys. Res. Atmos. 125, e2019JD031726 (2020).
Niemeier, U., Riede, F. & Timmreck, C. Simulation of ash clouds after a Laacher See-type eruption. Clim Past 17, 633–652 (2021).
Stenchikov, G. et al. How does a Pinatubo-size volcanic cloud reach the middle stratosphere? J. Geophys. Res. Atmos. 126, e2020JD033829 (2021).
Zanchettin, D. et al. Effects of forcing differences and initial conditions on inter-model agreement in the VolMIP volcpinatubo- full experiment. Geosci. Model Dev. 15, 2265–2292 (2022).
Marshall, L. et al. Exploring how eruption source parameters affect volcanic radiative forcing using statistical emulation. J. Geophys. Res. Atmos. 124, 964–985 (2019).
Zhuo, Z., Fuglestvedt, H. F., Toohey, M., & Kruger, K. Initial conditions control transport of volcanic volatiles, forcing and impacts, EGUsphere [preprint], https://doi.org/10.5194/egusphere-2023-2374 (2023).
Mills, M. J. et al. Global volcanic aerosol properties derived from emissions, 1990–2014, using CESM1 (WACCM). J. Geophys. Res. Atmos. 121, 2332–2348 (2016).
Kinnison, D. et al. Sensitivity of chemical tracers to meteorological parameters in the MOZART-3 chemical transport model. J. Geophys. Res. Atmos. 112, D20302 (2007).
Solomon, S., Kinnison, D., Bandoro, J. & Garcia, R. Simulation of polar ozone depletion: An update. J. Geophys. Res. Atmos. 120, 7958–7974 (2015).
Liu, X. et al. Description and evaluation of a new four-mode version of the modal aerosol module (MAM4) within version 5.3 of the Community Atmosphere Model. Geosci. Model Dev. 9, 505–522 (2016).
Guo, S., Bluth, G. J., Rose, W. I., Watson, I. M. & Prata, A. Re-evaluation of SO2 release of the 15 June 1991 Pinatubo eruption using ultraviolet and infrared satellite sensors. Geochem. Geophys. Geosyst. 5, Q04001 (2004).
Timmreck, C. et al. The interactive stratospheric aerosol model intercomparison project (ISA-MIP): motivation and experimental design. Geosci. Model Dev. 11, 2581–2608 (2018).
Óladóttir, B. A., Larsen, G., Thordarson, T. & Sigmarsson, O. The Katla volcano S-Iceland: Holocene tephra stratigraphy and eruption frequency. J.ökull. 55, 53–74 (2005).
Óladóttir, B. A., Sigmarsson, O., Larsen, G. & Thordarson, T. Katla volcano, Iceland: magma composition, dynamics and eruption frequency as recorded by Holocene tephra layers. Bull. Volcanol. 70, 475–493 (2008).
Labitzke, K. On the interannual variability of the middle stratosphere during the northern winters. J. Meteorol. Soc. Jpn. Ser. II 60, 124–139 (1982).
Butchart, N. et al. Multimodel climate and variability of the stratosphere. J. Geophys. Res. Atmos. 116, D05102 (2011).
Acknowledgements
This research is funded by the Research Council of Norway through the VIKINGS project (grant number 275191). The simulations were performed on resources provided by Sigma2 - the National Infrastructure for High Performance Computing and Data Storage in Norway. We thank Michael Sigl (University of Bern) for comments on the manuscript. Finally, we would like to thank our close colleague and key project contributor Frode Iversen for his enthusiasm and comments on this research.
Author information
Authors and Affiliations
Contributions
H.F.F. conducted the CESM2-WACCM6 simulations, analysed data, wrote the manuscript, and led the discussion with input from all authors. H.F.F., K.K. and Z.Z. designed the CESM2-WACCM6 experiments. M.T. and K.K. designed the MAECHAM5-HAM experiments, and M.T. conducted the simulations. K.K. initiated and led the research project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fuglestvedt, H.F., Zhuo, Z., Toohey, M. et al. Volcanic forcing of high-latitude Northern Hemisphere eruptions. npj Clim Atmos Sci 7, 10 (2024). https://doi.org/10.1038/s41612-023-00539-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41612-023-00539-4
