
Abstract
CO2 capture and storage in geological reservoirs have the potential to significantly mitigate the effects of anthropogenic gas emissions on global climate. Here, we report the results of the first laboratory experiments of CO2 injection in continental flood basalts of South America. The results show that the analyzed basalts have a mineral assemblage, texture and composition that efficiently allows a fast carbonate precipitation that starts 72 h after injection. Based on the availability of calcium, chemical monitoring indicates an estimated CO2 storage of ~ 75%. The carbonate precipitation led to the precipitation of aragonite (75.9%), dolomite (19.6%), and calcite (4.6%).
Similar content being viewed by others
Introduction
Mitigating CO2 emission levels is a crucial issue and has been linked to the life conditions of our times1,2,3,4. Carbon Capture and Storage (CCS) methods are one of the most promising technologies for reducing anthropogenic CO2 emissions5,6,7. Among the available strategies, storing carbon in soils and unstable geologic formations (i.e., exhausted hydrocarbon fields, saline aquifers) have critical difficulties related to the long-term stability of the reactions and physical states of CO2, unknown effects arising from geotechnical instabilities and constant risks of erosion and CO2 leakage1,5,7,8,9. In contrast, strategies based on basaltic formations have several advantages for carbon storage, including high reactivity of minerals with CO2, considerable storage potential due to the vast volume of subsurface basalt, and potential for rapid reaction kinetics and long-term storage of CO210,11,12,13.
Permanent CO2 geostorage or mineralization converts carbon dioxide into inert crystals by precipitating carbonate in subsurface basaltic rocks2,3,13. Flood basalts are common igneous rocks on the surface of continents, usually related to Large Igneous Provinces14. The high content of CaO (6–12 wt.%) and MgO (4–10 wt.%) turn basaltic rocks more reactive when compared to siliciclastic sedimentary rocks, mainly in the presence of surface fluids as carbonic acid and even meteoric waters1,12. Available studies demonstrate that natural reactions involving basalts on the Earth’s surface account for 30–35% of natural CO2 sequestration from the atmosphere by weathering15,16. In light of this, major CCS projects currently use basaltic rocks, like Carbfix projects in Iceland2,3,4 and Wallula project in the United States17.
In South America, there are few CCS projects currently in operation18. One region that may develop into a relevant site for CCS projects is the Paraná basin in southern Brazil, where voluminous layers of continental flood basalts19,20 cover an area of 1.2 × 106 km2. The studied basalts represent the main tholeiitic magmatism record in the south portion of the South American platform21,22,23,24, partially covering central and southern Brazil, Uruguay, and Paraguay19,20 (Fig. 1A,B). These basalt lavas are grouped into the Serra Geral Group, which reaches a thickness of 1750 m in the central north portion of the basin20,26,27. They are comprised of heterogeneous lava packages with thin sedimentary interbeds19,20. The more primitive basaltic layers stratigraphically overlie aeolian sandstones, followed by andesitic, dacitic and rhyolitic lavas; the upper stratigraphic unit is formed by a basaltic flow emplaced during the waning phase of volcanic activity24,25,28.

(A) Major geodynamic units of South America. (B) Geology map with the spatial distribution of the Paraná continental flood basalts in the southern portion of the South America platform24.
Although there are previous studies on the potential for CO2 storage in basalts of the Paraná Basin18,21, no published experimental study has monitored the interaction between a dissolved CO2 solution and basalt rock, especially concerning the precipitation of carbonates. Such a laboratory experiment would provide details on the time required for carbon mineralization, the textural and mineral relationships, the estimated CO2 storage yields, and the influence of parameters such as pH, pressure, and the mineral assemblage of basalts.
Methods
Setup for the CO2 mineralization experiment
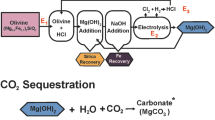
Carbon mineralization occurs when CO2 is dissolved in water and reacts with Ca-rich minerals from basalts to form stable carbonate minerals2,4,6,15. Based on this premise, the material and setup requirements for the experimental mineralization test (Fig. 2) were designed based on data available in the literature and results of the Carbfix projects2,3,5 and Wallula project Columbia River CO2 injections17. In this context, to set up the CO2 mineralization experiment, we used a high-pressure CO2 tank (25 kg) to deliver a pressure of 1 bar (approximately 14.5 psi) of CO2 into a closed water container (100 L) (Fig. 2, steps 1 and 2). The reaction of CO2 with H2O formed carbonic acid and bicarbonate solution according to the following reaction (Eq. 1):

Schematic diagram of the experimental setup to test the reactive potential of South American basalts to CO2 storage. The steps are described in the “Setup for the CO2 Mineralization experiment” section.
Afterwards, the carbonic acid solution was transferred from the pressurized container into a polyvinyl chloride tube (Fig. 2, step 3) previously filled with basalt grains of 800–1200 µm diameter. The carbonic acid solution that percolated through the spaces between the basalt grains was collected daily for analysis in two different ways. The first method was conducted early in the morning after the basaltic grains had interacted with the carbonic acid solution under static conditions for almost 24 h ("closed system"). The second collection was performed after flushing the basalt grains for an hour with a new carbonic acid solution under a flow rate of 1 L/h, 0.8–0.9 bar, and at room temperature (25 °C). We named this second sampling procedure dynamic fluid-rock interaction as the “open system”. In both cases, the solution was collected using a valve placed at the end of the experimental tube (Fig. 2, step 3) and stored in a container (Fig. 2, step 4). This above procedure was repeated for 30 days, after which the tube filled with basalt grains was opened and checked for the presence of precipitated material. The supplementary materials provide detailed information on the analytical procedures used to characterize the basalt before and after interacting with carbonic acid solution.
Sampling, field features and mineral assemblage of basalt before CO2 injection
Geological sampling in central Brazil was conducted to investigate and collect (see location in Fig. 1B) the representative rocks of the Paraná continental flood basalts. The basalts studied for the CO2 mineralization experiment ware collected at the northern limit of the Paraná continental flood basalts (Fig. 1A,B)20,26,27,28. The basalt outcrops display massive layers (Fig. 3A), usually interbedded with layers exhibiting high vesicular content (Fig. 3B). The massive basalt layers display pairs of NE and NW vertical and horizontal fractures (Fig. 3C). The mineral assemblage of the basalt layers includes plagioclase (45–55%), clinopyroxene (15–25%), olivine (2–5%) and Fe–Ti oxides (5–15%) as magnetite and ilmenite (Fig. 3D–F). The basalt samples used in the experiment do not have any veins or discrete carbonate crystals. Plagioclase with 12–16 wt.% CaO and clinopyroxene with 21–23 wt.% CaO (10–12 wt.% MgO) are the major calcium-bearing minerals (7.2–10.8 wt.% CaO in whole rock composition)20,28. Since these minerals represent 60–80% of the reactive surface of basalt (Fig. 3D), the studied crystalline basalts have a high potential for dissolution and release of Ca2+.

Field and petrographic features of studied basalts. (A–C) Basalt layers features in the Jataí region, central Brazil. (D–F) Detailed mineral textures and modal proportion of the basalt mineral assemblage.
Analysis of experimental results
Chemistry monitoring
Chemistry and pH analyses of the carbonic acid solution that percolated into the basalt grains (Fig. 2, step 3) were carried out daily to monitor the dissolution efficiency throughout 30 days (720 h) (Supplementary Data 1). For each analysis, 20 mL samples were collected. The pH analyses indicate that optimal conditions for basalt leaching were achieved at pH = 3.89 (± 0.1)3,29 during the continuous flow regime (pH 3.5–4.5 in the open mode) (Fig. 4A,B). In contrast, under the closed system conditions, we observed higher pH values of 4.6–6.1 (Fig. 4A,B).

pH values of the carbonic acid solution that percolated into the basalt grains during the CO2 injection experiment. An open system represents a constant flow of 1 L/h during 1 h, whereas a closed system represents a stationary mode for 23 h (without constant flow/outflow in the experiment).
Our chemical analyses reveal the release and transport of Ca, Mg and Sr with a leaching peak at 48 h during the open system mode (Fig. 5A–C). After 48 h, the concentrations of these elements in the open system were reduced by around 2–3 times. In contrast, the concentrations in the closed system mode became systematically higher compared to the open system (Fig. 5A–H). It is important to highlight that releasing elements (leaching), such as Ca and Mg, limits the effective CO2 mineralization in basalt4. Our results also indicate that carbonic acid hydrolysis and leaching of basalt were effective and fast (48 h). Only Fe showed long times for leaching (after 288 h, Fig. 5E), possibly because Fe-rich minerals (i.e., oxides and olivine) have slower leaching ratios when compared to Ca- and Mg-rich minerals (i.e., plagioclase and clinopyroxene)29. Another hypothesis is that the higher Fe concentrations recorded during the later phases do not necessarily result from slow dissolution kinetics. Instead, the Fe-rich phases may have either become more accessible to reaction during the latter stages of the experiment or were equally reactive during earlier stages, but secondary mineralization incorporated the dissolved Fe effectively such that Fe concentrations were relatively low during the earlier stages. It is still possible that Ca- and Mg-rich minerals were dissolved to the same degree during the later stages. Still, their lower concentrations during these stages may result from more effective secondary mineralization at this time. Additionally, it is also possible that secondary mineralization that incorporated Ca and Mg was sluggish initially but improved significantly as more nucleation sites became more prevalent throughout the basalt. Indeed, different dissolution and precipitation processes can occur as the fluid percolates through the basalt grains3,5,6. Lastly, an important fact is that on the 18th day, the experiment lost pressure (decompression from 1.0 to 0.5 bar) and, as a direct result, the removal of elements from basalt by carbonic acid was reduced, indicating that the dissolution of basaltic reservoirs increases with pressure (Supplementary Data 1).

Chemistry composition of the carbonic acid solution that percolated into the basalt grains during the CO2 injection experiment. An open system represents a constant flow of 1 L/h during 1 h, whereas a closed system represents a stationary mode for 23 h (without constant flow/outflow in the experiment).
Precipitation formation and characterization
After 72 h (3 days) of the experiment, the carbonic acid solution that percolated through the basalt began to present a thin layer of precipitate at the bottom of the reservoir (Fig. 2, step 4). After 432 h (18 days), a well-defined layer of light brown to beige precipitate was formed (Fig. 6A,B ). Indeed, the pH of this solution increased from 5.15 to 6.36 in the solution reservoir (Fig. 4B), favoring carbonate precipitation2,5,29,30. Furthermore, texture and crystallographic analysis in an optical microscope (Fig. 6C) and scanning electron microscopy (SEM) (Fig. 6D–G) revealed that the precipitate consisted mainly of trigonal calcite crystals with very high birefringence surrounded by a matrix of cryptocrystalline aragonite crystals (Fig. 6D–G). Most crystals were twinned growths of individual crystals that formed pseuohexagonal trilling (Fig. 6F,G). Most precipitated crystals were generally hexagonally shaped due to the twinning like that described for the aragonite-calcite precipitation mechanisms31. After 720 h (30 days) of continuous experiment, the tube filled with basalt grains (Fig. 2, step 3) was opened, and the same precipitate textures were observed on the surface of the basalt grains (Fig. 7A–F). This result was expected since during the closed mode (without continuous flow percolation), the pH usually reached values above 5.5, which allowed carbonate precipitation.

(A–C) Solution reservoir (Fig. 2, step 4) with a thin layer of precipitate after 18 days. (D–G) Backscattered electron images showing that the precipitate consists mainly of trigonal and orthorhombic crystals surrounded by a matrix of cryptocrystalline crystals.

Fourier transform infrared spectrophotometer (FTIR) and X-ray diffraction (XRD)
The FTIR patterns obtained of the precipitated material in the solution reservoir show two well-defined signatures with strong absorption bands between 3020 and 2875, 2626, 1743, 1636, 1418, 730 and 713 cm−1 (Fig. 8A) (Supplementary Data 2). These patterns are compatible with the presence of aragonite + calcite and dolomite32,33. Compared to aragonite + calcite, dolomite displays characteristic FTIR absorptions at 3020 cm−1, 2626 cm−1 and 730 cm−1; the presence of these absorption bands helps indicate the presence of dolomite34 (Fig. 8A). Specifically, the band at 730 cm−1 is related to the in-plane bending mode of CO32−33,34.

(A) FTIR spectra of precipitate formed by aragonite and dolomite. The absorption features at 3020, 2626 and 730 cm−1 are characteristic of dolomite, whereas the other absorption values are characteristics of combined calcite-aragonite and dolomite33,34. The strong absorption bands observed at 1017–990 cm−1 may be associated with glass-fiber wool fragments in the precipitate (*fragments of glass wool used to immobilize the basalt grains were found with the mineral precipitate). (B) X-ray diffractogram of the CO2 precipitate. The black balls represent the experimental points, the solid red line represents the refinement obtained using the Rietveld method, and the solid green line represents the difference between the experimental points and the fit. The bars represent the Bragg angles for the phases found. RP, RWP and REXP represent the goodness of fit.
The X-ray diffraction analyses revealed the presence of at least 6 crystalline phases in the experiment precipitate (Fig. 8B) (Supplementary Data 3 and 4): 36.02% and 6.19% of SiO2 (two quartz phases), 11.18% of CaMg(CO3)2 (dolomite), 43.95% of Ca(CO3) (aragonite), 2.15% and 0.51% of Ca(CO3) (two calcite structures). The quartz (SiO2, 42.21%) is related to the glass wool used during the experiment to retain the basaltic grains in the tube (Fig. 2, step 3). FTIR and XRD results are consistent with the aragonite-calcite and dolomite patterns (Fig. 8A,B)35. Therefore, the CO2 capture experiment yielded a precipitate comprising 75.95% aragonite, 19.34% dolomite, and 4.60% calcite mineral assemblage.
Discussion
Basalt dissolution and CO2 carbonation
Laboratory and field studies have shown that basalt formations could be secure repositories for anthropogenic CO2 emissions1,4,6. Specifically, the CarbFix project mineralized over 60% of the injected CO2 within four months of injection5. Furthermore, increased greenhouse gas injection rates also accelerated the rates of CO2 mineralization4,5,11. In basaltic reservoirs, mineral dissolution rates increase dramatically under low pH conditions near the CO2 injection point of mineral carbonation experiments1,2,4,5. In general, dissolution rates in aluminium-rich minerals and rocks (i.e., labradorite and basalts) are slower at neutral pH and increase again at higher pH3,5,6,36. On the other hand, the rates of aluminium-free minerals, such as olivine (forsterite) and pyroxene (diopside), continuously decrease with increasing pH, so the dissolution of these minerals is slow under conditions in which carbonates tend to precipitate3,5.
Our results also indicate that crystalline basalts have high reactivity due to primary magmatic minerals such as Ca-rich plagioclase and clinopyroxene being more easily dissolved by carbonic acid than supergene minerals4,5,6,11. This implies that supergene and alteration minerals reacted with hydrothermal or weathering fluids, consequently losing the capacity for the maximum release of cations. Basalt's natural porosity, combined with physical stimulation related to CO2 injection, increases the basalt dissolution rates due to increased surface contact between the carbonic acid and host rock10,11. According to the literature, temperature plays a significant role. For instance, an increase from 0 to 100 °C implies a CO2 dissolution ratio increase from 4.5 to 60 times1,4,36. Regarding porosity, the basaltic lava layers have an average porosity of 8%, with higher values at the base and top layers (up to 45%) and lower values at the central portions of the layers (5%)6,20,37.
The general decrease in element contents under a closed system in the first days of the experiment (i.e., without constant fluid percolation) indicates that dissolution of basaltic minerals tends to decrease with increasing pH from 3.6 to 6.23. This suggests that reduction of the carbonic acid flow promotes a balance between the injected solution and host basalt, raising the pH. Since high pH values (5.5 and 6.5) are ideal for carbonate precipitation1,2,37, this process occurs away from the CO2 injection site and under lower fluid pressures3.
Petrographic and XRD analyses indicate that 20% of the precipitate is made of large Mg-calcite crystals (10–30 µm) immersed in a mass made of 80% cryptocrystalline aragonite (< 5 µm) (Figs. 6, 7, 8). These analyses also suggest that aragonite and calcite have quite distinct precipitation kinetics, i.e., the growth rate of aragonite was much higher than that of calcite, as previously reported by other studies31,38. The higher proportion (~ 80%) of cryptocrystalline aragonite crystals (< 5 µm) compared to larger trigonal-rhombohedral dolomite crystals (~ 20%) (10–30 µm) possibly occurs due to aragonite precipitating faster at 25 °C31,38 (Fig. 6D–G). In this reaction, it is also frequent to incorporate divalent cations (i.e., Sr2+) into aragonite38. The incorporation of Mg occurs preferentially in overgrowth crystals of calcite (magnesium calcite) independent of the precipitation rate38. Incorporation of Mg into calcite increases in low-saline (low NaCl content) solutions such as those used during the experiment, which may explain the high concentrations of Ca-dolomite to high Mg-calcite transition38. Indeed, dolomite is more stable than Mg-calcite as pH declines39. Lastly, as mineral composition reflects the susceptibility to basalt dissolution, clinopyroxene leaching can increase the availability of magnesium ions for carbonate precipitation5,6.
CO2 storage yield
The solubility constant (Ksp) of aragonite (6.0 × 10–9) is higher compared to its polymorph, calcite (3.31 × 10–9)40. Therefore, as the molar concentration of Ca during the experiment oscillated from 0.109 to 6.012 mg/L (Supplementary Data 1), a higher proportion of aragonite (75.4%) compared to calcite (4.6%) in the precipitate was not expected (Fig. 8A). However, the coexisting presence of Mg in solution reduces the reaction kinetics of calcite precipitation41,42, which explains the higher volume of aragonite in the precipitate. According to the expected reactions (Fig. 2, step 3), precipitation of aragonite or calcite requires 1 mol of calcium to 2 mol of bicarbonate. Since the aqua-carbonic solution interacted with the basalt, the limiting factors for carbonate precipitation were pH and Ca2+ ions in the solution (minimum Ca values to initiate precipitation were reached 48 h after carbonic acid injection; Fig. 5A). Likewise, with the pH around 3.89 at 1 bar, the mean concentration of CO2 is 1.22 mg/L; therefore, considering this solubility of CO2, the Ca content (removed from basalt) required to initiate precipitation is around 6500 mg/L. Any Ca content near this value tends to initiate carbonate precipitation. Therefore, the higher Ca content in the system did not exceed 6.069 ppm even with the continued input of calcium into the reservoir (Fig. 5A). Calcium availability is a key (limiting factor) issue regarding CO2 mineralization in basalts4, given the continuous injection of CO2. In our experiment, the difference between Ca content under high pressure conditions (6069 mg/L) and low-pressure conditions (1527 mg/L), may be used to estimate the amount of precipitated carbonate. For estimation of the yield, we have considered the following assumptions: (i) Ca2+ ions were the limiting component in the precipitation process; (ii) precipitation of carbonate occurred only after the reaction of Ca2+ and HCO3- ions (approach for chemical estimation purposes); and (iii) yield was estimated based on the difference of Ca2+ ion per unit volume after extraction (initially at 6069 mg/L) and carbonate precipitation (depletion to 1527 mg/L). It implies that at least 74.8% of dissolved Ca reacted with HCO3− to precipitate carbonate 48 h after starting the experiment.
Regarding dolomite, although it presents a lower Ksp (mean of 8.12 × 10–18), this mineral constitutes a volume of 19.6% in the CO2 precipitate, reflecting its more complex kinetics (known as the “dolomite problem”)42 combined to Mg availability (2.43 × 10–3 to 1.803 mg/L; Supplementary Data 1) during the experiment for its formation (Ca2+ + Mg2+ + 2CO32− ⇌ CaMg(CO3)2). Uncertainty in the determination of Ksp and the scarcity of present-day dolomite formation is to be expected because of the known kinetic inhibition of precipitation of dolomite at low temperatures41,42.
CO2 storage potential in South America
The dissolution of the CO2 in water before or during injection results in immediate solubility trapping1,4,5,43. Since the injected gas-charged fluid is denser than the CO2-free water, it tends to sink rather than rise to the surface, reducing the risk of leaks2,13,37,43. Although CO2 dissolution requires a significant amount of water (Eq. 1), the method is simple and cost-effective2,4,7. Carbonic acid is a natural solution formed elsewhere on Earth by reacting water and dissolved CO2. Indeed, carbonic acid is found on rainwater and plays a major role in weathering processes and the mobility of metals on the Earth’s surface43,44. Furthermore, throughout the history of the Earth, approximately 99.9% of CO2 has been removed from the atmosphere through the weathering of rocks44,45. The bicarbonate solutions resulting from this weathering are transported by rivers to the sea, where marine organisms convert them into carbonate rocks44,45,46. Likewise, the higher the pressure conditions, the lower the mass of water required and the higher the CO2 dissolution rates, which accelerates the mineral carbonation process3,4,5,10,13.
Geological factors such as reactivity, interconnectivity, fracture systems, pre-existing fluid (water or hydrocarbon), and structural and stratigraphic traps are determinants for CO2 storage in basaltic reservoirs1,2,3,6,10. Thus, the first experimental results show that Paraná continental flood basalts have mineralogical, chemical, and petrophysical properties that are efficient for rapid and definitive CO2 geostorage. Indeed, the CarbFix results indicate that 72 ± 5% of the injected CO2 was mineralized to carbonate minerals8. Lastly, according to our experiment, Ca-rich mineral assemblage (~ 75% of the basalt), formed mainly by plagioclase and Ca-rich clinopyroxene, releases Ca2+ under pH values around 3.89, 1.0–0.5 bar, and at 25 °C. We further show that carbonate precipitation occurs at pH values between 5.52 and 6.14 under low CO2 pressure conditions.
Conclusion
The carbonic acid reacted with basalt and formed stable carbonate minerals such as aragonite-calcite (CaCO3) (80%) and dolomite (MgCa(CO3)2) (20%). Thus, the proposed experiment efficiently converted CO2 from a gas phase into a crystalline solid permanently stored in the subsurface. Furthermore, our findings demonstrate that converting CO2 into carbonate minerals within basalt rocks takes only a few days. Likewise, this is the first experiment demonstrating the safe long-term storage potential of anthropogenic CO2 emissions through precipitation of carbonates in the Paraná continental flood basalts of South America.
References
Gislason, S. R. & Oelkers, E. H. Carbon storage in basalt. Science 344(6182), 373–374 (2014).
Matter, J. et al. Rapid carbon mineralization for permanent disposal of anthropogenic carbon dioxide emissions. Science 352, 1312–1314 (2016).
Snæbjörnsdóttir, S. Ó. et al. Carbon dioxide storage through mineral carbonation. Nat. Rev. Earth Environ. 1, 90–102 (2020).
Gunnarsson, I. et al. The rapid and cost-effective capture and subsurface mineral storage of carbon and sulfur at the CarbFix2 site. Int. J. Greenh. Gas Control 79, 117–126 (2018).
Clark, D. E. et al. CarbFix2: CO2 and H2S mineralization during 3.5 years of continuous injection into basaltic rocks at more than 250 °C. Geochim. Cosmochim. Acta 279, 45–66 (2020).
Menefee, A. H., Giammar, D. E. & Ellis, B. R. Permanent CO2 trapping through localized and chemical gradient-driven basalt carbonation. Environ. Sci. Technol. 52(15), 8954–8964 (2018).
Raza, A., Glatz, G., Gholami, R., Mahmoud, M. & Alafnan, S. Carbon mineralization and geological storage of CO2 in basalt: Mechanisms and technical challenges. Earth-Sci. Rev. 229, 104036 (2022).
Pogge von Strandmann, P. A. E. et al. Rapid CO2 mineralisation into calcite at the CarbFix storage site quantified using calcium isotopes. Nat. Commun. 10, 1983 (2019).
Rosenqvist, M. P. et al. Reservoir properties and reactivity of the Faroe Islands Basalt Group: Investigating the potential for CO2 storage in the North Atlantic Igneous Province. Int. J. Greenh. Gas Control 123, 103838 (2023).
Matter, J. & Kelemen, P. Permanent storage of carbon dioxide in geological reservoirs by mineral carbonation. Nat. Geosci. 2, 837–841 (2009).
Bashir, A. et al. Comprehensive review of CO2 geological storage: Exploring principles, mechanisms, and prospects. Earth-Sci. Rev. 249, 104672 (2024).
Jia, J. et al. Ab Initio molecular dynamics study of carbonation and hydrolysis reactions on Cleaved Quartz (001) surface. J. Phys. Chem. C 2019(123), 4938–4948 (2019).
Gadikota, G. Carbon mineralization pathways for carbon capture, storage and utilization. Commun. Chem. 4, 23 (2021).
Prasanth, M. P. M., Shellnutt, J. G. & Lee, T.-Y. Secular variability of the thermal regimes of continental flood basalts in large igneous provinces since the Late Paleozoic: Implications for the supercontinent cycle. Earth-Sci. Rev. 226, 103928 (2022).
Gaillardet, J., Dupré, B., Louvat, P. & Allègre, C. J. Global silicate weathering and CO2 consumption rates deduced from the chemistry of the large rivers. Chem. Geol. 159, 3–30 (1999).
Dessert, C., Dupré, B., Gaillardet, J., Francois, L. M. & Allègre, C. J. Basalt weathering laws and the impact of basalt weathering on the global carbon cycle. Chem. Geol. 202, 257–273 (2003).
White, S. K. et al. Quantification of CO2 mineralization at the Wallula basalt pilot project. Environ. Sci. Technol. 54(22), 14609–14616 (2020).
Pelissari, R. M., Relva, G. S., Peyerl, D. Possibilities for carbon capture, utilization, and storage in Brazil. In Energy Transition in Brazil. The Latin American Studies Book Series (eds. Peyerl, D. et al.) (Springer, 2023).
Hawkesworth, C., Kelley, S., Turner, S., Le Roex, A. & Storey, B. Mantle processes during Gondwana break-up and dispersal. J. Afr. Earth Sci. 28(1), 239–261 (1999).
Rossetti, L. et al. Lithostratigraphy and volcanology of the Serra Geral Group, Paraná-Etendeka Igneous Province in Southern Brazil: Towards a formal stratigraphical framework. J. Volcanol. Geotherm. Res. 355, 98–114 (2018).
Ketzer, J. M., Machado, C. X., Rockett, G. X., Iglesias, R.S. Brazilian Atlas of CO2 Capture and Geological Storage. (EDIPUCRS, 2015).
Almeida, F. F. M., Brito Neves, B. B. & Carneiro, C. D. R. The origin and evolution of the South American Platform. Earth-Sci. Rev. 50, 77–111 (2000).
Armijo, R., Lacassin, R., Coudurier-Curveur, A. & Carrizo, D. Coupled tectonic evolution of Andean orogeny and global climate. Earth-Sci. Rev. 143, 1–35 (2015).
Ferreira, A. C. D., Conceição, R. V. & Mizusaki, A. M. P. Mesozoic to Cenozoic alkaline and tholeiitic magmatism related to West Gondwana break-up and dispersal. Gondwana Res. 106, 15–33 (2022).
Thiede, D. S. & Vasconcelos, P. M. Paraná flood basalts: Rapid extrusion hypothesis confirmed by new 40Ar/39Ar results. Geology 38, 747–750 (2010).
Janasi, V. A., Freitas, V. A. & Heaman, L. H. The onset of flood basalt volcanism, northern Paraná Basin, Brazil: A precise U-Pb baddeleyite/zircon age for a Chapecó-type dacite. Earth Planet. Sci. Lett. 302(1–2), 147–153 (2011).
Rocha-Júnior, E. V. et al. Re-Os isotope and highly siderophile element systematics of the Paraná continental flood basalts (Brazil). Earth Planet. Sci. Lett. 337–338, 164–173 (2012).
Peate, D. W., Hawkesworth, C. J. & Mantovani, M. S. Chemical stratigraphy of the Paraná lavas (South America): Classification of magma types and their spatial distribution. Bull. Volcanol. 55(1–2), 119–139 (1992).
Gudbrandsson, S., Wolff-Boenisch, D., Gíslason, S. R. & Oelkers, E. H. An experimental study of crystalline basalt dissolution from 2 < pH < 11 and temperatures from 5 to 75°C. Geochim. Cosmochim. Acta 75, 5496–5509 (2011).
Yadav, K., Sircar, A. & Bist, N. Carbon mitigation using CarbFix, CO2 plume and carbon trading technologies. Energy Geosci. 4(1), 117–130 (2023).
Giampouras, M. et al. On the controls of mineral assemblages and textures in alkaline springs, Samail Ophiolite, Oman. Chem. Geol. 533, 119435 (2020).
Farmer, V. C. Infrared spectra of minerals. In Mineralogical Society Monograph No. 4 (ed. Farmer, V.C.) 399 (Mineralogical Society, 1974).
Ji, J., Ge, Y., Balsam, W., Damuth, J. E. & Chen, J. Rapid identification of dolomite using a Fourier Transform Infrared Spectrophotometer (FTIR): A fast method for identifying Heinrich events in IODP Site U1308. Mar. Geol. 258, 60–68 (2009).
Nguyen, T. T., Janik, L. J. & Raupach, M. Diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy in soil studies. Aust. J. Soil Res. 29, 49–67 (1991).
Hossain, M. S. & Ahme, S. Crystallographic characterization of naturally occurring aragonite and calcite phase: Rietveld refinement. J. Saudi Chem. Soc. 27(3), 101649 (2023).
Oelkers, E. H., Gislason, S. R. & Matter, J. Mineral carbonation of CO2. Elements 4(5), 333–337 (2008).
Alfredsson, H. A. et al. The geology and water chemistry of the Hellisheidi, SW-Iceland carbon storage site. Int. J. Greenh. Gas Control 12, 399–418 (2013).
Flügel, E. & Munnecke, A. Microfacies of Carbonate Rocks: Analysis, Interpretation and Application 2nd edn. (Springer, 2010).
Nash, M. C., Adey, W. & Harvey, A. S. High magnesium calcite and dolomite composition carbonate in Amphiroa (Lithophyllaceae, Corallinales, Rhodophyta): Further documentation of elevated Mg in Corallinales with climate change implications. J. Phycol. 57(2), 496–509 (2021).
Ball, J. W., Nordstrom, D. K. User’s manual with revised thermodynamic data base and test cases for calculating speciation of major, trace and redox elements in natural waters, U.S.G.S. Open-File Report 90-129 (1991).
Antao, S. M., Mulder, W. H., Hassan, I., Crichton, W. A. & Parise, J. B. Cation disorder in dolomite, CaMg(CO3)2, and its influence on the aragonite + magnesite ↔ dolomite reaction boundary. Am. Mineral. 89, 1142–1147 (2004).
Bénézeth, P., Berninger, U.-N., Bovet, N., Schott, J. & Oelkers, E. H. Experimental determination of the solubility product of dolomite at 50–253 °C. Geochim. Cosmochim. Acta 224, 262–275 (2018).
Olsson, J., Stipp, S. L. S., Makovicky, E. & Gislason, S. R. Metal scavenging by calcium carbonate at the Eyjafjallajökull volcano: A carbon capture and storage analogue. Chem. Geol. 384, 135–148 (2014).
Stewart, E. M. et al. Carbonation and decarbonation reactions: Implications for planetary habitability. Am. Mineral. 104(10), 1369–1380 (2019).
Kump, L. R., Brantley, S. L. & Arthur, M. A. Chemical weathering, atmospheric CO2, and climate. Annu. Rev. Earth Planet. Sci. 28, 611–667 (2000).
Dunsmore, H. E. A geological perspective on global warming and the possibility of carbon dioxide removal as calcium carbonate mineral. Energ. Convers. Manage 33, 565–572 (1992).
Acknowledgements
This project was supported by Repsol Sinopec Brasil company. This article is part of the first author’s post-doctoral studies developed at the Universidade de Brasília. The authors would like to acknowledge the XRD laboratory at the Universidade de Brasília (http://drxif.unb.br/) for providing the equipment and technical support for X-Ray Diffraction experiments. We gratefully acknowledge support of the RCGI—Research Centre for Gas Innovation, hosted by the University of São Paulo (USP) and sponsored by FAPESP—São Paulo Research Foundation (2014/50279-4 and 2020/15230-5) and Repsol Sinopec Brasil, and the strategic importance of the support given by ANP (Brazil’s National Oil, Natural Gas and Biofuels Agency) through the R&D levy regulation.
Author information
Authors and Affiliations
Contributions
The authors, A.F., R.V.S., T.S.A., M.A.C., J.A.F., C.R.M., S.T.A.P., A.D.T.B., C.C.G.T., V.A.R. and G.G.C., accept and declare the availability of data so that it can be upload in Scientific Reports system as well as the manuscript text. All data generated or analysed during this study are included in this published article and its supplementary information files.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferreira, A., Santos, R.V., de Almeida, T.S. et al. Unraveling the rapid CO2 mineralization experiment using the Paraná flood basalts of South America. Sci Rep 14, 8116 (2024). https://doi.org/10.1038/s41598-024-58729-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-58729-w
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
