
Abstract
Herein, we report the synthesis of ZnFe2O4@SiO2@L-lysine@SO3H as a green, novel magnetic nanocatalyst, containing the sulfuric acid catalytic sites on the surface of zinc ferrite as the catalytic support. The physical and chemical properties of raw and modified samples (ZnFe2O4@SiO2@L-lysine@SO3H) were characterized by TGA, EDX, PXRD, Map, and FTIR analyses. The prepared nanocatalyst has excellent catalytic activity in synthesizing the oxidation of sulfides to the sulfoxides and Synthesis of pyrazolyl (Bis(pyrazolyl)methane) derivatives under green conditions. This designed nanocatalyst offers several advantages including the use of inexpensive materials and high yield, simple procedure, and commercially available. The synthesized mesoporous nanocatalyst was recovered and reused in five continuous cycles without considerable change in its catalytic activity.
Similar content being viewed by others

Introduction
In recent years, the development of green and environmentally friendly catalytic methods, along with a unique design to improve the reaction process, has attracted the attention of scientists1,2,3,4. In modern research, the recovery and reusability of catalysts is an important challenge because the used catalysts are often very expensive or economically and medicinally valuable5,6. Despite the widespread use of these catalysts, the leaching of toxic or expensive metals is one of the negative aspects of using heterogeneous metal-based catalysts in sustainable catalysis phenomena7,8,9. To overcome this problem, the coupling of green catalysts with heterogeneous magnetic materials as catalytic supports for organic reactions seems to be a suitable solution10,11. In the past decade, the use of magnetic nanoparticles as catalytic support in the preparation of catalysts in green methods has been considered by scientific researchers12,13. As a main member of the ferrite family, ZnFe2O4 has promising potential for use as novel catalytic support14,15,16. The ZnFe2O4 MNPs have attracted much attention due to their magnetic properties, abundant resources, environmental intimacy, nontoxicity, and phase stability17,18,19.
Most solid-state acids are heterogenized organic acids and transition metal complexes or acidic ion-exchange polymer resins. In recent years, various types of solid-acid catalysts, i.e. silica sulfuric acid (SSA), magnetic silica sulfuric acid, and boehmite silica sulfuric acid, have been developed using Zolfigol's method20.
Recently, pyrazoles and their derivatives have received great attention due to a broad spectrum of pharmacological and biological activities21,22. One of the most important ring systems pyrazoles is created in the composition of five-membered rings containing two groups of nitrogen23.
Diphenyl sulfides and their derivatives are important in medicinal chemistry, biologically active molecules, and intermediates in organic synthesis24. It should be noted that sulfide compounds have wide applications in the treatment of diseases such as Alzheimer's, Parkinson's, cancer, and HIV25,26,27.
In this paper, regarding the advantages of magnetic nanocomposite and their high efficiency in the synthesis of organic compounds, we report the synthesis of ZnFe2O4@SiO2@L-lysine@SO3H NPs. Also, we introduced a novel, reusable, eco-friendly, and green magnetically ZnFe2O4@SiO2@L-lysine@SO3H composite as a recoverable magnetic catalyst for the efficient oxidation of sulfides and synthesis of pyrazolyl derivatives in short reaction times.
Experimental
Materials
All required materials for the synthesis of catalysts, reagents, and solvents have been purchased from Merck or Fluka.
Preparation of ZnFe2O4@SiO2@L-lysine@SO3H
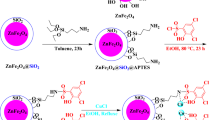
The ZnFe2O4 and ZnFe2O4@SiO2 MNPs were prepared according to our previous methods respectively28,29. In the next step, the ZnFe2O4@SiO2 (0.5 g) was dispersed in 60 mL DI (H2O) by sonication for 45 min. After vigorous stirring for 45 min, 1.5 mmol of L-lysine was added to the reaction mixture which was stirred at 60 °C degrees for 22 h. The product was separated by an external Neodymium magnet and washed with Ethanol and H2O and dried in an oven at 65 °C degrees to give ZnFe2O4@SiO2@L-lysine composite. Finally, to prepare ZnFe2O4@SiO2@L-lysine@SO3H, the obtained ZnFe2O4@SiO2@L-lysine (1 gr) were added to the flask and dispersed ultrasonically for 30 min in dry hexane (35 mL). Chlorosulfonic acid (0.4 mL) was added dropwise to a cooled ice-bath dispersion of ZnFe2O4@SiO2@L-lysine for 35 min. Chlorosulfonic acid was slowly added to the reaction mixture at cool temperature. Then, the reaction mixture was subjected to continuous stirring for 24 h, while the residual HCl was eliminated by suction. The product was then separated from the reaction mixture by an external Neodymium magnet and washed several times with dried hexane. Finally, ZnFe2O4@SiO2@L-lysine@SO3H was dried under vacuum at 60 °C (Fig. 1).

Synthesis of ZnFe2O4@SiO2@L-lysine@SO3H.
Synthesis procedure for pyrazolyl derivatives
A mixture of ethyl acetoacetate (2 mmol), phenylhydrazine (2 mmol), and substituted aromatic aldehydes (1 mmol) and ZnFe2O4@SiO2@L-lysine@SO3H (0.015 g) at 80 °C under solvent-free conditions for 15 min. After completion of the reaction (checked by TLC), the reaction mixture was diluted with hot EtOH to dissolve the organic products, the catalyst was separated using a magnet and the resultant unrefined pyrazolyl products were further purified through recrystallization in the EtOH Fig. 2.

The preparation of the pyrazolyl model reaction.
A general procedure for the oxidation of sulfides
A combination of sulfide (0.5 mmol) and H2O2 (0.15 mL) containing ZnFe2O4@SiO2@L-lysine@SO3H composite as catalyst (0.03 g) was stirred under solvent-free conditions at 25 °C. The progress of the reaction was monitored by TLC. Upon the completion of the reaction, the ZnFe2O4@SiO2@L-lysine@SO3H were separated by a magnet, and the products were extracted by DI (H2O) and EtOAc. The organic phase was dried with Na2SO4 (Fig. 3).

Oxidation of sulfides to sulfoxides catalyzed by ZnFe2O4@SiO2@L-lysine@SO3H.
Selected NMR data
4,4'-(Pyridin-3-ylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.32 (m, 6H), 4.85 (s, 1H), 6.24–7. 69 (m, 14H), 14.06 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 11.9, 33.6, 104.5, 118.6, 120,3, 125.5, 129.5, 135.2, 137.5, 139.1, 142.5, 147.5, 155.8, 156.1, 158.2 ppm. FT-IR (KBr) cm−1: 756, 1032, 1427, 1497, 1609, 3486.
4,4'-(Thiophen-2-ylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.30 (m, 6H), 4.89 (s, 1H), 6.80–7. 75 (m, 13H), 14.06 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 10.8, 34.6, 121.0, 124.1, 124.9, 126.6, 127.1, 128.1, 128.3, 129.2, 138.0, 142.5, 146.1, 149.0 ppm. FT-IR (KBr) cm−1: 753, 1380, 1504, 1575, 1600, 2835, 3065.
4,4'-((4-Nitrophenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.32 (m, 6H), 5.10 (s, 1H), 6.80–8.15 (m, 14H), 13.95 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 11.0, 35.4, 120.4, 123.3, 126.2, 128.9, 130.0, 138.2, 146.2, 147.0, 150.7 154.8, 155.4, 158.3, 159.9 ppm. FT-IR (KBr) cm−1: 756, 1345, 1520, 1605, 2930,3075, 3475.
4,4'-((3-Nitrophenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol
1H NMR (250 MHz, DMSO): 2.29 (m, 6H), 5.24 (s, 1H), 7.29 (t, J = 7.5 Hz, 2H), 7.44 (m, 5H), 7.56 (m, 2H), 7.61 (t, J = 7.5 Hz, 3H), 8.03 (s, 2H), 13.99 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 10.5, 34.1, 120.5, 120.9, 122.4, 126.7, 129.7,137.3, 137.6 137.9, 139, 145.6, 147.8, 148.3, 157.4 ppm. FT-IR (KBr) cm−1: 698, 762, 1351, 1528, 1604, 3086, 3464. Mass analysis: Calculated: M/Z = 481.18, Obtained: M/Z = 481.0
4,4'-((3-Fluorophenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.34 (m, 6H), 5.26 (s, 1H), 7.05–7.59 (m, 14H), 14.20 (s, br, 2H) ppm13C NMR (62.5 MHz, DMSO): 11.4, 33.7, 120.0, 123.2, 1235.1, 125.3, 129.4,129.8, 135.1, 139.1, 145.2, 147.6, 148.8, 150.4, 155.0 ppm.. FT-IR (KBr) cm−1:750, 1267, 1423,1495, 1607, 2930, 3078, 3375.
4,4'-((3-Methoxyphenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.19 (m, 6H), 3.84 (s, 3H), 5.22 (s, 1H), 7.15–7. 27 (m, 8H), 7. 43 (t, J = 7.5, 4H), 7.72 (t, J = 7.5, 2H), 14.44 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 13.7, 35.1, 58.2, 111.4, 112.5, 120.2, 120.7, 126.0, 126.9, 128.9, 130.4, 137.9, 142.0, 142.7, 149.1, 156.2 ppm.
4,4'-((3-Bromophenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.30 (m, 6H), 4.66 (s, 1H), 7.29–7. 56 (m, 14H), 14.04 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 11.7, 35.1, 120.3, 120.4, 125.3, 126.5, 128.1, 130.2, 130.4, 137.9, 146.1, 149.3, 156.2, 156.5, 157.0 ppm.. FT-IR (KBr) cm−1:761, 1053, 1427, 1497, 1590, 3087, 3453.
4,4'-(Phenylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.32 (m, 6H), 4.64 (s, 1H), 7.36–7. 58 (m, 15H), 14.00 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 10.2, 35.9, 105.5, 120.0, 125.9, 127.4, 128.5, 129.7, 134.8, 137.4, 142.6, 147.0, 155.1, 157.0, 158.3 ppm.
4,4'-((4-Hydroxyphenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.30 (m, 6H), 4.58 (s, 1H), 6.82–7. 57 (m, 13H), 8.80 (s, 1H) 14.00 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 9.0, 34.6, 110.5, 115.4, 119.9, 120.3, 125.2, 127.1, 129.1, 132.5, 137.9, 146.2, 156.4, 157.1 ppm. FT-IR (KBr) cm−1: 761, 1503, 1582, 1601, 3177, 3409.
4,4'-((2,4-Dichlorophenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.25 (m, 6H), 5.07 (s, 1H), 7.24–7.67 (m, 13H), 13.80 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 10.6, 33.9, 120.6, 125.1, 127.0, 128.2, 129.5, 131.9, 132.2, 137.1, 139.3, 147.1, 155.5, 157.2, 158.5, 159.8 ppm.
4,4'-((2-Methoxyphenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.29 (m, 6H), 4.49 (s, 1H), 6.79–7.68 (m, 14H), 14.08 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 11.8, 35.3, 110.8, 114.2, 118.7, 1201.4, 126.4, 129.2, 134.4, 137.5, 144,6, 146.7, 151.1, 156.3, 16.3 ppm. FT-IR (KBr) cm-1: 754, 1031, 1502, 1605, 3464.
4,4'-(p-Tolylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.25 (m, 6H), 4.87 (s, 1H), 7.11–7.66 (m, 14H), 13.91 (s, br, 2H) ppm. FT-IR (KBr) cm−1:751, 805, 1027, 1289, 1498, 1606, 3460.
4,4'-((4-Methoxyphenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.29 (m, 6H), 3.69 (s, 3H), 4.68 (s, 1H), 6.82–7.68 (m, 14H), 13.91 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 13.0, 34.6, 57.0, 112.8, 122.2, 122.7, 125.5, 127.3, 129.9, 134.5, 137.3, 141.9, 156.2, 158.2, 160.1 ppm.. FT-IR (KBr) cm−1:758, 1045, 1278, 1502, 1606, 3074, 3522.
4,4'-((4-Chlorophenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.30 (m, 6H), 4.56 (s, 1H), 7.28–7.69 (m, 14H), 13.92 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 11.8, 34.1, 118.5, 121.0, 126.1, 128.3, 129.1, 137.5, 138.5, 147.0, 1455.9, 157.3, 158.1, 159.0 ppm. FT-IR (KBr) cm−1: 750, 1293, 1415, 1498, 1607, 3499.
4,4'-((3-Chlorophenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.26 (m, 6H), 5.12 (s, 1H), 7.25–7.54 (m, 14H), 13.93 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 10.8, 32.5, 120.3, 121.1, 126.8, 127.2, 128.8, 129.2, 130.3, 131.5, 132.2, 137.6, 139.5, 146.4, 157.4 ppm.FT-IR (KBr) cm−1:694, 751, 1363, 1405, 1501, 1557, 1610, 2810, 3528.
4,4'-((3,4-Dimethoxyphenyl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.26 (m, 6H), 4.84 (s, 3H), 4.84 (s, 1H), 7.01–7.99 (m, 12H), 8.83 (s, 1H), 14.12 (s, br, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 11.1, 35.6, 57.8, 112.0, 116.3, 121.3, 124.6, 125.6, 127.4, 130.6, 131.4, 135.8, 148.6, 139.8, 147.3, 149.6, 152.4 ppm.
4,4'-((1H-indol-3-yl)methylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol)
1H NMR (250 MHz, DMSO): 2.40 (m, 6H), 4.18 (s, 1H), 6.87–8.34 (m, 15H), 10.05 (s, 1H), 13.56 (s, 2H) ppm. 13C NMR (62.5 MHz, DMSO): 13.7, 31.1, 112.9, 114.4, 119.0, 122.4, 123.9, 124.3, 127.4, 128.8, 129.1, 130.0, 133.8, 137.2, 138.6, 140.6, 152.0, 153.4 ppm.
Benzyl(phenyl)sulfane
1H NMR (250 MHz, DMSO) δ = 4.02 (m, 1H), 4.21 (m, 1H), 7.05–7.66 (m, 10H), ppm. Mass analysis: Calculated: M/Z = 216.06, Obtained: M/Z = 216.1
(Ethylsulfinyl)benzene
1H NMR (250 MHz, DMSO) δ = 2.64 (m, 3H), 3.18 (m, 2H), 7.53–7.93 (m, 5H), ppm. 13C NMR (62.5 MHz, DMSO): 10.5, 34.5, 122.0, 125.3, 126.8, 129.0 ppm.
(Methylsulfinyl)benzene
1H NMR (250 MHz, DMSO) δ = 4.22 (s, 3H), 7.38–7.83 (m, 5H) ppm. 13C NMR (62.5 MHz, DMSO): 43.8, 124.6, 129.2, 133.7, 146.7 ppm.
(Sulfinylbis(methylene))dibenzene
1H NMR (250 MHz, DMSO) δ = 3.76 (s, 2H), 4.17 (s, 2H), 6.90–7.17 (m, 10H) ppm. 13C NMR (62.5 MHz, DMSO): 57.4, 128.1, 129.3, 130.6, 133.6, ppm.
1-(Butylsulfinyl)butane
1H NMR (250 MHz, DMSO) δ = 0.97 (m, 6H), 1.40 (s, 4H), 1.53 (s, 4H), 3.04 (m, 10H) ppm. 13C NMR (62.5 MHz, DMSO): 13.9, 22.0, 25.6, 51.3 ppm.
(Methylsulfinyl)methane
1H NMR (250 MHz, DMSO) δ = 3.56 (m, 6H) ppm. 13C NMR (62.5 MHz, DMSO): 44.8 ppm.
Result and discussion
Catalyst characterization
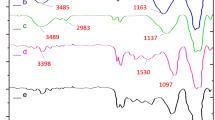
Using FT-IR spectroscopy, the synthesis of zinc ferrite nanoparticles (ZF-NPS) was confirmed. The absorption band at 582 cm−1 is assigned to the stretching vibrations of the zinc-oxygen bond30,31. In Fig. 4a, the bending and stretching vibration of hydroxyl groups on the surface of the nanoparticles at 1655 and 3442 cm−1 are respectively assigned29. Figure 4b confirms the condensation reaction between hydroxyl groups of ZnFe2O4 (MNPs) and the alkoxysilane molecules of tetraethyl orthosilicate (TEOS) as the first layer. Absorbed peaks at 3460 cm−1 were specified as hydroxide stretching vibration mode30. The two absorption peaks around 1103, and 606 cm−1 were indicated the presence of silicon-oxygen )Si–O-Si( asymmetric and symmetric stretching vibrations and bending vibration mode of silicon-oxygen (Si–O-Si), as well as a small peak around 1647 cm−1, was assigned to hydroxide stretching vibration of Silicon-hydroxyl group and twisting vibration of adsorbed H–O-H in a silica shell32. In Fig. 4c, ZnFe2O4@SiO2@L-lysine, the bands in the range of 2912 to 3000mc−1 correspond to the bending vibration of CH2 confirming the attachment of L-lysine molecules to the surface. Then, the presence of broad band at 2500–3700 cm−1 in FTIR spectra of ZnFe2O4@SiO2@L-lysine@SO3H (Fig. 4d) confirms the successful functionalization of ZnFe2O4@SiO2 with the SO3H groups28.

FTIR spectra of (a) ZnFe2O4 (b) ZnFe2O4@SiO2 (c) ZnFe2O4@SiO2@L-lysine (d) ZnFe2O4@SiO2@L-lysine@SO3H.
The PXRD spectra of the ZnFe2O4@SiO2@L-lysine@SO3H nanostructures are recorded in a range of Bragg's angle (2θ = 20°–70°) at room temperature (Fig. 5). The PXRD pattern of the prepared ZnFe2O4@SiO2@L-lysine@SO3H shows seven characteristic peaks at 30°, 35°, 36°, 43°, 54°, 57°, and 63°, corresponding to the (2 2 0), (3 1 1), (2 2 2), (4 0 0), (4 2 2), (5 1 1), and (4 4 0) which confirms the crystal structure of ZnFe2O4@SiO2@L-lysine@SO3H33.

PXRD spectrum of ZnFe2O4@SiO2@L-lysine@SO3H.
The PXRD analysis for the used catalyst was provided and the results were compared to the fresh catalyst, which shows high stability of the prepared catalyst under optimized reaction conditions (Fig. 6).

PXRD spectrum of recovered ZnFe2O4@SiO2@L-lysine@SO3H nanoparticles.
Figure 7 shows the TGA curves for ZnFe2O4 MNPs, ZnFe2O4@SiO2, ZnFe2O4@SiO2@L-lysine, and ZnFe2O4@SiO2@L-lysine@SO3H. In all samples, the first step of weight loss (below 200 °C) is owing to the removal of physically absorbed water and organic solvents (Fig. 7a–d). The decomposition of the organic layer on ZnFe2O4 has occurred in the TGA curve of the catalyst from 200 to 500 °C. Meanwhile, weight loss of about 2% and 8% from 200 to 500 °C occurred for SiO2 and L-lysine, respectively (Fig. 7b and c). Figure 7d illustrates two weight loss steps in the TGA curve of ZnFe2O4@SiO2@L-lysine@SO3H. The first weight loss (10%) between 25 and 250 °C is occurred due to the removal of adsorbed moisture and organic solvents. The next weight loss (50%) from 250 to 600 °C is due to the degradation of organic moieties and the chemisorbed sulfuric acid groups on the surface of the ZnFe2O4 core. Based on the results of the TGA–DSC curve, the well grafting of organic groups on the ZnFe2O4 magnetic nanoparticles is verified34.

TGA of (a) ZnFe2O4, (b) ZnFe2O4@SiO2 (c) ZnFe2O4@SiO2@L-lysine, (d) ZnFe2O4@SiO2@L-lysine@SO3H.
The distribution, size, surface morphology, particle shape, and fundamental physical properties of ZnFe2O4@SiO2@L-lysine@SO3H nanoparticles were investigated using the SEM technique (Fig. 8). The ZnFe2O4@SiO2@L-lysine@SO3H composite is spherical with an almost homogenous size distribution. In addition, the SEM image shows that the size of the nanoparticles is about ≈ 81 nm (Fig. 8).

SEM spectrum of ZnFe2O4@SiO2@L-lysine@SO3H.
In another investigation, EDX analysis confirmed the presence of Zn, C, O, Si, N, Fe, and S elements in the synthesized ZnFe2O4@SiO2@L-lysine@SO3H. As shown in Fig. 9, the presence of Si species confirmed the successful bonding of the SiO2 shell on the ZnFe2O4 catalytic support. The high purity of the synthesized nanocatalyst was confirmed by these observations. It can be concluded that the target catalyst has been successfully synthesized according to this EDX spectrum (Fig. 9).

EDS spectrum of ZnFe2O4@SiO2@L-lysine@SO3H.
The X-ray mapping of ZnFe2O4@SiO2@L-lysine@SO3H shows the scattering of elements in the ZnFe2O4@SiO2@L-lysine@SO3H (Fig. 10). This analysis confirms the presence of Si, Fe, S, N, C, Zn, and O elements in the synthesized nanoparticle with a suitable and homogeneous dispersity throughout the ZnFe2O4 surface.

X-ray map spectrum of ZnFe2O4@SiO2@L-lysine@SO3H.
Using TEM images, the core–shell structure of ZnFe2O4@SiO2@L-lysine@SO3H cubic nanoparticles was investigated. From Fig. 11, we can see the cubic nanoparticles of the ZnFe2O4@SiO2@L-lysine@SO3H composites. The TEM micrograph showed agglomeration of many ultrafine cubic particles which display gray magnetite (ZnFe2O4) cores surrounded by a SiO2@L-lysine@SO3H shell. It is very interesting that the TEM image again verifies the yolk-shell microstructure in ZnFe2O4@SiO2@L-lysine@SO3H, and it is clear that dense silica layers and L-lysine@SO3H were formed around ZnFe2O4 nanocores (Fig. 11).

TEM micrograph of ZnFe2O4@SiO2@L-lysine@SO3H.
The surface area and size distribution of ZnFe2O4@SiO2@L-lysine@SO3H acid is studied by N2 adsorption–desorption isotherms analysis. Regarding the N2 adsorption–desorption isotherms technique, the obtained surface area of ZnFe2O4@SiO2@L-lysine@SO3H is 6.42 (m2/g) based on the BET method. Also, the total pore volume and average pore diametere of ZnFe2O4@SiO2@L-lysine@SO3H are obtained by the BET technique and the values are 0.07 cm3 g−1, and 44 nm, respectively (Fig. 12).

N2 adsorption/desorption isotherms, of the ZnFe2O4@SiO2@L-lysine@SO3H.
Using the back titration method, the acid strength of the synthesized catalyst, that is, the surface density of SO3H groups, was investigated and determined. First, 0.1 g of synthesized catalyst was added to the 50 mL water and stirred for 1 h, then 10 mL NaOH (0.1 N) was added to the mixture and was stirred as long as the pH did not change the as-synthesized catalyst was separated using an external magnet. Then, two drops of phenolphthalein were added to the mixture and were tittered with 1.9 mL HCl (0.1 N). Thus 1 g of catalyst has 8.1 mmol of the acidic groups.
The magnetic behavior of ZnFe2O4 (a) and ZnFe2O4@SiO2@L-lysine@SO3H (b) composite was investigated with the vibrating sample magnetometer (VSM) (Fig. 13). The ZnFe2O4 nanoparticles exhibited almost zero coercivity and remanence with no hysteresis loop, approving the high permeability in magnetization and good magnetic responsiveness. Magnetic measurements showed saturation magnetization values of 41 and 22 emu/g for ZnFe2O4 and ZnFe2O4@SiO2@L-lysine@SO3H complex nanocomposite, respectively. The results showed that the magnetization of ZnFe2O4 decreases after the coating of L-lysine@SO3H on its surface, indicating the successful immobilization of the L-lysine@SO3H on ZnFe2O4.

The magnetic properties of (a) ZnFe2O4 and (b) ZnFe2O4@SiO2@L-lysine@SO3H.
Catalytic study
Checking catalytic activity of ZnFe2O4@SiO2@L-lysine@SO3H for the synthesis of pyrazolyl
In the next step, after the successful synthesis and characterization of ZnFe2O4@SiO2@L-lysine@SO3H, its catalytic activity was considered for the synthesis of pyrazolyl derivatives and oxidation of sulfides.
In early research to obtain optimal reaction conditions, after structural characterization of the prepared nanocatalyst (ZnFe2O4@SiO2@L-lysine@SO3H), its catalytic activity was investigated in the synthesis of pyrazolyl (Table 1). The reaction between benzaldehyde (1 mmol), phenylhydrazine (2 mmol), and ethyl acetoacetate (2 mmol), was selected as the model reaction, and the influence of various parameters including amounts of catalyst, reaction temperature, and solvent were examined. The model reaction did not take place in the absence of the ZnFe2O4@SiO2@L-lysine@SO3H. After optimizing the catalyst’s amount, the effect of temperatures and several solvents was checked. The best results were obtained in solvent-free conditions using 0.03 g ZnFe2O4@SiO2@L-lysine@SO3H at 80 °C.
After determining the optimal conditions, to identify the performance and generality of ZnFe2O4@SiO2@L-lysine@SO3H, the synthesis of diverse derivatives such as pyrazolyl was tested by various arylaldehydes (Table 2). As can be observed in this table, all arylaldehydes worked well in the reaction and it was observed that the synthesis of pyrazolyl in the presence of this catalyst afforded excellent yields with short reaction times.
The mechanism for the synthesis of pyrazolyl in the presence of ZnFe2O4@SiO2@L-lysine@SO3H has been depicted in Fig. 14. At the beginning of the reaction, ZnFe2O4@SiO2@L-lysine@SO3H composite activates C=O groups in the ethyl acetoacetate, and then phenylhydrazine attacks the C=O groups to afford pyrazolone 1 and was further rearranged into tautomer 2. Next, a Knoevenagel-type reaction takes place between activated aldehydes and tautomer 2 followed by the liberation of an H2O molecule to form intermediate 3. Then, a Michael addition reaction between intermediate 3 and tautomer 2 is facilitated to form intermediate 4. In the final step, the corresponding products are formed by tautomerization and aromatization of intermediate 435.

Proposed reaction mechanism.
After characterization of the synthesized heterogeneous ZnFe2O4@SiO2@L-lysine@SO3H nanocatalyst was examined in the oxidation of sulfide to understand the catalytic activity of the prepared material. First, the reaction of the Ph–S-Me with H2O2 was selected as a model reaction and carried out in the presence of ZnFe2O4@SiO2@L-lysine@SO3H Hat different conditions, including different temperatures and amounts of nanocatalyst, and the results showed that the catalyst showed high activity in solvent-free conditions at 25 °C at 120 min. In the next step, the effect of different solvents (EtOAc, n-Hexane, Ethanol, H2O) and also the conditions without solvent were investigated. It should be noted that in solvent-free conditions, the best yield was obtained in 120 min. The study of the amount of catalyst showed that the 0.03 g of nanocatalyst gave a high yield of product (Table 3, entries 1–5). The oxidation didn’t occur in the absence of ZnFe2O4@SiO2@L-lysine@SO3H even after 4 h (Table 3).
In the next step, after the completion of optimization, the catalytic activity of ZnFe2O4@SiO2@L-lysine@SO3H in the oxidation of a wide range of sulfide derivatives was examined under the optimized conditions. It is necessary to mention that all sulfoxides were produced with high yields, which showed the excellent catalytic activity of the synthesized nanoparticles (Table 4). This catalytic system is a suitable method in terms of the efficiency of conditions.
The proposed mechanism for the oxidation of sulfide to the corresponding sulfoxide is shown in Fig. 15. The efficiency of the oxidation can be explained by the interaction between the ZnFe2O4@SiO2@L-lysine@SO3H and H2O2. The OH moiety of the ZnFe2O4@SiO2@L-lysine@SO3H forms a strong hydrogen bond with H2O2 and increases the electrophilic ability of a peroxy oxygen atom of H2O2. In these reaction conditions hydrogen bonding may be assisting in controlling the chemoselectivity, because the hydrogen bond between the catalyst and the oxygen of the sulfoxides could decrease the nucleophilicity of the sulfur atom of the sulfoxides and prevent further oxidation of the sulfoxides. One explanation for this transformation is the in-situ formation of peroxy acid using the reaction of ZnFe2O4@SiO2@L-lysine@SO3H with Hydrogen peroxide, followed by the oxygen transfer to the organic substrate (Fig. 15a). Another explanation is that ZnFe2O4@SiO2@L-lysine@SO3H acts as protic acid, which polarizes the oxygen–oxygen bond in hydrogen peroxide to produce the reactive oxygen transfer agent (Fig. 15b)40.

Possible mechanism for the oxidation of sulfide.
Hot filtration
In this part, with optimal reaction conditions in hand, to confirm the heterogeneous nature of the ZnFe2O4@SiO2@L-lysine@SO3H in the synthesis of pyrazolyl compounds hot filtration experiment was performed using benzaldehyde as a model reaction. At the half time of reaction, the corresponding product was obtained in 55% of the yield. Next, when the reaction mixture was run in another half-time in the absence of nanocatalyst, the reaction afforded no augmentation in its yield. It can be concluded from this point that the catalyst can be considered a true heterogeneous nanocatalyst. Moreover, the stability of the L-lysine@SO3H complex on the surface of ZnFe2O4 confirms the heterogeneous nature of the as-prepared nanocatalyst.
Reusability of ZnFe2O4@SiO2@L-lysine@SO3H
The recoverability of ZnFe2O4@SiO2@L-lysine@SO3H catalyst was investigated for oxidation of sulfides (series 1) and Synthesis of pyrazolyl (series 2) derivatives. In this study, the recovery of the nanocatalyst from the reaction mixture was successfully carried out, which could be easily separated with a neodymium magnet and washed several times with EtOAc and DI (H2O). Then the recovered nanocatalyst was used in the next run. The results showed that recycled catalysts can be employed at both of the reactions up to five times, with insignificant loss of catalyst activity (Fig. 16).

Recyclability of ZnFe2O4@SiO2@L-lysine@SO3H for the preparation of oxidation of methyl phenyl sulfide (series 1) and 4,4'-(phenylmethylene)bis(pyrazole) (series 2).
Comparison of the catalyst
The comparative study of different catalytic for the synthesis of pyrazolyl derivatives (Table 5), with several previously reported methods, is presented. In the present research, the products were obtained in higher yields over faster times in the presence of ZnFe2O4@SiO2@L-lysine@SO3H. In addition, this catalyst is environmentally friendly and has several advantages in terms of sustainability, price, separation, and non-toxicity.
Conclusions
In this research project, we have successfully synthesized ZnFe2O4@SiO2@L-lysine@SO3H nanoparticles as an effective and recoverable nanocatalyst. Wide active surface area, reusability, suitable stability, excellent heterogeneity, and substantial magnetic behavior have distinguished this catalytic system as an instrumental tool for the synthesis of organic compounds. This research reported a novel route for the synthesis of an extensive range of synthesis of pyrazolyl derivatives and oxidation of sulfides with high yields and purity. The wondrous features of this protocol are novelty, no use of harmful solvents, simple synthesis procedure, short reaction time, facile filtration, and reusability. In addition, the as-synthesized magnetic nanocatalyst could be separated easily using a external magnet and reused several times without significant loss of its catalytic activity.
Data availability
All data generated or analyzed during this study are included in this published article [and its Supplementary Information Files].
References
Nguyen, N. T. T., Nguyen, T. T. T., Nguyen, D. T. C. & Tran, T. V. Green synthesis of ZnFe2O4 nanoparticles using plant extracts and their applications: A review. Sci. Total Environ. 872, 162212 (2023).
Ragu, S., Kim, B., Chen, S.-M., Ishfaque, A. & Kang, K.-M. N-substituted CQDs impregnated by Fe3O4 heterostructure: Bifunctional catalyst for electro-catalytic and photo-catalytic detection of an environmental hazardous organic pollutant. Chemosphere 311, 137168 (2023).
Han, Q. et al. Polyethylene glycol functionalized Fe3O4@MIL-101(Cr) for the efficient removal of heavy metals from Ligusticum chuanxiong Hort. Arab. J. Chem. 16, 104635 (2023).
Chen, M.-N., Mo, L.-P., Cui, Z.-S. & Zhang, Z.-H. Magnetic nanocatalysts: Synthesis and application in multicomponent reactions. Curr. Opin. Green Sustain. Chem. 15, 27–37 (2019).
Zhang, M., Liu, Y.-H., Shang, Z.-R., Hu, H.-C. & Zhang, Z.-H. Supported molybdenum on graphene oxide/Fe3O4: An efficient, magnetically separable catalyst for one-pot construction of spiro-oxindole dihydropyridines in deep eutectic solvent under microwave irradiation. Catal. Commun. 88, 39–44 (2017).
Ghasemzadeh, M. A. & Ghaffarian, F. Preparation of core/shell/shell CoFe2O4/OCMC/Cu (BDC) nanostructure as a magnetically heterogeneous catalyst for the synthesis of substituted xanthenes, quinazolines and acridines under ultrasonic irradiation. Appl. Organomet. Chem. 34, 1–10 (2020).
Poonia, K. et al. Recent advances in metal organic framework (MOF)-based hierarchical composites for water treatment by adsorptional photocatalysis: A review. Environ. Res. 222, 115349. https://doi.org/10.1016/j.envres.2023.115349 (2023).
Zhang, H.-Y. et al. A magnetic metal–organic framework as a highly active heterogeneous catalyst for one-pot synthesis of 2-substituted alkyl and aryl(indolyl)kojic acid derivatives. N. J. Chem. 41, 7108–7115 (2017).
Gao, G., Di, J.-Q., Zhang, H.-Y., Mo, L.-P. & Zhang, Z.-H. A magnetic metal organic framework material as a highly efficient and recyclable catalyst for synthesis of cyclohexenone derivatives. J. Catal. 387, 39–46 (2020).
Huang, X. et al. Space-confined growth of nanoscale metal-organic frameworks/Pd in hollow mesoporous silica for highly efficient catalytic reduction of 4-nitrophenol. J. Colloid Interface Sci. 629, 55–64 (2023).
Cai, W., Zhang, W. & Chen, Z. Magnetic Fe3O4@ZIF-8 nanoparticles as a drug release vehicle: pH-sensitive release of norfloxacin and its antibacterial activity. Colloids Surf. B Biointerfaces 223, 113170 (2023).
Öztürk, D. & Mıhçıokur, H. Removal of lansoprazole one of the most prescribed drugs in Turkey from an aqueous solution by innovative magnetic nanomaterial Tween 85®PEI@Fe3O4. J. Water Process Eng. 52, 103527 (2023).
Al-husseiny, R. A., Kareem, S. L., Naje, A. S. & Ebrahim, S. E. Effect of green synthesis of Fe3O4 nanomaterial on the removal of cefixime from aqueous solution. Biomass Convers. Biorefinery https://doi.org/10.1007/s13399-023-03921-7 (2023).
Jinxi, W. et al. Tailoring of ZnFe2O4-ZrO2-based nanoarchitectures catalyst for supercapacitor electrode material and methanol oxidation reaction. Fuel 334, 126685 (2023).
Doiphode, V. et al. Solution-processed electrochemical synthesis of ZnFe2O4 photoanode for photoelectrochemical water splitting. J. Solid State Electrochem. 25, 1835–1846 (2021).
Agyemang, F. O. & Kim, H. Electrospun ZnFe2O4-based nanofiber composites with enhanced supercapacitive properties. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 211, 141–148 (2016).
Zhang, X. et al. ZnFe2O4 nanospheres decorated residual carbon from coal gasification fine slag as an ultra-thin microwave absorber. Fuel 331, 125811 (2023).
Sanko, V., Şenocak, A., Tümay, S. O. & Demirbas, E. A novel comparative study for electrochemical urea biosensor design: Effect of different ferrite nanoparticles (MFe2O4, M: Cu Co, Ni, Zn) in urease immobilized composite system. Bioelectrochemistry 149, 108324 (2023).
Gandomi, F. et al. ROS, pH, and magnetically responsive ZnFe2O4@l-Cysteine@NGQDs nanocarriers as charge-reversal drug delivery system for controlled and targeted cancer chemo-sonodynamic therapy. Inorg. Chem. Commun. 150, 110544 (2023).
Marandi, A., Kolvari, E., Gilandoust, M. & Zolfigol, M. A. Immobilization of –OSO3H on activated carbon powder and its use as a heterogeneous catalyst in the synthesis of phthalazine and quinoline derivatives. Diamond Relat. Mater. 124, 108908 (2022).
Malysheva, S. et al. Phosphine chalcogenides and their derivatives from red phosphorus and functionalized pyridines, imidazoles, pyrazoles and their antimicrobial and cytostatic activity. Bioorgan. Chem. 132, 106363 (2023).
Ayman, R., Abusaif, M. S., Radwan, A. M., Elmetwally, A. M. & Ragab, A. Development of novel pyrazole, imidazo[1,2-b]pyrazole, and pyrazolo[1,5-a]pyrimidine derivatives as a new class of COX-2 inhibitors with immunomodulatory potential. Eur. J. Med. Chem. 249, 115138 (2023).
Roney, M. et al. Identification of pyrazole derivatives of usnic acid as novel inhibitor of SARS-CoV-2 main protease through virtual screening approaches. Mol. Biotechnol. https://doi.org/10.1007/s12033-023-00667-5 (2023).
Wang, J.-L. et al. Four unprecedented V14 clusters as highly efficient heterogeneous catalyst for CO2 fixation with epoxides and oxidation of sulfides. Sci. China Chem. 66, 107–116 (2023).
Zhang, J. et al. Preparation of core/shell-structured ZnFe2O4@ZnIn2S4 catalysts and its ultrafast microwave catalytic reduction performance for aqueous Cr(VI). Chem. Eng. J. 451, 138182 (2023).
Sadeghi, Z. & Hajiarab, R. New nanoparticles of NaY, Ni-NaY, and Mn-NaY zeolites: Highly efficient catalysts for the oxidation of sulfides to sulfoxides. Phosphorus Sulfur Silicon Relat. Elem. https://doi.org/10.1080/10426507.2023.2174983 (2023).
Wan, W. L. et al. Samarium oxide as efficient and non-endangered metal for synthesis of sulfones from sulfides: An elemental sustainability concept. J. Taibah Univ. Sci. 17, 2174485 (2023).
Khanmohammadi-Sarabi, F., Ghorbani-Choghamarani, A., Aghavandi, H. & Zolfigol, M. A. l-Methionine-Zr complex supported on magnetic ZnFe2O4 as a novel, green, and efficient heterogeneous magnetic nanocatalyst for the synthesis of 1H-tetrazole and polyhydroquinoline derivatives. N. J. Chem. https://doi.org/10.1039/D2NJ04071A (2023).
Khanmohammadi-Sarabi, F., Ghorbani-Choghamarani, A., Aghavandi, H. & Zolfigol, M. A. ZnFe2O4@SiO2-ascorbic acid: Green, magnetic, and versatile catalyst for the synthesis of chromeno[2,3-d] pyrimidine-8-amine and quinazoline derivatives. Appl. Organomet. Chem. 36(8), e6768 (2022).
Aghavandi, H. & Ghorbani-Choghamarani, A. ZnFe2O4@l-Arginine-Ni: A novel, green, recyclable, and highly versatile catalyst for the synthesis of 1H-tetrazoles and oxidation of sulfides to the sulfoxides. J. Phys. Chem. Solids 170, 110952 (2022).
Ghorbani-Choghamarani, A., Aghavandi, H. & Talebi, S. M. A new copper-supported zinc ferrite as a heterogeneous magnetic nanocatalyst for the synthesis of bis(pyrazolyl)methanes and oxidation of sulfides. Sci. Rep. 12, 20775 (2022).
Aghavandi, H. & Ghorbani-Choghamarani, A. Preparation and application of ZnFe2O4@SiO2–SO3H, as a novel heterogeneous acidic magnetic nanocatalyst for the synthesis of tetrahydrobenzo[b]pyran and 2,3-dihydroquinazolin-4(1H)-one derivative. Res. Chem. Intermed. https://doi.org/10.1007/s11164-022-04890-8 (2022).
Andhare, D. D. et al. Structural and chemical properties of ZnFe2O4 nanoparticles synthesised by chemical co-precipitation technique. J. Phys. Conf. Ser. 1644, 012014 (2020).
Mohammadi, M. & Ghorbani-choghamarani, A. Hercynite silica sulfuric acid : a novel inorganic sulfurous solid acid catalyst for one-pot cascade organic transformations. RSC Adv. 12, 26023–26041. https://doi.org/10.1039/d2ra03481f (2022).
Aghavandi, H., Ghorbani-Choghamarani, A. & Mohammadi, M. Mesoporous SBA-15@Tromethamine-Pr: Synthesis, characterization and its catalytic application in the synthesis of Bis(Pyrazolyl)Methanes. Polycycl. Aromat. Compd. https://doi.org/10.1080/10406638.2022.2147202 (2022).
Filian, H., Kohzadian, A., Mohammadi, M., Ghorbani-Choghamarani, A. & Karami, A. Pd(0)-guanidine@MCM-41: A very effective catalyst for rapid production of bis (pyrazolyl)methanes. Appl. Organomet. Chem. https://doi.org/10.1002/aoc.5579 (2020).
Anizadeh, M. R., Torabi, M., Zolfigol, M. A. & Yarie, M. Catalytic application Fe3O4@SiO2@(CH2)3-urea-dithiocarbamic acid for the synthesis of triazole-linked pyridone derivatives. J. Mol. Struct. 1277, 134885 (2023).
Kordnezhadian, R. et al. Polyethylene glycol-bonded triethylammonium l-prolinate: A new biodegradable amino-acid-based ionic liquid for the one-pot synthesis of bis(pyrazolyl)methanes as DNA binding agents. N. J. Chem. 44, 16995–17012 (2020).
Molaei, S. & Ghadermazi, M. A green methodology for thioether formation reaction and synthesis of symmetrical disulfides over new heterogeneous Cu attached to bifunctionalized mesoporous MCM-41. Microporous Mesoporous Mater. 319, 110990 (2021).
Mirfakhraei, S., Hekmati, M., Eshbala, F. H. & Veisi, H. Fe3O4/PEG-SO3H as a heterogeneous and magnetically-recyclable nanocatalyst for the oxidation of sulfides to sulfones or sulfoxides. N. J. Chem. 42, 1757–1761 (2018).
Tayebi, S., Baghernejad, M., Saberi, D. & Niknam, K. Sulfuric Acid ([3-(3-Silicapropyl)sulfanyl]propyl)ester as a Recyclable Catalyst for the Synthesis of 4,4′-(Arylmethylene)bis(1H-pyrazol-5-ols). Chin. J. Catal. 32, 1477–1483 (2011).
Khazaei, A., Abbasi, F. & Moosavi-Zare, A. R. Tandem cyclocondensation-Knoevenagel-Michael reaction of phenyl hydrazine, acetoacetate derivatives and arylaldehydes. N. J. Chem. 38, 5287–5292 (2014).
Acknowledgements
This work was supported by the research facilities of Bu-Ali Sina University, Hamedan, Iran.
Author information
Authors and Affiliations
Contributions
Amir Ghanbarpour: laboratory work, Investigation, Methodology. Arash Ghorbani-Choghamarani: Resources, Writing-review & editing, Conceptualization, Supervision. Hamid Aghavandi: Methodology, Investigation, Writing-original draft, Validation. Ahmad Jafari: laboratory work, Methodology.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghanbarpour, A., Ghorbani-Choghamarani, A., Aghavandi, H. et al. ZnFe2O4@SiO2@L-lysine@SO3H: preparation, characterization, and its catalytic applications in the oxidation of sulfides and synthesis of Bis(pyrazolyl)methanes. Sci Rep 14, 7449 (2024). https://doi.org/10.1038/s41598-024-57317-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-57317-2
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
