
Abstract
Herpesviruses are large double-stranded DNA viruses that cause infections in animals and humans with a characteristic of latent infectious within specific tissues. Bats are natural hosts of variety human-infecting viruses and recently have been described as hosts for herpesviruses in several countries around the world. In this study we collected 140 insectivorous bats in the neighboring urban areas of Wuhan City, Hubei Province in the central China between 2020 and 2021. Nested PCR targeting the dpol gene sequence indicated that a total of 22 individuals (15.7% of the sample) tested positive for herpesvirus with 4 strains belonging to the genus Betaherpesvirus and the remaining 18 strains classified as Gammahersvirus. Furthermore, the herpesvirus prevalence in Rhinolophus pusillus was higher at 26.3%, compared to 8.4% in Myotis davidii. The RP701 strain from R. pusillus was the predominant gammaherpesvirus strain detected in bats, accounting for 94.4% (17/18) of all strains. The variations in γ-herpesviruses genomic sequences was evident in phylogenetic tree, where RP701 strain was clustered together with ruminant γ-herpesviruses, while MD704 strain formed a distinct clade with a hedgehog γ-herpesvirus. Four betaherpesviruses exclusively identified from M. davidii, with nucleotide identities ranging from 79.7 to 82.6% compared to known betaherpesviruses. Our study provided evidence that M. davidii can sever as natural host for β-herpesviruses, which extended the host species range. In conclusion, we found that bats from central China harbored novel β-herpesviruses and γ-herpesviruses which were phylogenetically related to ruminant γ-herpesvirus and hedgehog γ-herpesvirus. Our study indicates that bats are natural hosts of β- and γ-herpesviruses and further studies are needed to determine whether there is cross-species transmission of herpesviruses between bats and other animals, or humans.
Similar content being viewed by others

Introduction
Outbreaks of emerging zoonotic diseases have posed significant threats to public health and caused substantial disruptions to the global economy in recent years. Due to their widely distribution and unique social habitats, bats serve as a reservoir for a large variety of pathogens. Cross-species transmission of bat-borne viruses such as SARS-CoV, lyssaviruses, filoviruses, henipaviruses, and Ebola viruses1,2,3,4,5,6,7,8,9 pose significant threats to both livestock health and human well-being.
Herpesviruses, belonging to the family Herpesviridae, are enveloped viruses with a linear double-stranded DNA genome ranging from 124 to 295 kbp. Herpesviruses have been found in mammals, birds, reptiles, amphibians, fishes, and mollusks10,11. All herpesviruses are able to remain latent infection in their natural hosts12,13. Based on biological properties and genome sequence similarities, mammal herpesviruses are classified into three subfamilies, namely Alpha(α)herpesvirinae, Beta(β)herpesvirinae, and Gamma(γ)herpesvirinae, along with a total of 17 genera. The Gammaherpesvirinae subfamily is further divided into Macavirus, Percavirus, Lymphocryptovirus, Bossavirus, Manticavirus, Patagivirus, and Rhadinovirus genera11,14. The Betaherpesvirinae is divided into five genera: Cytomegalovirus, Muromegalovirus, Proboscivirus, Roseolovirus, and Quwivirus14. Betaherpesviruses differ from alpha- and gammaherpesviruses in their restricted host range and long infection cycle. Herpesviruses can cause significant human diseases. The human β-herpesvirus, including human cytomegalovirus (HCMV), human herpesvirus (HHV) 6A, HHV-6B, and HHV-7, can cause severe diseases including encephalitis and cognitive decline in immune-compromised and immune-naive populations15,16. Besides, γ-herpesvirus mainly targeted lymphoid cell lineage causing neoplasias, including human γ-herpesvirus 4 (also known as Epstein-Barr virus) and human γ-herpesvirus 8 (also known as Kaposi’s sarcoma-associated herpesvirus)17.
The first study of bat herpesviruses dated back to 1996, in which Tandler described cytomegalovirus-like particles in salivary glands of the little brown bat (Myotis lucifugus) by light and electron microscopy18. It was not until 2007 that the sequence of the bat herpesvirus was initially characterized12. Since then, many bat herpesviruses sequences have been obtained from diverse bat species worldwide19,20,21,22,23,24. To date, evidences existed that bat γ-herpesviruses, such as the Myotis γ-herpesvirus 8 (BGHV8), could replicate and cause cytopathic effects in bat and other mammalian cells25. The replication of the bat β-herpesvirus was restricted in specific types of cells like kidney cells22.
In this study, we investigated herpesviruses in insectivorous bats collected from Hubei Province in the central China. Upon identifying the herpesviruses, subsequent phylogenetic analyses of the sequences obtained were conducted to unravel epidemiological characteristics.
Materials and methods
Bat collection and bat DNA extraction
Bats were collected from Karst caves in the neighboring urban areas of Wuhan City, including Jingzhou and Xianning, Hubei Province in central China from 2020 to 2021 as described previously26. Bat species were initially identified morphologically27 and the accuracy of species identification was subsequently confirmed by DNA sequencing the PCR amplified cytochrome b (cytB) genes of representative bats of each species28.
Genomic DNA was extracted from intestine and liver tissues of each individual bat using DNA extraction kits (Qiagen, Valencia, CA). The DNA extraction process was carried out according to the manufacturer’s instructions. The experimental procedure could be briefly as follows: after thorough homogenizing the tissue samples, proteinase K and buffer ATL were successively added into the mixture, followed by digestion and incubation processes. Subsequently, the resultant mixture was transferred into a spin column for centrifugation, obtaining a solution containing the genomic DNA. The amount and purity of the DNA were estimated using NanoDrop One equipment (Thermo Scientific, Rockford, CA) and stored at −20 ºC until use.
Detection of bat herpesviruses
Nested PCR assay was performed with degenerate primers targeting herpesviruses DNA polymerase gene (dpol) (approximately 200 bp) for molecular herpesvirus identification29. Further steps were taken employing purpose-designed primers (Round 1: dpol-F11 5′-CGCTAATGAGCTGGCACAAG-3′, dpol-F12 5′-CKSCKWAGACARTCWCCACA-3′, dpol-R11 5′-GAGATGGTCATGTGTGGCGG-3′. Round 2: dpol-F21 5′- HGGGTCTGGRTASGGMARR-3′, dpol-R21 5′-CAGGCTGTTAGTGCCAATGT-3′). To further characterize the herpesviruses, nested PCR targeting 500 bp herpesvirus glycoprotein B gene (gB) were carried out30. PCR products were electrophoresed and purified from 1.5% agarose gels with a gel extraction kit (Tsingke Biotechnology, Beijing, China). The purified PCR products were sequenced bidirectionally using Sanger sequencing. The 5’- and 3’- ends of the sequences derived from primers were trimmed.
Phylogenetic analysis
A BLAST search was conducted (https://blast.ncbi.nlm.nih.gov/Blast.cgi), and the most similar herpesvirus sequences were aligned using the ClustalW algorithm through MEGA X software. Nucleotide sequences were subjected to phylogenetic analysis. The phylogenetic trees were constructed using the Maximum Likelihood (ML) method of the Kimura 2-parameter model in MEGA X31. The bootstrap method (1,000 replicates) was applied to assess the reliability of the tree.
Ethical statement and permits
The study was conducted with the approval of the Ethics Committee of the Medical School, Wuhan University (WHU2020-YF0023), and was in accordance with the ARRIVE guidelines (https://arriveguidelines.org). We confirm that all methods were performed in accordance with the relevant guidelines and regulations. All efforts were made to minimize discomfort to the animals.
Results
Prevalence of herpesvirus in bats
During the period of 2021 to 2022, a total of 140 bats were sampled from the neighboring areas around Wuhan City, including Xianning and Jingzhou, in Hubei Province, China. We investigated the prevalence of herpesvirus in tissue samples (liver and intestines) collected from these insectivorous bats (Table 1), which consisted of two different species, Myotis davidii (n = 83, 59.3%) and Rhinolophus pusillus (n = 57, 40.7%). Within Xianning District, 121 bats specimens were collected with M. davidii constituting 52.9% and R. pusillus accounting for 47.1%. The remaining 19 individuals of M. davidii originated from Jingzhou District.
Nested PCR with dpol primers showed that 15.7% (22/140) bats were herpesvirus positive, including 8.4% (7/83) Myotis davidii and 26.3% (15/57) Rhinolophus pusillus (Table 1). These herpesviruses were further classified into γ-herpesviruses (81.8%, 18/22) and β-herpesviruses (18.2%, 4/22) (Table 2). Among all samples obtained from M. davidii tested positive for herpesvirus, there was only one specimen (MD704) originated from Jingzhou District.
Seven partial dpol gene sequences of herpesviruses were obtained from intestine samples. The sequences were analyzed using the BLASTN and BLASTX algorithms of the NCBI database. Out of the seven bats that tested positive for herpesvirus, three individuals were identified as R. pusillus, whereas the other four were M. davidii. BLAST analysis of three dpol gene sequences from R. pusillus revealed that these sequences were 100% identical to each other, and were classified as the subfamily Gammaherpesvirinae. Interestingly, these sequences were identical to a γ-herpesvirus RB/13YF97 from bat R. blythi in Guangdong Province, which is located in southern China (GenBank: KR261894)32. The other four sequences were obtained from M. davidii with nucleotide identity of > 97% to each other and belonged to the subfamily Betaherpesvirinae. They were most identical to M. emarginatus β-herpesvirus 1 detected in Spain (GenBank: KR608285) with nucleotide identity ranged from 79.7 to 82.6%, showing 69.2 to 82.7% amino acid identity with corresponding protein sequences.
Seventeen partial dpol gene sequences were obtained from bat liver specimens, which belonged to the subfamily Gammaherpesvirinae. According to the pairwise alignment between these sequences, it showed that MD704 strain detected in M. davidii from Jingzhou District shared approximately 80% nucleotide identity with other 16 sequences. MD704 was most identical to the BtHVNeoV4 (GenBank: MF579869) from South Africa, showing 75.2% identity at the nucleotide level and 82.6% amino acid identity, respectively. The remaining 16 dpol gene sequences represented by RP701 was almost identical with each other (nucleotide identities: 99.3%–100%), indicating these bats were infected with the same viral strain. At the same time, this strain was most identical to the γ-herpesvirus RB/13YF9732 (nucleotide identities > 99%). Among these bats, herpesviruses were both identified in the intestine and liver tissues of only two specific individuals, namely RP739 and RP752. In this study, β-herpesviruses obtained only from the intestine tissues of M. davidii.
For samples positive for the dpol gene, we proceeded to amplify the herpesvirus glycoprotein B gene (gB) using nested PCR. The results showed that only two samples (RP701 and RP716) were PCR positive for gB gene (Table 2). BLAST analysis of the gB nucleotide sequences showed that they were almost identical with each other (identity > 99%) and mostly identical (> 98%) to a herpesvirus from the lesser Asiatic yellow house bat in China (GenBank: KR261912).
Phylogenetic analyses
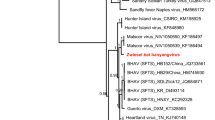
Phylogenetic analysis based on dpol gene sequences showed that γ-herpesviruses from bats in this study were divided into two distinct clades (Fig. 1). Seventeen strains of γ-herpesviruses from this study formed a clade with five known bat γ-herpesviruses from Guangdong Province and Hainan Province, which are located in southern China. It was evident that the γ-herpesviruses were clustered within a same clade, while their corresponding hosts were from different bat species (Rhinolophidae, Vespertilionidae and Hipposideridae) widely distributed across the host phylogenetic tree (Fig. 1b). Additionally, these bat-borne γ-herpesviruses exhibited close evolutionary relationship with ruminant herpesviruses based on the phylogenetic tree (Fig. 1a). It indicated that bat herpesviruses potentially shared a common evolutionary ancestor with herpesviruses from other species, specifically ruminants. MD704 was distinct from all other bat γ-herpesviruses and independently formed a clade with a hedgehog herpesvirus with approximately 73% nucleotide identity. In general, the branches of bat γ-herpesviruses were cross-distributed with human γ-herpesviruses, ruminant γ-herpesviruses, and rodent γ-herpesviruses, indicating the interactive evolution of these γ-herpesviruses among different species.

Phylogenetic trees of Gammaherpesvirus and bats. The tree was constructed based on the dpol gene sequences (160 bp) with 30 representatives of γ-herpesviruses from the GenBank database using the Maximum Likelihood method with 1,000 bootstrap repetitions. Human herpesvirus 7 served as an outgroup. Sequences generated in this study were highlighted in bold (a). The phylogeny of host bats was derived from the cytB sequences (1140 bp) of 31 bats from 6 bat families (Hipposideridae, Rhinolophidae, Phyllostominae, Pteropodidae, Molossidae, and Vespertilionidae) with Dromiciops bozinovici cytB sequences as an outgroup (b). Silhouette images were downloaded from PhyloPic (http://phylopic.org), an open-access database that stores reusable silhouette images of organisms.
In contrast to the γ-herpesviruses, four β-herpesviruses identified from M. davidii were clearly distinct from currently known Vespertilionidae bat β-herpesviruses. Three of four β-herpesviruses displayed a nucleotide identity approximately 98% and formed a clade with higher bootstrap support (Fig. 2). In addition, the cluster of Vespertilionidae bat HV included 2 different clades, one clade mainly containing sequences derived from Myotis sp. and the other one containing viral sequences detected in other Vespertilionidae genera.

Betaherpesvirus phylogenetic tree based on partial dpol gene sequences. The virus detected in this study was highlighted in bold. The tree was constructed using the Maximum Likelihood method with 1000 bootstrap repetitions. Human herpesvirus 7 and human betaherpesvirus 6A served as outgroups.
We further analyzed the phylogeny of the bat γ-herpesviruses based on the gB gene sequences (Fig. 3). The results showed that the gB gene sequences of two γ-herpesviruses (RP701 and RP716) belonged to the genus Lymphocryptovirus, clustered with other known Rhinolophidae and Hipposideridae bat herpesviruses. In addition, these herpesviruses exhibited a close evolutionary relationship as evidenced by the high bootstrap value. Through comparison examination of their gB and dpol sequences, as well as analysis of the phylogenetic trees, it was evident that the two viral strains (RP701 and RP716) were almost identical, strongly suggesting that these two bats were likely infected by the same viral strain.

Phylogenetic analysis based on partial gB gene sequences (150 bp) of Gammaherpesvirus from bats. The virus detected in this study was highlighted in bold. The tree was constructed using the Maximum Likelihood method with 1000 bootstrap repetitions. Mountain gorilla alphaherpesvirus served as an outgroup.
Discussion
In this study, we detected herpesvirus dpol gene sequences in M. davidii and R. pusillus bats collected from neighboring areas around Wuhan City in central China. Nested PCR methods targeting well-conserved genes, such as dpol gene, were designed in order to study the bat-borne herpesviruses. Conserved genes exhibit a high degree of sequence similarity among different viral strains or species. By focusing on these genes, the PCR assays can detect a broad range of relative herpesviruses, increasing the likelihood of identifying known and potentially novel viral strains infecting bats from the central China. Previous studies have reported herpesviruses identified in bats which predominantly inhabit in southern regions of China such as Hainan and Guangdong32, and in the northern regions such as Beijing20, whereas there is limited reporting on their presence in the central China. Our study has verified the existence of multiple γ-herpesvirus and β-herpesviruses in the bat population in the central regions. It extended the geographic distribution of bat herpesviruses.
Currently, Myotis davidii is a predominant bat species in China, with its distribution spanning from central to northern of the country33. The least horseshoe bat, R. pusillus is widely distributed throughout the Indomalayan realm34. The prevalence of γ-herpesviruses in M. davidii (3.6%, 3/83) was lower than that in R. pusillus (21.7%, 15/57) based on dpol gene sequences in this study. Meanwhile, the β-herpesviruses were only found in M. davidii. It indicated that the prevalence and species of herpesviruses in bats might be related to the bat species genetic and habitat. The discovery of γ-herpesviruses and β-herpesviruses in M. davidii and R. pusillus extended the host species of bat herpesvirus.
According to the results of pairwise alignment and phylogenetic analysis of γ-herpesviruses dpol gene sequences, RP701 strain was the major epidemic γ-herpesvirus strain in the central regions, which constituted 94.4% of γ-herpesvirus strains identified in bats in this study. In addition, RP701 was identical to a bat γ-herpesvirus reported previously in Guangdong Province and Hainan Province in southern China32, indicating RP701 has a broad distribution from central China to southern China. It was evident that γ-herpesvirus RP701 strain widely circulating among bat population in the country clustered with ruminant herpesvirus, suggesting these viruses shared a common evolutionary ancestor. By comparing the genomic sequence of MD704 strain and analysis of the phylogenetic tree, it was found that the dpol gene sequence exhibited lower identity with other γ-herpesvirus sequences in the study. The reason for this outcome might be attributed to the different geographic locations and the host species. Each virus in the family Herpesviridae had a restricted host range35, while the branches of bat γ-herpesviruses were distributed among viral sequences from various species. It provided strong indirect evidence supporting that these herpesviruses might have undergo cross-species transmission.
The viral sequences of β-herpesviruses derived from M. davidii were confirmed in the same clade with other herpesviruses from different Vespertilionidae bats based on the phylogenetic analysis. It could be explained by the fact that these novel β-herpesviruses had more than one primary host among the Vespertilionidae, probably caused by close inter-species contact in roosts. Distinct from γ-herpesvirus, β-herpesviruses exhibited a relatively restricted host range, demonstrating a degree of host specificity.
In an extensive comparative analysis of herpesvirus in different host tissues, it revealed that there were organ tropisms for bat herpesvirus. Notably, β-herpesvirus nucleotide sequences were mainly obtained from the intestine tissues. In the case of different tissue samples from the same individual bat, viral sequences could be obtained in liver tissues, but not in the intestinal tissues, and the vice versa. It might stem from two main causes: Firstly, the outcome of detection could be influenced by the methods of sampling and laboratory procedure, such as the site of tissue sampling, storage conditions, and the sensitivity of the detection techniques applied. Secondly, disparities in the distribution and replication dynamics of the virus within different tissues can lead to substantial variations in viral loads among these tissues, meaning that in certain tissues with higher viral loads, detection is more feasible, while in those with lower viral loads, detecting the virus presents greater difficulty.
For samples that tested positive for the dpol gene, we attempted to obtain the corresponding gB gene segments but was only able to successfully obtain two gB gene sequences among the 22 positive samples. It illustrated that the gB gene segments displayed lower sensitivity compared to the dpol gene during the detection process. In addition, co-infections with beta- or gammaherpesviruses were not obtained in this study, but have been reported in primates36. Due to the limitation of sample size, the positive rate and species of bat herpesvirus might be different from the real situation.
In conclusion, we have found that bats from central China harbored β-herpesviruses and γ-herpesviruses similar to ruminant γ-herpesvirus and hedgehog γ-herpesvirus.
Data availability
These sequences of bat β-herpesviruses and γ-herpesviruses obtained in this study were deposited in the GenBank with accession numbers from OP793817 to OP793840. Additional data and information are available from the corresponding author on reasonable request.
References
Leroy, E. M. et al. Fruit bats as reservoirs of Ebola virus. Nature. 438(7068), 575–576 (2005).
Freuling, C. M. et al. Novel lyssavirus in Natterer’s bat, Germany. Emerg. Infect. Dis. 17(8), 1519–1522 (2011).
Lau, S. K. et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. 102(39), 14040–14045 (2005).
Field, H. E., Mackenzie, J. S. & Daszak, P. Henipaviruses: Emerging paramyxoviruses associated with fruit bats. Curr. Top. Microbiol. Immunol. 315, 133–159 (2007).
Li, W. D. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 10(5748), 676–679 (2005).
Han, H. J. et al. Bats as reservoirs of severe emerging infectious diseases. Virus Res. 205, 1–6 (2015).
Weatherman, S., Feldmann, H. & de Wit, E. Transmission of henipaviruses. Curr. Opin Virol. 28, 7–11 (2018).
Mahalingam, S. et al. Hendra virus: An emerging paramyxovirus in Australia. Lancet Infect. Dis. 12(10), 799–807 (2012).
Annan, A. et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 19(3), 456–459 (2013).
Whitley, R.J. Herpesviruses. In Medical Microbiology, 4th edn (ed Baron, S.). Galveston (TX): University of Texas Medical Branch at Galveston; 1996. Chapter 68.
Davison, A. J. Herpesvirus systematics. Vet. Microbiol. 143(1), 52–69 (2010).
Wibbelt, G. et al. Discovery of herpesviruses in bats. J. Gen. Virol. 88(Pt 10), 2651–2655 (2007).
Brock, A. P. et al. A novel gammaherpesvirus in a large flying fox (Pteropus vampyrus) with blepharitis. J. Vet. Diagn. Invest. 25(3), 433–437 (2013).
Gatherer, D. et al. ICTV virus taxonomy profile: Herpesviridae 2021. J. Gen. Virol. 102(10), 001673 (2021).
De Bolle, L., Naesens, L. & De Clercq, E. Update on human herpesvirus 6 biology, clinical features and therapy. Clin. Microbiol. Rev. 18(1), 217–245 (2005).
Sausen, D. G., Reed, K. M., Bhutta, M. S., Gallo, E. S. & Borenstein, R. Evasion of the host immune response by betaherpesviruses. Int. J. Mol. Sci. 22(14), 7503 (2021).
Wada, Y. et al. Detection of novel gammaherpesviruses from fruit bats in Indonesia. J. Med. Microbiol. 67(3), 415–422 (2018).
Tandler, B. Cytomegalovirus in the principal submandibular gland of the little brown bat, Myotis lucifugus. J. Comp. Pathol. 114(1), 1–9 (1996).
Razafindratsimandresy, R. et al. Partial molecular characterization of alphaherpesviruses isolated from tropical bats. J. Gen. Virol. 90(Pt 1), 44–47 (2009).
Wu, Z. Q. et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 86(20), 10999–11012 (2012).
Escalera-Zamudio, M. et al. Bats, primates, and the evolutionary origins and diversification of mammalian gammaherpesviruses. MBio. 7(6), e01425-e1516 (2016).
Zhang, H. J. et al. A novel bat herpesvirus encodes homologues of major histocompatibility complex classes I and II, C-type lectin, and a unique family of immune-related genes. J. Virol. 86(15), 8014–8030 (2012).
Noguchi, K. et al. The complete genomic sequence of Rhinolophus gammaherpesvirus 1 isolated from a greater horseshoe bat. Arch. Virol. 164(1), 317–319 (2019).
Holz, P. H. et al. Virus survey in populations of two subspecies of bentwinged bats (Miniopterus orianae bassanii and oceanensis) in south-eastern Australia reveals a high prevalence of diverse herpesviruses. PLoS ONE. 13(5), e0197625 (2018).
Shabman, R. S. et al. Isolation and characterization of a novel gammaherpesvirus from a microbat cell line. MSphere. 1(1), e0070-e115 (2016).
Li, Z. M. et al. Human-pathogenic relapsing fever borrelia found in bats from central China phylogenetically clustered together with relapsing fever borreliae reported in the New World. PLoS Negl. Trop Dis. 15(3), e0009113 (2021).
Wu, Y. et al. Morphometric varation in the pusillus group of the genus Rhinolophus (Mammalia: Chiroptera: Rhinolophidae) in East Asia. Zool. Sci. 29(6), 396–402 (2012).
Ishii, A. et al. A nairovirus isolated from African bats causes haemorrhagic gastroenteritis and severe hepatic disease in mice. Nat. Commun. 5, 5651 (2014).
Van Devanter, D. R. et al. Detection and analysis of diverse herpesviral species by consensus primer PCR. J. Clin. Microbiol. 34(7), 1666–1671 (1996).
James, S., Donato, D., de Thoisy, B., Lavergne, A. & Lacoste, V. Novel herpesviruses in neotropical bats and their relationship with other members of the Herpesviridae family. Infect. Genet. Evol. 84, 104367 (2020).
Tamura, K. et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28(10), 2731–2739 (2011).
Zheng, X. Y. et al. High prevalence and diversity of viruses of the subfamily Gammaherpesvirinae, family Herpesviridae, in fecal specimens from bats of different species in southern China. Arch. Virol. 161(1), 135–140 (2016).
You, Y. Y. et al. Pleistocene glacial cycle effects on the phylogeography of the Chinese endemic bat species, Myotis davidii. BMC Evol. Biol. 10, 208 (2010).
Wang, J. Y., Zhao, A. & Sun, H. J. The complete mitochondrial genome of the least horseshoe bat (Rhinolophus pusillus). Mitochondrial DNA B Resour. 5(1), 881–882 (2020).
Azab, W., Dayaram, A., Greenwood, A. D. & Osterrieder, N. How host specific are herpesviruses? Lessons from herpesviruses infecting wild and endangered mammals. Annu. Rev. Virol. 5(1), 53–68 (2018).
Prepens, S., Kreuzer, K. A., Leendertz, F., Nitsche, A. & Ehlers, B. Discovery of herpesviruses in multi-infected primates using locked nucleic acids (LNA) and a bigenic PCR approach. Viro J. 4, 84 (2007).
Acknowledgements
This project was supported by grants from the National Natural Science Funds of China (81971939).
Author information
Authors and Affiliations
Contributions
Data curation: S.D., Z.L. Formal analysis: S.D.. Funding acquisition: X.-J.Y. Supervision: X.-J.Y. Visualization: X.Z. Writing-original draft: S.D.. Writing-review & editing: X.Z., X.-J.Y. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Duan, S., Li, Z., Zhang, X. et al. Novel betaherpesviruses and gammaherpesviruses in bats from central China. Sci Rep 14, 10651 (2024). https://doi.org/10.1038/s41598-024-61290-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-61290-1
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
