
Abstract
Resistance to insecticides and adaptation to a diverse range of environments present challenges to Anopheles gambiae s.l. mosquito control efforts in sub-Saharan Africa. Whole-genome-sequencing is often employed for identifying the genomic basis underlying adaptation in Anopheles, but remains expensive for large-scale surveys. Reduced coverage whole-genome-sequencing can identify regions of the genome involved in adaptation at a lower cost, but is currently untested in Anopheles mosquitoes. Here, we use reduced coverage WGS to investigate population genetic structure and identify signatures of local adaptation in Anopheles mosquitoes across southern Ghana. In contrast to previous analyses, we find no structuring by ecoregion, with Anopheles coluzzii and Anopheles gambiae populations largely displaying the hallmarks of large, unstructured populations. However, we find signatures of selection at insecticide resistance loci that appear ubiquitous across ecoregions in An. coluzzii, and strongest in forest ecoregions in An. gambiae. Our study highlights resistance candidate genes in this region, and validates reduced coverage WGS, potentially to very low coverage levels, for population genomics and exploratory surveys for adaptation in Anopheles taxa.
Similar content being viewed by others


Introduction
The Anopheles gambiae species complex is marked by incredible genetic diversity1,2 that has likely contributed to its adaptation to a diverse range of naturally-occurring and anthropogenic ecologies across sub-Saharan Africa. For example, An. coluzzii often breed in water sources associated with human activity (e.g. irrigation ditches, reservoirs, rice fields), and can have elevated pollution-tolerance3,4,5. By contrast, An. gambiae prefer humid environments, are highly anthropophilic, and tend to breed in transient, rain-dependent habitats6. Even within species of the An. gambiae complex, local genetic and ecological adaptation7,8 and subsequent variation in epidemiologically important traits, such as, resilience to aridity7,9,10,11, host preference and resting behaviour12, seasonality and preference of breeding site6,7 all have important implications for the design and implementation of vector surveillance and control programmes13. Moreover, knowledge of how environmental and geographic factors constrain gene flow can help to predict the spread of insecticide resistance (IR)14,15 or genetic control (e.g. gene drives) through a population16.
Genomic signatures of local adaptation often manifest as regions of the genome or polymorphisms displaying elevated genetic differentiation (Fst) among populations17. Whole-genome-sequencing (WGS) enables identification of Fst outlier regions in genome scans18 which, in Anopheles spp., have been instrumental in identifying the genomic determinants of epidemiologically critical traits such as insecticide resistance19, introgression at IR loci between Anopheles taxa15,20, environmental adaptation9,21, and cryptic speciation22,23. However, broad-range surveys indicate that Anopheles populations, particularly those in West Africa, are exceptionally diverse and exhibit little structure over vast spatial scales1,2, suggesting that inference of population genetic structure over fine scales will be difficult. As such, it is possible that with sample sizes sufficiently large to capture representative allele frequencies of massive, diverse, populations, the expense of WGS may often be prohibitive. Low-coverage WGS (lcWGS) is an approach that, by reducing per-individual sequencing coverage, enables sequencing of more individuals and therefore capture of more accurate population-level allele frequencies24,25, while retaining individual information for many analyses that can use genotype information inferred from likelihood-based methods26,27,28 (e.g. PCA, ADMIXTURE, relatedness, inference of inbreeding). lcWGS offers promise especially for analyses relying on allele-frequency estimation—for example, in the exploratory analysis of population structure, and identification of regions of the genome under selection29,30 (e.g. in response to insecticide selection pressures, or local adaptation to environment) as a prelude to further investigation of specific genotypes and populations using deeper WGS. For example, lcWGS was used to identify rapid adaptation in response to fisheries-induced size selection in Atlantic silversides31, and environmental local adaptation at polymorphic inversions in the seaweed fly32. To date, however, this approach has remained untested in Anopheles populations.
Local adaptation and ecological divergence are often associated with transitions between biomes and environmental heterogeneity in Anopheles gambiae5,21, 33,34,35, as well as other mosquitoes. For example in An. funestus, local adaptation to breeding in irrigated rice fields versus natural swamps is concentrated in chromosomal inversions36, as is adaptation to aridity in An. gambiae9,10, 21. Insecticide pressures specific to certain habitats (e.g. land-use) may also confer habitat-specific signals of local adaptation that are related to ecology4,5. For example, different agricultural practices are associated with variations in the frequencies of resistance mechanisms in An. gambiae s.l. in Côte d’Ivoire37. In southern Ghana, differentiation at microsatellite loci between the four main ecozones in the region38: mangrove, savannah, and deciduous and rainforest ecoregions has been reported33, suggesting that local adaptation to ecology may be occurring in this region, though whether this is due to adaptation to the environment, or to selection pressures associated with differential pesticide use, is currently unknown.
Here, we conduct a reduced coverage WGS study of Anopheles coluzzii and Anopheles gambiae samples collected from the four main ecoregions in southern Ghana38 to answer the questions: (1) are there genomic signatures of adaptation to specific ecoregions? (2) Do insecticide resistance loci show signs of structuring by ecoregion? (3) And does lcWGS represent a viable option toward reduced-cost vector WGS studies?
Results
Population structure
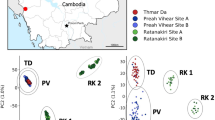
A total of 314 An. gambiae s.l. larvae were sampled from across the four main ecoregions of southern Ghana38, (Fig. 1): Rainforest (RF), Deciduous Forest (DF), Coastal Savannah (CS) and Mangrove Swamp (MS). (N.B. due to a lack of An. gambiae samples from MS, An. gambiae analyses were restricted to CS, DF and RF). Samples were whole-genome-sequenced to a median per-sample depth-of-coverage of ~ 13.7× (Min: 2.8×, Max 52.464×, see Supplementary Table S1). Species PCR (see “Materials and methods” section), followed by PCA correction 7 of mis-assigned samples, and the identification of one potential hybrid (Fig. 2A), identified 162 Anopheles coluzzii individuals and 151 Anopheles gambiae individuals (Supplementary Table S1). Sample counts per-species and per-ecoregion are detailed in Table 1. PCA and ADMIXTURE analysis of all 314 samples disclosed two major clusters corresponding to species assignment (Fig. S1), with one small outgroup of unknown origin.

Sample collection scheme for Anopheles coluzzii (LH Panel) and Anopheles gambiae (RH panel) in Southern Ghana. X axis indicates longitude, y axis indicates latitude, scale bar indicates 200 km distance. Point colour stands for ecoregion (CS coastal savannah, DF deciduous forest, MS mangrove swamp, RF rainforest, respectively).

Panels (A–C) denote population structure of An. coluzzii in Southern Ghana. Principal component analysis (PCA) plots for principal components (PCs) 1 versus 2 (A) and 3 versus 4 (B), with points coloured by sample ecoregion. Panel (C) depicts the most likely value of K in an ADMIXTURE analysis; the Y axis is the admixture proportion of a given cluster (colour) for a single individual (X axis), from one of the four ecoregions sampled (defined in Fig. 1). Panels (D–F) indicate Population structure of An. gambiae in Southern Ghana. Principal component analysis (PCA) plots for principal components (PCs) 1 versus 2 (D) and 3 versus 4 (E), with points coloured by sample ecoregion. Panel (F) depicts the most likely value of K in an ADMIXTURE analysis, whereby the Y axis is the admixture proportion of a given cluster (colour) for a single individual (X axis), from one of the four ecoregions sampled.
To explore population structure and possible correspondence to ecoregion within the two species, we ran PCA and ADMIXTURE on An. coluzzi (Fig. 2A–C) and An. gambiae (Fig. 2D–F) samples. Anopheles coluzzii exhibited a single large cluster corresponding to the majority of the samples from all 4 ecoregions. PC1 and PC2, and PC3 and PC4, show outlier individuals and small subclusters, the most divergent of which came from the DF zone (Fig. 2A,B). ADMIXTURE analysis of chromosome arm 3L GLs from An. coluzzii supported the presence of one major and one minor cluster, with no apparent clustering pattern by ecoregion (Fig. 2C), and only 5 individuals predominantly belonging to Cluster 2, corresponding to the DF outroup samples in 2A and 2B. PCA of An gambiae 3L GLs showed most individuals belonging to one large cluster, with outlying subclusters again corresponding to samples from the DF zone (Fig. 2D,E). ADMIXTURE analysis of chromosome arm 3L GLs from An. gambiae supported the presence of one major and two minor clusters, with no apparent clustering pattern by ecoregion. Unlike An. coluzzii, the minor admixture clusters did not correspond to outlying subgroups in the PCA. (Fig. 2F).
Diversity and differentiation by ecology and species
We characterised genomewide differentiation (Fst) within An. coluzzii and An. gambiae between ecological zones. (Table 2A,B). Each pairwise Fst between An. gambiae ecoregions was higher than those in An. coluzzii (Table 2). In An. coluzzii, Fst between mangrove and non-mangrove ecoregions (Table 2A), tended to be higher than between non-mangrove ecoregions (Table 2A). In An. gambiae, the highest Fst was between reainforest and coastal savannah (0.0271). We calculated the mean genomewide Tajima’s D and π, along with estimated Ne, which are shown in Table 1. Values of mean π varied between 0.019 (An. gambiae DF) and 0.017 (An. coluzzii DF, CS & MS), and those for mean Tajima’s D between -1.099 (An. coluzzii DF) and -1.438 (An. coluzzii CS). Estimates of mean Ne varied between approximately 1.74 million (An. coluzzii MS) and 2.24 million (An. gambiae CS). Anopheles coluzzii π and Ne were generally lower than An. gambiae, and Tajima’s D consistently less negative across comparable ecoregions (Table 1).
We identified a total of 9161 genes located in outlier regions of relatively increased Fst (see “Materials and methods” section) between ecoregions in An. coluzzii and An. gambiae combined (Table S2 The majority (5755–62.8%) of these genes were located as part of a general elevation of Fst in the 2La inversion, making it difficult to identify specific genes therein which are potentially under selection. We identified numerous peaks centred on, or close to, genes implicated in insecticide resistance. Though windows in peaks with the highest Fst were not always those containing IR genes, which were sometimes close or adjacent, we considered it likely that the IR genes might typically be targets of selection (see “Materials and methods” section). On An. gambiae chromosome 2R, we observed a sharp outlier peak centred on the Ace1 locus in all 3 ecoregions, particularly striking for the RF comparisons, and at the Cyp6P complex of genes, between all 3 ecoregions and highest for CS comparisons (Fig. 3). We were unable to identify any specific genes underlying the peak between DF:CS at ~ 40 Mb on chromosome 2R, or in the pericentromeric region between ~ 46–60 Mb. On An. gambiae chromosome 2L, the signal was dominated by the 2La inversion with the highest Fst between CS and RF, suggesting strong contrast in inversion polymorphism frequencies. On 2L, we noted other striking peaks, centred on the Vgsc gene at approximately 2 Mb, and at genes of unknown function (AGAP005300 and AGAP005787) in the comparisons between forest and non-forest regions. On chromosome 3R an outlier Fst peak—at approximately 28 Mb—contained a number of genes of unknown function but is located approximately 20 kb from the epsilon Gst cluster containing Gste2 (Fig. 3). Additional peaks between RF and non-RF zones on chromosome arm 3R are located in the pericentromeric region that contain a range of genes, including potential IR candidates acyl-coA synthetase and Cyp303a1. Clear peaks were less evident on chromosome arm 3L but on chromosome X, two major peaks were obvious, one centred on the diacylglycerol kinase (ATP-dependent) gene at approx 9 Mb, and the other outlier Fst peak centred on the Cyp9k1 gene at approx 15 Mb (Fig. 3). This was the highest region of differentiation genomewide for any of the three ecoregion comparisons in An. gambiae.

Fst genome scan (10 kb windows with 5 kb step) between An. gambiae larvae from deciduous forest (DF), coastal savannah (CS), and rainforest (RF) from southern Ghana. The x-axis indicates chromosomal position (bp). The y-axis indicates Fst, orange point colour indicates that a given window was designated as an outlier. A chromosome ideogram with IR gene positions is plotted at the top. Red shaded region indicates position of the 2La inversion.
For Anopheles coluzzii, (Fig. 4) there were fewer Fst peaks between ecoregions than in An. gambiae. Generally, there appeared to be very few peaks of differentiation between forest (DF:RF) ecoregions (Fig. 4). On chromosome 2R, there were consistent peaks at approx 28 Mb around the Cyp6-complex containing region, and less consistently a peak at ~ 3.4 Mb around the Ace1 locus (Fig. 4). On chromosome 2L, we found a striking peak on 2L between CS and non-CS zones centred on a window containing the Pyruvate dehydrogenase E1 component subunit alpha and alpha-tocopherol transfer protein-like genes (Fig. 4). Against a backdrop of generally higher Fst between CS:RF and other ecoregion comparisons, we found relatively small peaks along 2L, including Vgsc and, notably, no 2La differentiation. In addition to the Vgsc peak, we also saw a small peak between MS and CS at approx. ~ 38 Mb at the AGAP029693 and amiloride-sensitive sodium channel genes. There were very few Fst outlier peaks on 3R and 3L, with outlier regions mainly concentrated in broad peaks in the pericentromeric regions (Fig. 4). The most distinct peak on 3R was a peak at approximately 32 Mb that contained a number of odorant receptor (Or18–53) genes, as well as a small peak at ~ 4.3 Mb that contained a complex of Cyp12F genes)—this peak appeared between CS and non-CS zones in An. coluzzii but was not designated as an outlier in other comparisons outwith CS:RF (Fig. 4). The most notable Fst outlier peaks were consistently between forest (DF/RF) and nonforest (MS/CS) ecoregions at the Cyp9k1 locus, with much lower peaks between DF and RF or MS and CS.

Fst genome scan (10 kb windows with 5 kb step) between An. coluzzii larvae from deciduous forest (DF), coastal savannah (CS), mangrove swamp (MS), and rainforest (RF) from southern Ghana. The x-axis indicates chromosomal position (bp). The y-axis indicates Fst, orange point colour indicates that a given window was designated as an outlier. A chromosome ideogram with IR gene positions is plotted at the top. Red shaded region indicates position of the 2La inversion.
Selection
Fst can identify signatures of selection by looking between populations. We further investigated genomic selection within populations by using H123 scans of per-species and per-ecoregion phased GLs (see “Materials and methods” section). In Anopheles coluzzii, elevated H123 was apparent in the Cyp6-containing region on chromosome arm 2R, at the Vgsc and Rdl—containing regions on chromosome arm 2L, in the epsilon Gst cluster containing Gste2, on 3R, and the Cyp9k1—containing region on chromosome X (Fig. 5A). In H123 scans of An. gambiae, (Fig. 5B) notable peaks included the windows containing the Cyp6 cluster on chromosome 2R, Vgsc, Rdl and Coeaef on chromosome 2L, the Gst-epsilon containing region on chromosome 3R, and the Cyp9k1 and the diacylglycerol kinase (ATP-dependent) genes on chromosome X. Whilst signatures of elevated H123 largely recapitulated the Fst results from Figs. 3 and 4, peak sizes were more consistent across the ecoregions, than when comparing between ecoregion types with Fst, e.g. at the Cyp9k1 locus.

Garud’s H12 scans (in windows of 100 phased GLs) in (A) An. coluzzii and An. gambiae (B) larvae from deciduous forest (DF), coastal savannah (CS), mangrove swamp (MS), and rainforest (RF) (row wise panels) from southern Ghana. The x-axis indicates chromosomal position (bp). The y-axis indicates Garud’s H123. A chromosome ideogram with IR gene positions is plotted at the top. Red shaded region indicates position of the 2La inversion.
Variation at the Cyp9k1 locus
The strongest signals of selection were present at the Cyp9k1 gene. Upon further investigation, we found 27 possible polymorphic sites spanning the locus in An. coluzzi An. gambiae (Table S3). These were a mix of 3’UTR, 5’UTR, synonymous, intronic variants, with two missense variants, (p.Asn224Ile and pVal325Leu) (Table S3).
Geographic population structure
We attempted to identify signatures of geographic population structure through the decay in between-sample kinship with geographic distance (isolation-by-distance) (Fig. S2). The estimated KING kinship coefficient between samples varied between − 2.066 and 0.293, with a median of − 0.250, for An. coluzzii, and − 1.067 and 0.370 with a median of − 0.265 for An. gambiae. We identified 9 full-sib pairs (see “Materials and methods” section) in An. coluzzii and 16 full-sib pairs in An. gambiae. We identified only one full-sib pair from the same site in An. coluzzii, and none in An. gambiae. Mantel tests for isolation-by-distance showed no statistically significant signal of isolation-by-distance for An coluzzi (r = 0.009 p = 0.49) or An gambiae (r = − 0.05 p = 0.90. In addition, we attempted to identify isolation-by-distance using between-site Fst (Fig. 6A), and found that, like relatedness, Mantel tests for isolation-by-distance were insignificant in An. coluzzii (r = 0.0008, p = 0.485) and An. gambiae (r = − 0.05, p = 0.898). However, in Mantel correlograms showing correlation between genetic and geographic distance classes (Fig. 6B), we found evidence of significant isolation-by-distance in An. coluzzii at a distance class between 50 and 100 km (r = 0.14, p = 0.02), but none in An. gambiae (Fig. 6C).

(A) Fst plotted against geographic distance between sites for An. gambiae (blue) and An. coluzzii (red). Mantel correlograms showing spatial correlation values over spatial distance classes for An. coluzzii (B) and An. gambiae (C).
Discussion
The results presented in this study suggest that irrespective of ecoregion there are high levels of gene flow and genetic diversity among relatively unstructured populations of An. gambiae and An. coluzzii across southern Ghana. Isolation by distance was almost absent, with the only significant result coming from comparisons in the 50–100 km bracket in An. coluzzii, and no relationship between kinship and distance in either species. Selection at IR loci appears ubiquitous across all four ecoregions in both species. Observed values of π and Tajima’s D, as well as an apparent lack of genetic structure at a cross-country-wide spatial scale, are consistent with previous WGS data from West African Anopheles gambiae species complex1,2. However, some previous studies indicated much stronger differentiation between ecoregions in the An. gambiae species pair—for example between mangrove and non-mangrove in Ghana33 and between forest and savannah in An. coluzzii2 and An. gambiae34. We find that genomewide differentiation between ecoregions (including between the mangrove swamp ecoregion and other areas) is on the whole low, with differentiation concentrated in specific genomic regions.
We observed numerous signatures of selection localised in specific genomic regions, often linked to genes involved in IR. Most of the outlier Fst-associated genes were concentrated in inversion 2La—a genomic region frequently implicated in environmental adaptation in Anopheles gambiae s.l.10,21 and in other Anopheles taxa36. Aside from this, the notable Fst outlier regions were detected in comparisons between multiple ecoregions in An. coluzzii and included outlier windows in peaks at the Vgsc between all ecoregions, the Cyp9k1 locus—particularly striking between forest and non-forest ecoregions, and the Cyp6 region on chromosome 2R (that was present between all ecoregions in An. coluzzii, but particularly in comparisons involving Coastal Savannah). Peaks in H123 were present around other IR loci (e.g. Gste2). In contrast to the ecoregion-specific outlier peaks; at these loci, the H123 scans showed signatures of selection at Vgsc, Cyp6, Cyp9k1 and Gst-epsilon (consistent with other studies in West Africa15,19, 20, 39, 40) across all four ecoregions in An. coluzzii, suggesting that different alleles of these genes may be selected in different ecoregions. In Anopheles gambiae, genomewide differentiation was higher in comparisons involving rainforest, most notably CS: RF, consistent with at least some structure between forest and savannah as suggested in previous analyses2,9, 34. Interestingly, whilst genomewide mean Fst was similar between RF and CS or MS in An. coluzzii, genomewide outlier profiles showed many more peaks in both species in the CS: RF comparison in both species suggesting possible commonality of selection pressures. RF:non-RF differentiation was particularly marked at the 2La inversion in An. gambiae , as well as at the Cyp9k1 and Ace1 containing regions. The cause of these differences in IR allele frequencies by environment is unknown, but it is noteworthy that the possible selection pressure responsible would likely need to be strong enough to counter the homogenising effect of high gene flow. In the absence of an IRS program, selection on markers responsible for carbamate or organophosphate resistance, notably Ace1, may indicate ecoregion- or geographically-specific selection pressures in response to agricultural usage of pesticides. In both species, the most striking signal was an elevation in Fst between forest and non-forest ecoregion populations, and in all samples for H123, in the genomic region containing the Cyp9k1 locus—a gene under selection in West African Anopheles, implicated in resistance to pyrethroid insecticides2,12, 19, 22, 39, 40, having undergone extensive copy number variation41. The data we present here suggest that although differentiation between ecoregions varies at Cyp9k1 in both species, this region appears to be undergoing a selective sweep in all populations and species.
Further systematic studies incorporating more per-site samples across different ecoregions (the per-site number of samples was generally quite low, between 1 and 10, making accurate sitewise SFS estimation difficult), will enable identification of specific sites where selection pressures may be acting, and studies employing sequencing modalities that enable the resolution of individual genotypes (as opposed to genotype-likelihoods and allele frequencies) will facilitate genotype-environment associations as well as resolution of specific haplotypes involved in IR at loci that appear to be under selection in this study. Reduced sequencing coverage in this study (~ 10× per-sample) prevented us from calling individual genotypes with confidence24. A lack of robustly called genotypes precluded us from investigating the frequency and environmental association of specific genotypes in regions that appeared to be under selection. However, the ability of reduced coverage data to resolve signatures of genomic differentiation and selection, including with phased haplotypes, makes the reduced WGS approach employed here promising for future analyses of vector population genomics where individual genotypes are of less interest. Examples of these may include: the identification of vector dispersal distance with close-kin-mark-recapture and other SFS-based approaches such as population demographic history with coalescent modelling2; interrogation of changes in diversity, population size and selection during vector control42; and exploratory surveys of vector population structure over space and time as a prelude to more in-depth sequencing studies that interrogate genomic regions under apparent selection. This is particularly pertinent given the growing role of genome sequencing in vector surveillance both for discovery of population structure and dynamics, and for control impact and insecticide resistance2,15, 19, 42.
Materials and methods
Sampling and sequencing
Mosquito larvae were collected using dippers from 34 study sites spanning the four major agro-climatic zones in southern Ghana38 (Table S1, Fig. 1). The sampled larvae were collected between April 2016 and October 2017 and raised to adults in an insectary. Genomic DNA was extracted individually from each mosquito using Nexttec kits following the manufacturer’s (nexttec™) protocol. Species characterizations into An. gambiae complexes were performed using the PCR protocol described by Scott et al.43 and further characterized into An. coluzzii and An. gambiae using protocols described by Santolamazza et al.44. The PCR products were visualised under ultraviolet light after electrophoresis using 2% agarose gel stained with Peqgreen dye manufactured by Peqlab Biotechnologie. Eight mosquito samples—of known species—were picked from each site, and in study sites where both An. coluzzii and An. gambiae were found, 16 samples (comprising eight of each species) were chosen. Genomic DNA was submitted for library preparation and WGS at SNPsaurus, Oregon, USA.
GL calling and bioinformatics
Data analyses were performed in R45, incorporating the ggplot2, and geosphere libraries46,47. Read trimming and mapping for all 384 sequenced samples were implemented in nextflow48. Reads were trimmed with fastp49, aligned to the AgamP3 PEST reference genome50 using bwa-mem51, and samtools52 commands sort, markdup, index. ANGSD53 was used to infer genotype likelihoods (GLs) with the samtools genotype likelihood model –GL 1 and the filters –maxDepth 6000 –minQ 30 –minInd 0.25, removing sites with a total depth of > 6000×, a phred-scaled quality score of < 30, and sitewise missingness of > 0.25, the SNP p value of > 0.05 (indicating probable lack of polymorphism), and a minor allele frequency of < 0.05. For π and Tajima’s D estimation, the above parameters were used but without filters for polymorphism (i.e. monomorphic sites were included), depth, or minor allele frequency. GL calling and filtering was performed by species and ecoregion for each of the 5 main chromosomes (2L, 2R, 3L, 3R and X). 70 individuals missing more than 25% of genotype-likelihoods were removed from subsequent analyses, leaving 314 individuals from both species.
Analysis of population structure, differentiation, and diversity
GLs from chromosome 3L were used as input for pcangsd26, which calculates covariance matrices and most likely individual admixture proportions. The covariance matrix was used for principal component analysis (PCA) with the eigen function in R.
Per-subpopulation (e.g. by species and ecoregion) site allele frequencies (saf) were used to calculate folded joint site-frequency spectra (SFS) in realSFS53, with pairwise comparisons between each species and ecoregion per-species to calculate the Fst in 10000 bp sliding windows with a 5000 bp step. One-dimensional (1d) SFS were calculated from the unfiltered GLs for each species and ecoregion using realSFS. Nucleotide diversity (π), theta (for Ne estimation), and Tajima’s D were inferred using the dothetas option. For the relatedness analysis, whole-genome GLs, (with genomic regions containing inversion polymorphisms masked, as these can lead to spurious estimates of relatedness19), were used as input for NGSRelate28. A pairwise relatedness (KING)54 matrix was extracted and compared with a pairwise geographic distance matrix (inferred with the distGeo function from the geosphere package in a Mantel test for isolation by distance with vegan55.
Identification of outlier genes
We identified genomic windows of potential selection by searching genome-scans of Fst for regions of greater than expected differentiation18. Outlier windows were designated according to the approach described here19, but briefly, for each ecoregion:ecoregion comparison, we took the difference between the smallest Fst value, and the modal Fst, and designated as outliers any window with an Fst more than three times this distance away from the mode on the of the right hand side of the Fst distribution. We then intersected outlier windows with the AgamP4 PEST annotation track50,56. We reported IR genes located at, or close to, the window of highest Fst contained in an outlier peak. Windows with the highest Fst in peaks were not always those containing IR genes but, as selection on these genes in response to insecticide pressures is ubiquitous in the region, and occasionally genomic window analysis misses these genes despite their association with resistance phenotypes19, we considered it most likely that these are the genes responsible for the peak and reported them as such. A full list of genes is available at Table S2.
Selection and haplotype analysis
Genotype-likelihoods for each species condition (see GL calling) were phased using BEAGLE v457. The resulting phased GLs were used to calculate Garud’s H12358 in scikit-allel59. Phased GLs from the region containing the Cyp9k1 gene on the X chromosome (AgamP4_X:15,240,572–15,242,864) were analysed for potential functional effects using SNPEff v.4.160, and frequencies of each variant and ecoregion calculated using scikit-allel.
Maps
Maps were plotted in R45, using the rnaturalearth61, and sf62 libraries.
Data availability
The Docker container and Conda env containing all dependencies and versions for read trimming, mapping, and QC are located at https://github.com/tristanpwdennis/basicwgs. Scripts for GL calling, SFS estimation and all subsequent analyses are available at: https://github.com/tristanpwdennis/td_je_angam_2022. Raw reads are deposited in the European Nucleotide Archive under project accession number PRJEB71887.
References
The Anopheles gambiae 1000 Genomes Consortium et al. Genome variation and population structure among 1142 mosquitoes of the African malaria vector species Anopheles gambiae and Anopheles coluzzii. Genome Res. 30, 1533–1546 (2020).
Miles, A. et al. Genetic diversity of the African malaria vector Anopheles gambiae. Nature 552, 96–100 (2017).
Kudom, A. A. Larval ecology of Anopheles coluzzii in Cape Coast, Ghana: Water quality, nature of habitat and implication for larval control. Malar. J. 14, 447 (2015).
Kamdem, C. et al. Anthropogenic habitat disturbance and ecological divergence between incipient species of the malaria mosquito Anopheles gambiae. PLoS ONE 7, e39453 (2012).
Kamdem, C., Fouet, C., Gamez, S. & White, B. J. Pollutants and insecticides drive local adaptation in African malaria mosquitoes. Mol. Biol. Evol. 34, 1261–1275 (2017).
Coetzee, M. et al. Anopheles coluzzii and Anopheles amharicus, new members of the Anopheles gambiae complex. Zootaxa 3619, 246–274 (2013).
Coluzzi, M., Sabatini, A., Petrarca, V. & Di Deco, M. A. Chromosomal differentiation and adaptation to human environments in the Anopheles gambiae complex. Trans. R. Soc. Trop. Med. Hyg. 73, 483–497 (1979).
Ayala, D. et al. Chromosome inversions and ecological plasticity in the main African malaria mosquitoes. Evol. Int. J. Org. Evol. 71, 686–701 (2017).
Cheng, C. et al. Ecological genomics of Anopheles gambiae along a latitudinal cline: A population-resequencing approach. Genetics 190, 1417–1432 (2012).
Cheng, C., Tan, J. C., Hahn, M. W. & Besansky, N. J. Systems genetic analysis of inversion polymorphisms in the malaria mosquito Anopheles gambiae. Proc. Natl. Acad. Sci. 115, E7005–E7014 (2018).
Weetman, D. et al. Candidate-gene based GWAS identifies reproducible DNA markers for metabolic pyrethroid resistance from standing genetic variation in East African Anopheles gambiae. Sci. Rep. 8, 2920 (2018).
Main, B. J. et al. The genetic basis of host preference and resting behavior in the major African malaria vector, Anopheles arabiensis. PLoS Genet. 12, e1006303 (2016).
Ferguson, H. M. et al. Ecology: A prerequisite for malaria elimination and eradication. PLoS Med. 7, e1000303 (2010).
Ismail, B. A. et al. Temporal and spatial trends in insecticide resistance in Anopheles arabiensis in Sudan: Outcomes from an evaluation of implications of insecticide resistance for malaria vector control. Parasit. Vectors 11, 122 (2018).
Clarkson, C. S. et al. The genetic architecture of target-site resistance to pyrethroid insecticides in the African malaria vectors Anopheles gambiae and Anopheles coluzzii. Mol. Ecol. 30, 5303–5317 (2021).
Lukindu, M. et al. Spatio-temporal genetic structure of Anopheles gambiae in the Northwestern Lake Victoria Basin, Uganda: Implications for genetic control trials in malaria endemic regions. Parasit. Vectors 11, 246 (2018).
Lewontin, R. C. & Krakauer, J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 74, 175–195 (1973).
Lotterhos, K. E. & Whitlock, M. C. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol. Ecol. 24, 1031–1046 (2015).
Lucas, E. R. et al. Genome-wide association studies reveal novel loci associated with pyrethroid and organophosphate resistance in Anopheles gambiae and Anopheles coluzzii. Nat. Commun. 14, 4946 (2023).
Grau-Bové, X. et al. Evolution of the insecticide target Rdl in African anopheles is driven by interspecific and interkaryotypic introgression. Mol. Biol. Evol. 37, 2900–2917 (2020).
Love, R. R. et al. Chromosomal inversions and ecotypic differentiation in Anopheles gambiae : The perspective from whole-genome sequencing. Mol. Ecol. 25, 5889–5906 (2016).
Tennessen, J. A. et al. A population genomic unveiling of a new cryptic mosquito taxon within the malaria-transmitting Anopheles gambiae complex. Mol. Ecol. 30, 775–790 (2021).
Crawford, J. E. et al. Evolution of GOUNDRY, a cryptic subgroup of Anopheles gambiae s.l., and its impact on susceptibility to Plasmodium infection. Mol. Ecol. 25, 1494–1510 (2016).
Nielsen, R., Paul, J. S., Albrechtsen, A. & Song, Y. S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 12, 443–451 (2011).
Lou, R. N., Jacobs, A., Wilder, A. P. & Therkildsen, N. O. A beginner’s guide to low-coverage whole genome sequencing for population genomics. Mol. Ecol. 30, 5966–5993 (2021).
Meisner, J. & Albrechtsen, A. Inferring population structure and admixture proportions in low-depth NGS data. Genetics 210, 719–731 (2018).
Vieira, F. G., Fumagalli, M., Albrechtsen, A. & Nielsen, R. Estimating inbreeding coefficients from NGS data: Impact on genotype calling and allele frequency estimation. Genome Res. 23, 1852–1861 (2013).
Hanghøj, K., Moltke, I., Andersen, P. A., Manica, A. & Korneliussen, T. S. Fast and accurate relatedness estimation from high-throughput sequencing data in the presence of inbreeding. GigaScience https://doi.org/10.1093/gigascience/giz034 (2019).
Andrews, K. R. et al. Whole genome resequencing identifies local adaptation associated with environmental variation for redband trout. Mol. Ecol. 32, 800–818 (2023).
Wilder, A. P., Palumbi, S. R., Conover, D. O. & Therkildsen, N. O. Footprints of local adaptation span hundreds of linked genes in the Atlantic silverside genome. Evol. Lett. 4, 430–443 (2020).
Therkildsen, N. O. et al. Contrasting genomic shifts underlie parallel phenotypic evolution in response to fishing. Science 365, 487–490 (2019).
Mérot, C. et al. Locally adaptive inversions modulate genetic variation at different geographic scales in a seaweed fly. Mol. Biol. Evol. 38, 3953–3971 (2021).
Yawson, A. E., Weetman, D., Wilson, M. D. & Donnelly, M. J. Ecological zones rather than molecular forms predict genetic differentiation in the malaria vector Anopheles gambiae s.s. in Ghana. Genetics 175, 751–761 (2007).
Printo, J. et al. Geographic population structure of the African malaria vector Anopheles gambiae suggests a role for the forest-savannah biome transition as a barrier to gene flow. Evol. Appl. 6, 910–924 (2013).
Hemming-Schroeder, E. et al. Ecological drivers of genetic connectivity for African malaria vectors Anopheles gambiae and An. arabiensis. Sci. Rep. 10, 19946 (2020).
Small, S. et al. Standing genetic variation and chromosome differences drove rapid ecotype formation in a major malaria mosquito. PNAS 120, 11 (2023).
Kouadio, F.-P.A. et al. Relationship between insecticide resistance profiles in Anopheles gambiae sensu lato and agricultural practices in Côte d’Ivoire. Parasit. Vectors 16, 270 (2023).
Rhebergen, T. et al. Climate, soil and land-use based land suitability evaluation for oil palm production in Ghana. Eur. J. Agron. 81, 1–14 (2016).
Hearn, J. et al. Multi-omics analysis identifies a CYP9K1 haplotype conferring pyrethroid resistance in the malaria vector Anopheles funestus in East Africa. Mol. Ecol. 31, 3642–3657 (2022).
Vontas, J. et al. Rapid selection of a pyrethroid metabolic enzyme CYP9K1 by operational malaria control activities. Proc. Natl. Acad. Sci. 115, 4619–4624 (2018).
Lucas, E. R. et al. Whole-genome sequencing reveals high complexity of copy number variation at insecticide resistance loci in malaria mosquitoes. Genome Res. 29, 1250–1261 (2019).
Lynd, A. et al. LLIN evaluation in Uganda Project (LLINEUP)—Plasmodium infection prevalence and genotypic markers of insecticide resistance in Anopheles vectors from 48 districts of Uganda. medRxiv https://doi.org/10.1101/2023.07.31.23293323 (2023).
Scott, J. A. et al. IIdentification of single specimens of the Anopheles gambiae complex by the polymerase chain reaction. Am J Trop Med Hyg. 49(4), 520–529. https://doi.org/10.4269/ajtmh.1993.49.520 (1993).
Santolamazza, F. et al. Insertion polymorphisms of SINE200 retrotransposons within speciation islands of Anopheles gambiae molecular forms. Malar J. 7, 163. https://doi.org/10.1186/1475-2875-7-163 (2008).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, 2021). https://www.eea.europa.eu/data-and-maps/indicators/oxygen-consuming-substances-in-rivers/r-development-core-team-2006.
Wickham, H. Create Elegant Data Visualisations Using the Grammar of Graphics (Springer, 2016).
Hijmans, R. J. geosphere: Spherical Trigonometry (2022).
Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Holt, R. A. et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science 298, 129–149 (2002).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 15, 356 (2014).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Oksanen, J. et al. vegan: Community Ecology Package (R Foundation for Statistical Computing, 2000).
Giraldo-Calderón, G. I. et al. VectorBase: An updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 43, D707–D713 (2015).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Garud, N. R., Messer, P. W., Buzbas, E. O. & Petrov, D. A. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 11, e1005004 (2015).
Miles, A. et al. cggh/scikit-allel: v1.3.7. 10.5281/zenodo.8326460.
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms. SnpEff. Fly (Austin) 6, 80–92 (2012).
South, A., Michael, S., & Massicotte, P. rnaturalearthhires: High Resolution World Vector Map Data from Natural Earth used in rnaturalearth (2024)
Pebesma, E. & Bivand, R. Spatial Data Science: With Applications in R 1st edn, 314 (Chapman and Hall/CRC, 2023).
Acknowledgements
We are grateful to Emily Rippon and Dimitra Pipini for laboratory technical support.
Funding
J.E. was supported by a Wellcome Trust Training Fellowship to J.E (Grant No. 110236/Z/15/Z). M.V. and T.D. were supported by the European Research Council under the European Union’s Horizon 2020 Research and Innovation Programme, Grant No. 852957.
Author information
Authors and Affiliations
Contributions
J.E., A.E.Y and D.W. designed the study, J.E. collected the samples, T.D., J.E. and D.W. analysed the data, T.D., J.E., B.K.M., M.V. and D.W. interpreted the data, T.D. and J.E. wrote the manuscript. M.V., B.K.M and D.W. supervised the project. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dennis, T.P.W., Essandoh, J., Mable, B.K. et al. Signatures of adaptation at key insecticide resistance loci in Anopheles gambiae in Southern Ghana revealed by reduced-coverage WGS. Sci Rep 14, 8650 (2024). https://doi.org/10.1038/s41598-024-58906-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-58906-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
