
Abstract
We present the fabrication of a MoS2−xSex thin film from a co-sputtering process using MoS2 and MoSe2 commercial targets with 99.9% purity. The sputtering of the MoS2 and MoSe2 was carried out using a straight and low-cost magnetron radio frequency sputtering recipe to achieve a MoS2−xSex phase with x = 1 and sharp interface formation as confirmed by Raman spectroscopy, time-of-flight secondary ion mass spectroscopy, and cross-sectional scanning electron microscopy. The sulfur and selenium atoms prefer to distribute randomly at the octahedral geometry of molybdenum inside the MoS2−xSex thin film, indicated by a blue shift in the A1g and E1g vibrational modes at 355 cm−1 and 255 cm−1, respectively. This work is complemented by computing the thermodynamic stability of a MoS2−xSex phase whereby density functional theory up to a maximum selenium concentration of 33.33 at.% in both a Janus-like and random distribution. Although the Janus-like and the random structures are in the same metastable state, the Janus-like structure is hindered by an energy barrier below selenium concentrations of 8 at.%. This research highlights the potential of transition metal dichalcogenides in mixed phases and the need for further exploration employing low-energy, large-scale methods to improve the materials’ fabrication and target latent applications of such structures.
Similar content being viewed by others

Introduction
Layered molybdenum disulfide (MoS2) has been used as a semiconductor in the improvement of thin films for the development of hybrid solar cell prototypes. When mixing the material with organic molecules, the array 2H-MoS2/p-type organic semiconductor can achieve conversion efficiencies of up to 2.8%1. A recent study highlights remarkable mechanical properties of MoS2, denoted by a reported elastic modulus of E = 136 ± 2 GPa and hardness of H = 10.5 ± 0.1 GPa2. The fabrication by radio-frequency sputtering (RF-sputtering) reveals a vertical crystal growth alignment along the [101] direction corresponding to laminar colonies of grains. Moreover, recent advances in tuning the electronic and optical behavior of transition metal dichalcogenides (TMDC) by doping3,4 and heterostructure formation5,6 have expanded the interest in TMDC even further. From the approaches mentioned, heterostructure formation has stood as a potential route to attain new mixed phases with enhanced piezoelectric properties7, improved catalytic activity8, band gap engineering9, and reduced contact resistance. The latter, for example, has been determined by the electronic structure calculations of ITO-MoS2 with a n-type Schottky barrier height (Φn) of ~ 0.2 eV10. However, the research on mixed phases based on TMDC heterostructures still requires an extended understanding of their properties and potential improvements in their fabrication.
Most of the research on mixed MoS2−xSex phases starts with single- and bi-layer systems obtained by mechanical exfoliation and manual layer stacking11,12,13 which is a methodology suitable for fundamental research but not scalable for large-area and low-cost production and integration with devices. Most metastable phases reported in the literature described Janus-like heterojunctions based on TMDC—a Janus heterojunction required substituting half the original chalcogen composition with a different one, commonly substituting S by Se or vice versa, and this substitution needs to occur in only one atomic layer. Their fabrication is also limited to the critically controlled environments required for their fabrication, and from these reports, its long-term stability and scalability can be under question. In contrast, proof of large-scale fabrication of MoS2 thin films has been reported by Muratore et al.14, Ramos et al.2, and others15,16,17, by using RF-sputtering, which is a low-cost method and already employed in commercial manufacturing and attain laminar colonies of 2H MoS2 grains oriented vertically in the <001> basal plane or <101> plane direction.
Remarkably, Conca et al. achieved doping a nanocrystalline silicon thin film with boron via an RF-sputtering co-deposit process with and ease of control over the boron concentration and the resulting thin film crystallinity18. Furthermore, co-deposit by RF sputtering dates back decades, when Stupp improved the tribological properties of MoS2 coatings as a function of co-deposited metals by RF-sputtering19. This work has been followed recently by other authors to enhance the catalytic properties of silver-decorated MoS2 nanoflakes20 and the performance of biosensors21. In addition, theoretical calculations on Janus heterostructures and related metastable phases are needed to provide new insights into mixed phases and their performance, like electron–phonon relations22, heterostructure coupling9,23, photocatalyst capacity24, and especially, thermodynamic stability. To date, no theoretical study compares Janus and random MoS2−xSex mixed phase in terms of thermodynamic stability and basic electronic properties at different concentrations (up to 33.33 at. % of selenium) to attain a MoSSe phase.
This work contributes to the research on mixed phases based on TMDC by demonstrating the ease of fabrication of a MoSSe thin film mixed phase and correlating these experimental data with density functional theory-based calculations. We start presenting the characterization of a MoSSe thin film co-deposited by RF-sputtering. Raman spectroscopy, time-of-flight secondary ion mass spectrometry, and scanning electron microscopy indicate a random distribution of sulfur and selenium atoms within the thin film. Next, we present the computation of the thermodynamic stability of a MoS2−xSex mixed phase in a Janus-like and random distribution from 0 at.% up to 33.33 at.% selenium concentration. Our results indicate that lattice expansion is related to the observed Raman shift, and the formation process of random MoS2−xSex meta-phase is more suitable for practical applications as semiconductors, where long-term stability is the main concern. We believe this information is essential to boost improvements in the fabrication methods of large-area and scalable mixed thin films using RF-sputtering.
Results and discussion
Fabrication of the MoS2− xSex phase
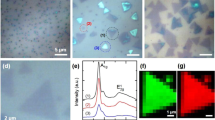
From the co-deposition process of MoS2−xSex phase by RF-sputtering, we obtained a thin film of approximately 200 nm sandwiched between ITO layers (being approx. 150 nm thick). ToF–SIMS measurements confirm that our multilayer array consists of four distinctly different regions with sharp interface formation (Fig. 1). The presence of sharp interfaces between the top and bottom ITO layers with the MoS2-xSex phase in between is specified by the signals for In2+, InSnO+, and SnO+ ions are distinguished and decay exponentially as the Mo+ signals start to appear as depicted in Fig. 1a implying no intermixing between the ITO and the MoS2−xSex phase. In terms of selenium and sulfur, these measurements detected a homogeneous distribution within the fabricated sample, denoted by the signals from S− and Se− ions (Fig. 1b). Cross-section scanning electron microscopy (SEM) images present visual aspects of the ITO/MoS2−xSex/ITO array (Fig. 1c), confirming a well-defined and sharp interface formation as previously reported10,25 and of comparable quality to those obtained by CVD methods26.

(a, b) Time-of-flight secondary ion mass spectrometry depth profile measurements for positive and negative ions, respectively. (c) Schematic description of the deposited multilayer arrangement and cross-section scanning electron microscopy image. (d) Raman spectra of the obtained MoSSe thin film showing the blue-shifted A1 and E1 corresponding to the MoSSe phase. Inset displays a schematic of the A1 and E1 Raman modes of MoSSe with gray balls denoting molybdenum atoms, and orange and yellow selenium and sulfur atoms, respectively.
The Raman spectroscopy reveals that the film is homogenous and presents a degree of crystallinity (Fig. 1d). The sample presents vibrational modes at 402 cm−1 and 374.8 cm−1, which correspond to the A1g and E12g vibrational modes of MoS2 and suggest the presence of pure Mo-S vibration in some regions (Supplemental Information Fig. S1). However, signals at 355 cm−1 and 255 cm−1 correspond to the blue-shifted E1 and A1 modes, respectively, due to the formation of the MoSSe phase27,28. The blue shift in the A1 vibrational mode is attributed to the out-of-plane change in symmetry caused by the integration of Se into the MoS2 matrix during the co-deposit, while the shift in E1 modes is related to the expansion of the lattice29 and will be discussed in the next section. Deconvolution of the Raman spectra shows a thin film with traces of MoSe2 signals, detectable by the A1g Raman mode before 250 cm−1 alongside the shifted A1 modes of the MoSSe phase (Supplemental Information Fig. S2). Grazing incidence x-ray diffraction (GIXD) spectroscopy matches with our Raman analysis on the degree of crystallinity of the sample, indicating the presence of peaks at 13°, 33°, 54° and 56° which corresponds to the Bragg’s angle (2θ) of the planes (002), (101), (008) and (100) of 2H MoS2 and 2H MoSe230,31,32 with P63/mmc-194 space group, as shown in Fig. S3 from the Supplemental Information. The average crystal size was calculated to be ~ 75 nm (Table S1) in agreement with previous reports2 by using the Scherrer equation. Future studies can exploit low-energy methods like laser annealing33, annealing in a controlled atmosphere15,26,34, or ink printing35, integrated into the one-step process to fabricate mixed MoS2−xSex thin films with improved crystal quality for applications in energy harvesting and optoelectronics devices.
Compared to previous reports on RF-sputtering of MoS2 and MoSe2, we report the fabrication of a co-sputtered MoSSe phase under a low-temperature and high-power conditions for an smooth and homogenous deposit knowing that high RF power tends to facilitate the crystallization of MoS232. The listed works in Table 1 also show a similar trend, indicating that for an optimal deposition of MoS2 by RF-sputtering, and probably most TMDC, enough kinetic energy is require to achieve the Mo+4 state and with this, an homogeneous crystallization36. This former argument is supported by our computational analysis presented in the next section. In our work, we increased the RF power for high deposition rates, but we also increased the working distance (see “Methods” section) to avoid the impact of high-energy sputtered material on the substrate. Furthermore, the literature also points out that deposition time has a crucial role in determining the crystallinity of the MoS2 thin film, as short deposition times (< 120 s) normally yield amorphous MoSx phases or minuscule crystalline structures30. Then, the current course in physical vapor deposition (PVD) of TMDC, either by RF or direct current (DC), is to ease the fabrication of thin films avoiding subsequent high-energy annealing and post-sputtered treatments, aiming for one-step direct processes.
Structural optimization of the MoS2−xSex phase
To obtain insights into the structural stability and possible formation routes, we considered a MoS2−xSex phase model using density functional theory (DFT) calculations. Details are provided in the “Methods” section. MoS2 crystallizes in the hexagonal P63/mmc space group, and its structure consists of two MoS2 sheets oriented parallel to the (001) plane. From these calculations, it is found that as the concentration of selenium increases, the initial MoS2 lattice needs to expand in the random and Janus-like situations to accommodate the foreign ions and reach the structural optimization convergence. In the MoS2−xSex random phase at 33.33 at. % of selenium, a relative volume expansion (ΔV) of almost 7% is noted, while in the Janus-like situation the ΔV results 5.8% (see Table 2). The random phase at 33.33 at. % of selenium approaches more closely to the lattice parameter of 0.329 nm for 2H MoSe240,41. The volume variation indicates a distortion from the pure MoS2 to the MoS2−xSex phase in the random situation and contrasts with the volume variation in the Janus-like, which is less abrupt. The volume variation in the Janus-like model is in line with previous reports, where a low lattice distortion occurs in the experimental Janus MoSSe monolayer42.
We attribute the increase in the volume of the supercell as a direct consequence of the change in the bonding distance between molybdenum and the chalcogen ions. In the random phase, the Mo-S bond distance (dMo-S), presents a contraction from 0.2406 nm to 0.2405 nm in most cases, a minor relative contraction of 0.04%. The Mo-Se bond distance (dMo-Se) shows contractions up to 0.08%, recalling the dMo-Se in pure MoSe2 resulted in 0.25322 nm. In the Janus distribution, an expansion of 0.17% of dMo-S resulted after the structural optimization, while the dMo-Se presents a contraction close to 0.1% concerning the pure 2H phase in both cases. However, as the dMo-Se is significantly larger than dMo-S, about 4%, we found a significant change in the resulting volume of the MoS2−xSex. This is attributed to the rearrangement of charge from disturbed Mo d orbitals and p orbitals as reported previously6,43 and possible electron density change in the 4d orbital due to lower electronegativity of Se over S. This uneven charge distribution may lead to local piezoelectricity as reported in other studies on Janus heterojunctions7,44,45.
This change in the bonding distance agrees to previous reports27 on Janus MoSSe systems and may be related to the resulting blue shift in the E1 and A1 Raman modes, which relates with our experimental observations presented earlier. The A1 Raman modes represent the out-of-plane vibrational modes, perpendicular to the (001) basal plane, but selenium and sulfur have different bonding distances with molybdenum, affecting the symmetry along such planes and thus, the resulting vibrational mode. Similarly, the in-plane E1 vibrational modes (alongside the atomic plane of the S-Mo-S distribution) will have differences compared to the pure 2H phase due to the included selenium. Even though in our case we focused on bulk structures, we expect a comparable explanation for monolayers and two-dimensional (2D) systems.
Thermodynamic stability of the MoS2−xSex phase
The formation energy (Eform) -for MoS2−xSex phase for various selenium concentrations- indicates that at low and high selenium concentrations, the lattice can favorably accommodate these ions in the random distribution. Figure 2 describes the atomistic model used for the DFT computations. For the Janus-type phase, at low selenium concentrations, the system reaches a low entropy state, and the process becomes endothermic. Under these circumstances, the lattice shows a lattice expansion situation quantified by a ΔV of 1.78%. However, after surpassing a selenium concentration of 8 at.% the Janus-like lattice reaches favorable conditions of formation, expressed by the exothermic values of Eform. The clear presence of a threshold value for the formation of the Janus-like phase indicates that it would not be a feasible process unless enough energy is provided to overcome this energy barrier. On the other hand, the random phase shows an exothermic tendency, regardless of the selenium concentration. This analysis indicates that a co-deposit process would favor the formation of the random MoS2−xSex phase.

(a) Schematic of the 2H-MoS2 supercell, (b) random MoS2−xSex supercell at x = 1, and (c) Janus-type MoS2−xSex supercell at x = 1. All MoS2-xSex models were created using a 2H-MoS2 4 × 4 × 1 supercell containing 32 molybdenum atoms and 64 sulfur atoms. Color code: yellow is sulfur, orange is selenium and grey corresponds to molybdenum atoms.
In terms of the site specific substitution of S by Se—denoted by the substitutional energy, Esubst—we observe that for the random phase, the process is endothermic in all ranges of selenium concentrations up to 33.3 at.% having energy demands below 0.3 eV, which is comparable to the proposed activation energy for MoS2 crystallization of 0.7 eV46. In this situation, as the concentration of selenium increases, the energy demand also increases as the systems need to accommodate foreign Se atoms into the initial matrix. For the Janus-like phase, the process is not favorable until a threshold value of 8 at.% selenium concentration is reached. In the latter situation, the presence of an energy barrier becomes noticeable; for concentrations below 8 at. % of selenium, the system has no tendency to exchange sulfur with selenium with Esubst values above 16 eV. After surpassing this threshold value, the process becomes favorable and behaves similarly to the random phase, with substitution energies below 0.3 eV. This suggests that subsequent substitution is self-maintained in the Janus-type phase. Previous reports have hinted towards the critical stability of Janus MoSSe monolayers between 700 °C and 800 °C47,48. Lu et al.27 report a randomized MoSSe phase achieved above 600 °C, which supports our statement that MoSSe Janus-type structures are indeed low entropy systems in a metastable state. The Janus-like and random phases reach almost an equal value of equilibrium energy (Fig. 3a), meaning both phases are located at the same low entropy point, and with this, the same metastable state located between pure 2H MoS2 and pure 2H MoSe2 as depicted in Fig. 3b. Nevertheless, the low entropy state of the Janus-type is determined by the highly ordered distribution of selenium and sulfur ions, related to the observed increase in Esubst as mentioned before.

(a) Computed equilibrium energy (Eq) for pure 2H MoS2, 2H MoSe2, Janus, and random MoS2−xSex at a Se concentration of 33.33 at.%. The random MoS2-xSex phase at x = 1 reaches almost the same entropy state as the Janus-like phase. Eq computed by Quantum Espresso code includes a one-electron energy part (eelec), the Hartree contribution (eH), the exchange–correlation energy (exc), the Ewald contribution (eewald), and the dispersion correction part by DFT + 3 (eDFT+D). (b) Estimated energy profile for the transition from pure MoS2 to pure MoSe2 having a random and Janus-like phase. The fabrication of MoSSe Janus-like would be limited by an energy barrier at 8 at. % of selenium. Arrows point to the location of their relative energy of formation.
Sulfur vacancies might be present in sputtered MoS249, consequently on MoSe2 as well, due to the higher vapor pressure of the chalcogen species46. To explore the role of S vacancies as lattice defects in the substitutional process of Se into MoS2, we computed the values of Esubst but added an energy contribution for sulfur vacancy formation. The computed formation energy of a sulfur vacancy (EV) is 7.09 eV, meaning it is an endothermic process and is within the range of defect formation in MoS237,50,51. The latter was added to the previous Esubst to get an energy of substitution with an S vacancy formation (Esubst-V) and expressed as \(E_{subst - V} = E_{seg} + n_{V} E_{V}\), where nV is the number of S vacancies needed to accommodate the targeted Se concentration. The difference in our computed energy of vacancy formation with previous studies reporting the effect of S vacancies in MoS2 is attributed to the addition of long-range dispersion correction in our study and the fact that we are using a crystal model rather than monolayer models. Besides, using crystal models helped to reach lower entropy phases compared to monolayers, i.e., lower surface energy. From this analysis, a clear difference between the Janus and the random MoS2−xSex phase becomes noticeable. For the Janus-like phase, the inclusion of Se atoms in a MoS2 lattice with nV sulfur vacancies is not favorable to occur below a Se concentration of 8.3 at. %, after this tipping point the process follows a self-maintained behavior (Table 2) and the energy demand for creating S vacancies is compensated. For the random MoS2-xSex phase, the contrary occurs, as the energy demand remains in the endothermic regime but is doable in terms of energy supply. This indicates that the fabrication of Janus-like phases is locked or inhibited by this energy barrier and explains the experiments performed by Lu et al.27 and Li et al.52 and the critical part of the sulfur vacancy creation and post-selenization process (Fig. 3).
Electronic structure of the MoS2−xSex phase
To get insights into the type of bonding between the chalcogen atoms and the molybdenum, we computed the electronic structure for the random and the Janus-like distribution. First, the projected density of states (PDOS) indicates the strong metallic influence around the Fermi level, characterized by the high contribution of the molybdenum d orbitals in all situations like in the case of MoS2 and MoSe210,41,53. Second, the contribution of the sulfur and selenium p orbitals in the random distribution has a pronounced overlap around the Fermi level, indicated by the close similarities in the sulfur and selenium DOS curves’ distribution. This overlap is less pronounced in the Janus-like distribution compared to the latter, especially at – 1 eV and after 2 eV (Fig. 4). The PDOS analysis suggests a higher degree of coupling between the sulfur and the selenium ions when the MoSSe phase is reached in the random distribution, correlating the resulting contraction in the dMo-S and dMo-Se as described in previous sections. Inside the Janus-like distribution, a strong peak at − 1.2 eV suggests a hybridization of sulfur and selenium p orbitals and molybdenum d orbitals, primordially attributed to the in-plane distribution of the chalcogen atoms and alignment of the px, py, and pz symmetries. Lastly, the computed band structure reveals that the MoSSe phase remains with its semiconducting properties and a reduced indirect band gap close to 0.9 eV in both situations. Such reduction in the band gap agrees with what was observed by Li and coworkers, where a graded MoS2(1−x)Se2x nanosheet showed a variable band gap ranging from 1.8 eV to 1.6 eV as the selenium concentration increased48. The latter potentially implies that the MoS2−xSex mixed phase could work as a research platform for bandgap engineering of TMDC thin films.

(a) Computed projected density of states (PDOS) for the MoSSe in a random and Janus-like distribution. The Fermi level is located at 0 eV. The MoSSe phase having a random distribution presents a higher degree of orbital hybridization between the S p- and Se p-orbital, indicated by the similarities in the curvatures of the pDOS around the Fermi level. (b) Computed band structure for the random and Janus-like distribution in the Γ-Μ-Κ-Γ path. The estimated band gap of both situations is close to 0.9 eV.
Conclusions
The co-deposited thin film exhibits blue-shifted A1 and E1 Raman modes at 355 cm−1 and 255 cm−1, which correspond to the Raman modes of the MoSSe phase and are evidence of lattice expansion. Time-of-flight secondary mass spectrometry shows an even distribution of selenium and sulfur ions throughout the thin film and evidenced the fabrication of sharp interfaces with the encapsulated layers. Our density functional theory calculations demonstrate that the formation of the random MoS2−xSex phase is thermodynamically favorable compared to the Janus-like phase, the latter hindered by an energy barrier below 8 at.% selenium concentrations. Interestingly, both mixed phases reached similar metastable states, denoted by their corresponding equilibrium energy. However, they presented different lattice expansion, above 7% for the random distribution and 5% for the Janus-like, attributed to different orbital’s reallocation in Mo-S and Mo-Se bonds. These results display outstanding ease of fabrication of MoS2-xSex mixed phase with a random distribution of chalcogens by RF-sputtering. We believe that this report provides critical insights that can enhance the fabrication methods of large-area and scalable MoS2−xSex mixed phase in the future.
Methods
RF-sputtering of MoS2− xSex phase
The co-deposition by RF magnetron sputtering of the MoS2−xSex phase was completed using a Kurt J. Lesker PVD-75 instrument, which is equipped with three holders and these can open simultaneously during the deposition process without breaking the vacuum. ITO, MoS2 and MoSe2 targets were mounted separately inside each holder. The substrate was a thermally oxidized Si/SiO2 with 25.4 mm in diameter mounted within a working distance of 20 cm and no heating was applied to the substrate during the co-deposition process. The system was ready for the deposition once a vacuum of 1 × 10–9 bar was reached. After this, a flow or Ar+ ions was introduced into the main chamber setting a working pressure of 4 × 10–6 bar. A layer of indium-tin-oxide (ITO) was first deposited to enhance the adhesion of the MoS2-xSex phase to the substrate using a commercial target of In2Sn2O7 (99.9% purity and 76.2 mm in diameter) for 1800 s at 145 W of RF power at a frequency of 13.56 MHz to achieve the ~ 150 nm layer thickness. Subsequently, the MoS2−xSex phase was deposited by simultaneous sputtering of MoS2 and MoSe2 targets (99.99% purity and 76.2 mm in diameter) for 1000 s at 275 W of RF power at a frequency of 13.56 MHz, aiming for a MoSSe phase with a layer thickness of approximately 200 nm. Finally, a layer of ITO was deposited on top of the MoSSe phase to protect the material from environmental degradation without breaking the vacuum with the same parameters described before.
Cross-section, time-of-flight secondary ion mass spectrometry, Grazing incidence x-ray diffraction, and Raman spectroscopy
Cross-section images were acquired using a scanning electron microscope ZEISS Auriga 60 high-resolution dual beam equipped with an ion gun to qualify the interface formation of our co-deposited thin film. Images were recorded using the InLens detector at 20 kV. A ToFSIMS5-100 (ION-TOF GmbH) instrument was used for the time-of-flight secondary ion mass spectrometry (ToF-SIMS) with the aim of resolving the elemental composition of the thin film. This spectrometer is equipped with a Bi cluster primary ion source (field emission from liquid Bi wetting a tungsten tip) and a reflectron type time-of-flight analyzer. Ultra-high vacuum (UHV) base pressure during analysis was < 3 × 10–8 mbar. For high mass resolution the Bi source was operated in the “high current bunched” mode providing short Bi+ primary ion pulses at 25 keV energy, a lateral resolution of approx. 4 μm, and a target current of 1.4 pA. The short pulse length of 1 ns allowed for high mass resolution (8000 m/∆m). The primary ion beam was scanned across a 250 µm2 × 250 µm2 field of view on the sample, and 64 × 64 data points were recorded. For depth profiling a dual beam analysis in interlaced mode was performed. The sputter gun (operated with Cs+ or O2+ ions, 2 keV, scanned over a concentric field of 500 µm2 × 500 µm2, and target current of 180 nA and 600 nA, respectively) was applied to erode the sample. No further sample preparation was required for this measurement. Grazing incidence x-ray diffraction (GIXD) spectroscopy was collected using a Panalytical X-Pert system with a CuKα radiation source at 40 kV and 35 mA at room temperature. The grazing incidence angle was set at 5° < θ < 60° and step size of 0.05° using a graphite flat crystal monochromator. Raman measurements were used to discern the crystallinity of the thin films and were run on a Renishaw inVia Raman microscope using a laser excitation wavelength (λe) of 532 nm, a laser power of approx. 2 mW, and a 100x NA0.85 objective lens. All Raman measurements were taken at room temperature as well.
Computational details
Our DFT calculations were carried out utilizing the Quantum Espresso54,55 package, which solves the Kohn–Sham equations by a plane-wave method. For the exchange-correlations term, the generalized gradient approximation (GGA) and the Perdew–Burke–Ernzerhof (PBE) options were chosen. The electrons’ distribution were described by the optimized Vanderbilt pseudopotentials56 provided through the SSSP package in its 1.2.1 version57. To account for the long-range forces inherently present in layered materials, we included the long-range dispersion correction DFT-D3 as described by Grimme et al.58 for an accurate description of the material. Structural visualization of the models was assisted by the VESTA59 and XCrysden60 codes. For the structural optimization calculations, the plane-wave cutoff energy was set to 470 eV, while the convergence criterion of ionic minimization was achieved when all forces were smaller than 5.1 × 10–2 eV nm−1 and the total energy changes less than 1.3 × 10–2 eV atom−1 in two consecutive self-consistent field steps. Additionally, a k-point set of 4 × 4 × 4 was used to sample in the Brillouin zone. During the structural optimization of the MoS2−xSex phases, all atoms were able to move freely, and the lattice dimensions were not fixed. For the electronic structure calculations, an increased plane-wave cutoff energy of 544 eV was used along with a denser k-point set of 10 × 10 × 10.
All models started with an optimized MoS2 unit cell with space group P63/mmc and lattice constant a = b = 0.3168 nm, c = 1.249067 nm, α = β = 90°, and γ = 120°, with a Mo-S bond distance (dMo-S) of 0.2406 nm. The ionic state of S and Se inside the lattice is as S2− and Se2−, both in a three-coordinate geometry bonded to three equivalent Mo4+ ions, the latter remaining in trigonal prismatic coordination. We created a 4 × 4 × 1 supercell from this model containing 32 molybdenum atoms and 64 sulfur atoms. This ensured modeling a wide variety of selenium concentrations avoiding self-interactions and achieved a robust and reliable model. For this study, we considered the evolution of the MoS2−xSex phase to occur from a selenium concentration of 4 at.% up to a 33.3 at. %, corresponding to a 1:1 relation between S and Se.
Two types of MoS2−xSex phases were considered, the first in a random configuration and the second in a Janus-type configuration. For the random MoS2−xSex phase, the selenium atoms substituted sulfur atoms and were distributed randomly throughout the supercell model. For the Janus MoS2-xSex phase, the selenium atoms replaced sulfur atoms in only one atomic layer of the MoS2 structure as depicted in Fig. 1. The stability analysis of these two atomic arrangements first considered the formation energy per atom, (Eform/eV atom−1), of the different MoS2−xSex phases, mimicking a co-deposit process, where all the atomic species converged to the formation of the targeted MoS2-xSex phase. This was computed using the formula:
where \(E_{{{\text{MoS}}_{{2 - {\text{x}}}} {\text{Se}}_{{\text{x}}} }}\) is the resulting enthalpy of the phase from the structural optimization calculations, N is the total number of atoms, and \(\mu_{i}\) is the chemical potential of the \(i\)th atom in the mentioned phase; this latter parameter was considered as the bulk energy of the atom. This approach considers that the distribution of selenium and sulfur atoms have an equal probability and that no other type of defect is left at the supercell afterward. Secondly, we were interested in the case where a possible inclusion of Se might occur into the MoS2 lattice considering initially a pristine MoS2 supercell and ultimately derive in the formation of the targeted MoS2-xSex phase. For this scenario, the substitutional energy per atom (Eseg/eV atom−1) was computed as follows:
where EMoS2 is the energy of the 4 × 4 × 1 defect-free MoS2 supercell. Here, µr is the chemical potential of the ith-atom that was substituted or released from its original position, and µs is the chemical potential of the ith-atom that substitutes the released atoms. As in the previous case, we consider that the occupation of sulfur positions by selenium atoms has an equal probability and that no other type of defect exists at the supercell before or afterwards.
Data availability
The data that support the findings of this report are available from the corresponding authors upon reasonable request.
References
Manuel, A. R. M. et al. Organic-semiconducting hybrid solar cell, US10727428B1, n.d. https://patents.google.com/patent/US10727428B1/en (2023).
Ramos, M. et al. Mechanical properties of RF-sputtering MoS2 thin films. Surf. Topogr. Metrol. Prop. 5, 025003. https://doi.org/10.1088/2051-672X/aa7421 (2017).
Zhang, K. et al. Manganese doping of monolayer MoS2: The substrate is critical. Nano Lett. 15, 6586–6591. https://doi.org/10.1021/acs.nanolett.5b02315 (2015).
Zhu, H. et al. Heteroatom doping of two-dimensional materials: From graphene to chalcogenides. Nano Today 30, 100829. https://doi.org/10.1016/j.nantod.2019.100829 (2020).
Chaudhary, R., Patel, K., Sinha, R. K., Kumar, S. & Tyagi, P. K. Potential application of mono/bi-layer molybdenum disulfide (MoS2) sheet as an efficient transparent conducting electrode in silicon heterojunction solar cells. J. Appl. Phys. 120, 013104. https://doi.org/10.1063/1.4955071 (2016).
López-Galán, O. A., Perez, I., Nogan, J. & Ramos, M. Determining the electronic structure and thermoelectric properties of MoS2 /MoSe2 type-I heterojunction by DFT and the Landauer approach. Adv. Mater. Interfaces 2023, 2202339. https://doi.org/10.1002/admi.202202339 (2023).
Yagmurcukardes, M., Sevik, C. & Peeters, F. M. Electronic, vibrational, elastic, and piezoelectric properties of monolayer Janus MoSTe phases: A first-principles study. Phys. Rev. B 100, 045415. https://doi.org/10.1103/PhysRevB.100.045415 (2019).
Guan, Z., Ni, S. & Hu, S. Tunable electronic and optical properties of monolayer and multilayer janus MoSSe as a photocatalyst for solar water splitting: A first-principles study. J. Phys. Chem. C 122, 6209–6216. https://doi.org/10.1021/acs.jpcc.8b00257 (2018).
Beshir, B. T., Obodo, K. O. & Asres, G. A. Janus transition metal dichalcogenides in combination with MoS 2 for high-efficiency photovoltaic applications: A DFT study. RSC Adv. 12, 13749–13755. https://doi.org/10.1039/D2RA00775D (2022).
López-Galán, O. A. et al. The electronic states of ITO-MoS2: Experiment and theory. MRS Commun. 12, 137 (2021).
Roy, T. et al. 2D–2D tunneling field-effect transistors using WSe2/SnSe2 heterostructures. Appl. Phys. Lett. 108, 083111. https://doi.org/10.1063/1.4942647 (2016).
Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V. & Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 6, 147–150. https://doi.org/10.1038/nnano.2010.279 (2011).
Wu, D. et al. Visualization of local conductance in MoS2/WSe2 heterostructure transistors. Nano Lett. 19, 1976–1981. https://doi.org/10.1021/acs.nanolett.8b05159 (2019).
Muratore, C. & Voevodin, A. A. Control of molybdenum disulfide basal plane orientation during coating growth in pulsed magnetron sputtering discharges. Thin Solid Films 517, 5605–5610. https://doi.org/10.1016/j.tsf.2009.01.190 (2009).
Imai, S. et al. Importance of crystallinity improvement in MoS2 film by compound sputtering even followed by post sulfurization. Jpn. J. Appl. Phys. https://doi.org/10.35848/1347-4065/abdcae (2021).
Hussain, S. et al. Large-area, continuous and high electrical performances of bilayer to few layers MoS2 fabricated by RF sputtering via post-deposition annealing method. Sci. Rep. 6, 30791. https://doi.org/10.1038/srep30791 (2016).
Zhong, W. et al. Feasible route for a large area few-layer MoS2 with magnetron sputtering. Nanomaterials 8, 590. https://doi.org/10.3390/nano8080590 (2018).
Conca, A., Ferreiro-Vila, E., Cebollada, A. & Martin-Gonzalez, M. Sputtering codeposition and metal-induced crystallization to enhance the power factor of nanocrystalline silicon. ACS Appl. Electron. Mater. https://doi.org/10.1021/acsaelm.2c01772 (2023).
Stupp, B. C. Synergistic effects of metals co-sputtered with MoS2. Thin Solid Films 84, 257–266. https://doi.org/10.1016/0040-6090(81)90023-7 (1981).
Solomon, G. et al. Decorating vertically aligned MoS2 nanoflakes with silver nanoparticles for inducing a bifunctional electrocatalyst towards oxygen evolution and oxygen reduction reaction. Nano Energy 81, 105664. https://doi.org/10.1016/j.nanoen.2020.105664 (2021).
Chia, H. L., Mayorga-Martinez, C. C. & Pumera, M. Doping and decorating 2D materials for biosensing: Benefits and drawbacks. Adv. Funct. Mater. 31, 2102555. https://doi.org/10.1002/adfm.202102555 (2021).
Pai, H.-C. & Wu, Y.-R. Investigating the high field transport properties of Janus WSSe and MoSSe by DFT analysis and Monte Carlo simulations. J. Appl. Phys. 131, 144303. https://doi.org/10.1063/5.0088593 (2022).
Zhao, N. & Schwingenschlögl, U. Transition from Schottky to Ohmic contacts in Janus MoSSe/germanene heterostructures. Nanoscale 12, 11448–11454. https://doi.org/10.1039/D0NR02084B (2020).
Singh, A., Jain, M. & Bhattacharya, S. MoS2 and Janus (MoSSe) based 2D van der Waals heterostructures: Emerging direct Z-scheme photocatalysts. Nanoscale Adv. 3, 2837–2845. https://doi.org/10.1039/D1NA00154J (2021).
Ramos, M. et al. Study of indium tin oxide–MoS2 interface by atom probe tomography. MRS Commun. 9, 1261–1266. https://doi.org/10.1557/mrc.2019.150 (2019).
Belete, M. et al. Dielectric properties and ion transport in layered MoS2 grown by vapor-phase sulfurization for potential applications in nanoelectronics. ACS Appl. Nano Mater. 1, 6197–6204. https://doi.org/10.1021/acsanm.8b01412 (2018).
Lu, A.-Y. et al. Janus monolayers of transition metal dichalcogenides. Nat. Nanotechnol. 12, 744–749. https://doi.org/10.1038/nnano.2017.100 (2017).
Petrić, M. M. et al. Raman spectrum of Janus transition metal dichalcogenide monolayers WSSe and MoSSe. Phys. Rev. B 103, 035414. https://doi.org/10.1103/PhysRevB.103.035414 (2021).
Han, Z. et al. Fabrication of patternable Janus transition-metal dichalcogenides assisted by electron beam irradiation. Appl. Phys. Lett. 120, 221901. https://doi.org/10.1063/5.0095650 (2022).
Tiwari, P., Jaiswal, J. & Chandra, R. Optical and electrical tunability in vertically aligned MoS2 thin films prepared by DC sputtering: Role of film thickness. Vacuum 198, 110903. https://doi.org/10.1016/j.vacuum.2022.110903 (2022).
Jaiswal, J., Das, A., Chetry, V., Kumar, S. & Chandra, R. NO2 sensors based on crystalline MoSe2 porous nanowall thin films with vertically aligned molecular layers prepared by sputtering. Sens. Actuat. B Chem. 359, 131552. https://doi.org/10.1016/j.snb.2022.131552 (2022).
Feng, L., Li, N. & Liu, Z. Effect of radio frequency power on composition, structure and optical properties of MoSex thin films. Phys. B Condens. Matter 444, 21–26. https://doi.org/10.1016/j.physb.2014.03.026 (2014).
Sirota, B., Glavin, N. & Voevodin, A. A. Room temperature magnetron sputtering and laser annealing of ultrathin MoS2 for flexible transistors. Vacuum 160, 133–138. https://doi.org/10.1016/j.vacuum.2018.10.077 (2019).
Chiappe, D. et al. Controlled sulfurization process for the synthesis of large area MoS2 films and MoS2/WS2 heterostructures. Adv. Mater. Interfaces 3, 1500635. https://doi.org/10.1002/admi.201500635 (2016).
Kim, T.-Y. et al. Transparent large-area MoS2 phototransistors with inkjet-printed components on flexible platforms. ACS Nano 11, 10273–10280. https://doi.org/10.1021/acsnano.7b04893 (2017).
Moser, J. & Lévy, F. Random stacking in MoS2−x sputtered thin films. Thin Solid Films 240, 56–59. https://doi.org/10.1016/0040-6090(94)90693-9 (1994).
Muratore, C. et al. Continuous ultra-thin MoS2 films grown by low-temperature physical vapor deposition. Appl. Phys. Lett. 104, 261604. https://doi.org/10.1063/1.4885391 (2014).
Choi, J. et al. Annealing temperature effect on the surface properties of the MoSe thin films. Phys. Status Solidi A 220, 2300477. https://doi.org/10.1002/pssa.202300477 (2023).
Wang, X. et al. Chemical vapor deposition growth of crystalline monolayer MoSe2. ACS Nano 8, 5125–5131. https://doi.org/10.1021/nn501175k (2014).
Schutte, W. J., De Boer, J. L. & Jellinek, F. Crystal structures of tungsten disulfide and diselenide. J. Solid State Chem. 70, 207–209. https://doi.org/10.1016/0022-4596(87)90057-0 (1987).
Jiang, H. Electronic band structures of molybdenum and tungsten dichalcogenides by the GW approach. J. Phys. Chem. C 116, 7664–7671. https://doi.org/10.1021/jp300079d (2012).
Zhang, J. et al. Janus monolayer transition-metal dichalcogenides. ACS Nano 11, 8192–8198. https://doi.org/10.1021/acsnano.7b03186 (2017).
López-Galán, O. A. & Ramos, M. Semimetal transition in curved MoS2/MoSe2 Van der Waals heterojunction by dispersion-corrected density functional theory. MRS Commun. https://doi.org/10.1557/s43579-022-00233-1 (2022).
Strasser, A., Wang, H. & Qian, X. Nonlinear optical and photocurrent responses in janus MoSSe monolayer and MoS 2 –MoSSe van der Waals heterostructure. Nano Lett. 22, 4145–4152. https://doi.org/10.1021/acs.nanolett.2c00898 (2022).
Rawat, A., Mohanta, M. K., Jena, N., Dimple, R. & Ahammed, A. D. S. Nanoscale interfaces of janus monolayers of transition metal dichalcogenides for 2D photovoltaic and piezoelectric applications. J. Phys. Chem. C 124, 10385–10397. https://doi.org/10.1021/acs.jpcc.0c02199 (2020).
Muratore, C., Voevodin, A. A. & Glavin, N. R. Physical vapor deposition of 2D Van der Waals materials: A review. Thin Solid Films 688, 137500. https://doi.org/10.1016/j.tsf.2019.137500 (2019).
Su, S.-H. et al. Band gap-tunable molybdenum sulfide selenide monolayer alloy. Small 10, 2589–2594. https://doi.org/10.1002/smll.201302893 (2014).
Li, H. et al. Lateral growth of composition graded atomic layer MoS2(1–x )Se 2 x nanosheets. J. Am. Chem. Soc. 137, 5284–5287. https://doi.org/10.1021/jacs.5b01594 (2015).
Ishihara, S. et al. Evaluation of sputtering deposited 2-dimensional MoS2 film by raman spectroscopy. MRS Proc. 1781, 11–16. https://doi.org/10.1557/opl.2015.563 (2015).
Wang, Y., Li, S. & Yi, J. Electronic and magnetic properties of Co doped MoS2 monolayer. Sci. Rep. 6, 24153. https://doi.org/10.1038/srep24153 (2016).
Komsa, H.-P., Kurasch, S., Lehtinen, O., Kaiser, U. & Krasheninnikov, A. V. From point to extended defects in two-dimensional MoS2: Evolution of atomic structure under electron irradiation. Phys. Rev. B 88, 035301. https://doi.org/10.1103/PhysRevB.88.035301 (2013).
Lin, Y.-C. et al. Low energy implantation into transition-metal dichalcogenide monolayers to form janus structures. ACS Nano 14, 3896–3906. https://doi.org/10.1021/acsnano.9b10196 (2020).
Gong, C., Colombo, L., Wallace, R. M. & Cho, K. The unusual mechanism of partial fermi level pinning at metal–MoS2 interfaces. Nano Lett. 14, 1714–1720. https://doi.org/10.1021/nl403465v (2014).
Giannozzi, P. et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502. https://doi.org/10.1088/0953-8984/21/39/395502 (2009).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 29, 465901. https://doi.org/10.1088/1361-648X/aa8f79 (2017).
Hamann, D. R. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 88, 085117. https://doi.org/10.1103/PhysRevB.88.085117 (2013).
Prandini, G., Marrazzo, A., Castelli, I. E., Mounet, N. & Marzari, N. Precision and efficiency in solid-state pseudopotential calculations. Npj Comput. Mater. 4, 72. https://doi.org/10.1038/s41524-018-0127-2 (2018).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104. https://doi.org/10.1063/1.3382344 (2010).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276. https://doi.org/10.1107/S0021889811038970 (2011).
Kokalj, A. XCrySDen—a new program for displaying crystalline structures and electron densities. J. Mol. Graph. Model. 17, 176–179. https://doi.org/10.1016/S1093-3263(99)00028-5 (1999).
Acknowledgements
Oscar A. López-Galán thanks the support from the KAAD from Germany for the granted postdoctoral fellowship, especially to Thomas Krüggeler, Renate Flügel, and Helen Meier for their helpful assistance. All authors thankfully acknowledge the high-performing computing resources and support provided by the Baden-Württemberg University Cluster 2.0 (bwUniCluster 2.0). All authors acknowledge the Karlsruhe Nano and Micro Facility of Karlsruhe Institute of Technology (KIT) for usage of time-of-flight secondary ion mass spectroscopy and electron microscopy equipment. OALG recognizes the support from Sergei Lebedkin from Institute of Nanotechnology (INT) at KIT during Raman measurements. Part of this work was performed at the Center for Integrated Nanotechnologies (CINT), an Office of Science User Facility operated by the U.S. Department of Energy (DOE) Office of Science. Sandia National Laboratories is a multi-program laboratory managed and operated by Sandia Corporation, a fully owned subsidiary of Lockheed Martin Corporation, for the U.S. Department of Energy’s National Nuclear Security Administration under contract DOE-AC04-94AL85000, and CINT proposal number 2022BC0102. Manuel Ramos and OALG thanks the program Sistema Nacional de Investigadores e Investigadoras from CONAHCyT-México, and Coordinación General de Investigación y Posgrado (CGIP) from Universidad Autónoma de Ciudad Juárez (UACJ) for project RIPI2020IIT5. All authors acknowledge the support from Subdirección de Vinculación e Intercambio Académico from UACJ for the agreement management between UACJ, CINT and KIT. All authors acknowledge the support of the KIT-Publication Fund of the KIT.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
OALG did computational calculations, and wrote the main manuscript. OALG and MR performed the fabrication and characterization of the thin films, and analyzed the results. JN assisted with the fabrication of thin films. TB and DC helped with the discussion, analysis of results, and SEM figures. AW did ToF-SIMS measurements and helped with data analysis. MH reviewed and helped with the discussion of the results. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
López-Galán, O.A., Boll, T., Nogan, J. et al. One-step sputtering of MoSSe metastable phase as thin film and predicted thermodynamic stability by computational methods. Sci Rep 14, 7104 (2024). https://doi.org/10.1038/s41598-024-57243-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-57243-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
